
Influence of the oxazole ring connection on the fluorescence of oxazoyl-triphenylamine biphotonic DNA probes†
Blaise
Dumat
ab,
Elodie
Faurel-Paul
ab,
Pauline
Fornarelli
ab,
Nicolas
Saettel
ab,
Germain
Metgé
c,
Céline
Fiorini-Debuisschert
c,
Fabrice
Charra
c,
Florence
Mahuteau-Betzer
*ab and
Marie-Paule
Teulade-Fichou
*ab
aInstitut Curie, PSL Research University, CNRS, INSERM, UMR9187/U1196, F-91405, Orsay, France. E-mail: florence.mahuteau@curie.fr; mp.teulade-fichou@curie.fr
bUniversité Paris Sud, Université Paris-Saclay, CNRS, UMR9187/U1196, F-91405 Orsay, France
cCEA-Saclay, DSM/IRAMIS/SPCSI, F-91191 Gif-sur-Yvette, France
First published on 18th November 2015
Abstract
On the basis of our previous work on DNA fluorophores derived from vinylpyridinium-triphenylamine, we explored the structure space around the electron-rich triphenylamine (TP) core by changing the vinyl bond to an oxazole ring. As 2,5-diaryloxazoles are known to be highly fluorescent and efficient two photon absorbers, we synthesized analogues with two different connections of the oxazole to the triphenylamine core: TP-Ox2Py and TP-Ox5Py sets. Since the benzimidazolium group was proven to be more effective in the TP series than the pyridinium, we also synthesized a TP-Ox5Bzim set. The TP-Ox5Py series retains the TP-Py properties: on/off behavior on DNA, good two-photon cross-section and bright staining of nuclear DNA by microscopy under both one or two-photon excitation. On the other hand, the TP-Ox2Py series does not display fluorescence upon binding to DNA. The TP-Ox5Bzim set is fluorescent even in the absence of DNA and displays lower affinity than the corresponding TP-Ox5Py. CD experiments and docking were performed to understand these different behaviors.
Introduction
Two–photon microscopy utilizes infra-red light to excite fluorescently-labeled samples through a non-linear two-photon absorption process leading to better light penetration in biological tissues, 3D confinement of the excitation and less photobleaching or photodamage of the samples.1–3 While these features are particularly promising for in-depth tissue and even small animal imaging,4–8 most usual fluorophores display poor two-photon absorption ability.9 Thus, the tremendous development of two-photon microscopy in the past decade has prompted chemists to design new fluorescent probes with enhanced two-photon cross-sections.10–19 The basic strategy to improve two-photon absorption is to increase the length of electronic conjugation and the charge-transfer during excitation.20–22 Despite the complex underlying theory, this is a quite straightforward design strategy but it soon meets its limitations due to the necessity to find the right balance between photophysical and biochemical properties which imposes additional constraints such as molecular weight limitations and water solubility. Adding to the difficulty, biological probes must often be targeted towards a specific biomolecule and/or cellular compartment and thus, suitable vectorization or binding specificity is required.We recently reported a series of cationic triphenylamine (TP) probes bearing two or three pyridinium groups with high two-photon absorption for cellular DNA imaging (TP-Py, Fig. 1).23 By combining the electron-rich TP core with two or three cationic acceptor groups through a conjugated linker, the TP scaffold allows building octupolar structures for the three-branched derivatives and V-shaped structures with a strong quadrupolar character for the two-branched derivatives.24 Due to both the charge transfer between donor and acceptor groups and the cooperative effect between the branches, such structures are expected to be efficient two-photon absorbers and are thus widely used in material science to build up non-linear optical compounds.25–30 The TP-Py set was however the first example of biocompatible TP dyes and the TP scaffold has since then been further used for the design of fluorescent probes for bioimaging.31–33 In order to improve the two-photon fluorescence properties of our TP dyes, we sequentially modified the donor core and the cationic acceptor groups of the original TP-Py. Replacing the TP core with a carbazole (Cbz-Py, Fig. 1) resulted in an increase of the fluorescence quantum yield due to the higher rigidity of the structure but decreased the two-photon absorption cross-section.34 On the other hand replacing the pyridinium acceptors by benzimidazolium groups resulted in a spectacular concomitant increase of the two-photon absorption cross-section and of the fluorescence quantum yield, affording one of the brightest two-photon probe to date among small organic fluorophores (TP-2Bzim, Fig. 1).35 All the different cationic TP dyes tested in fixed cell microscopy gave a specific staining of the nuclear DNA. Although the positive charge was shown to be necessary for DNA binding,36 the specific interaction with DNA and the observed AT sequence preference originate from the insertion of two branches of the TP dyes inside the DNA minor-groove.14
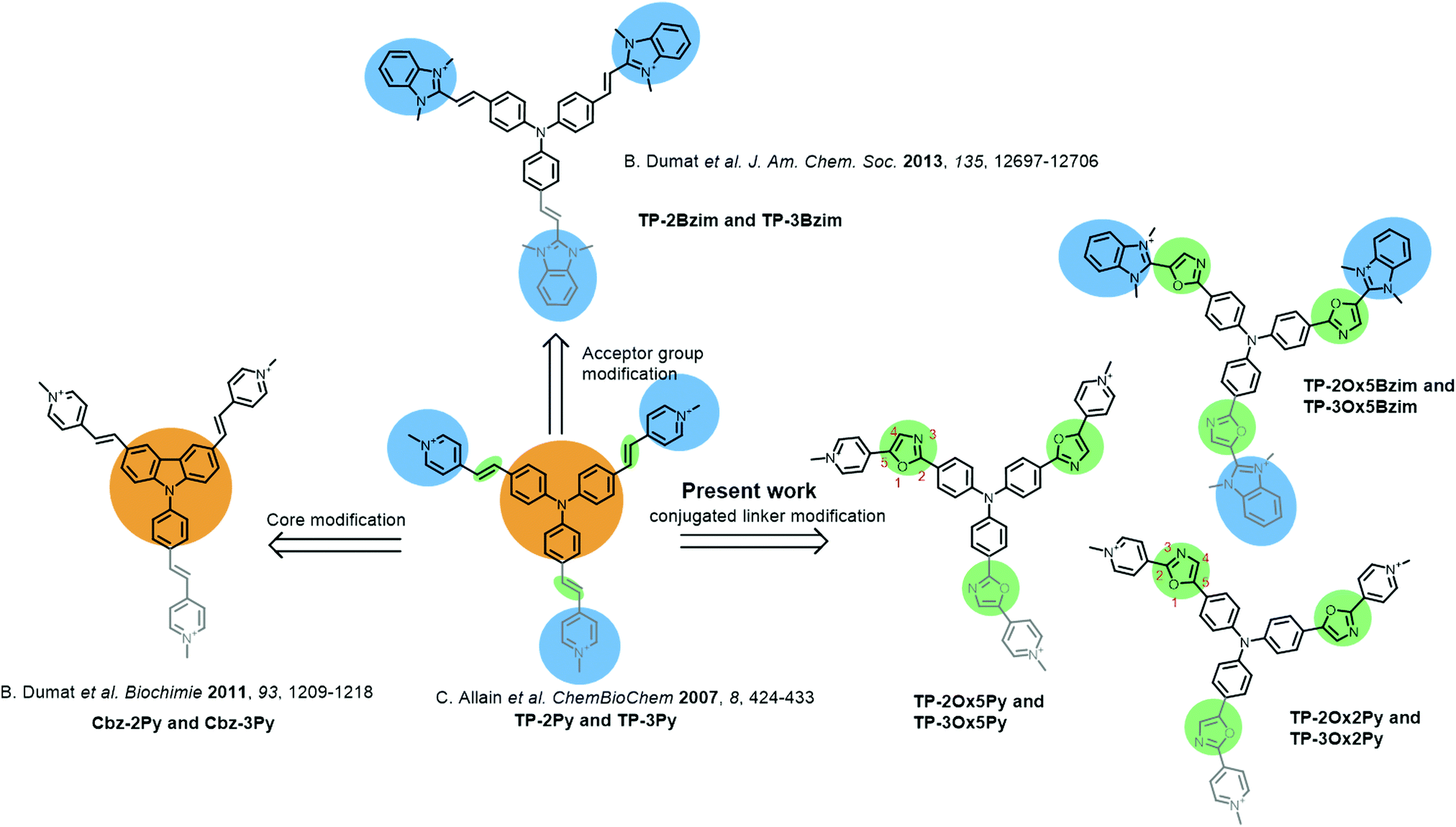 | ||
| Fig. 1 Structure of the different series of TP dyes exemplifying the successive modifications brought to the original TP-Py scaffold and showing the new TP-oxazole series reported herein. The third branch present only in compounds bearing 3 acceptor groups, TP-3X or Cbz-3X where X is the pyridinium (Py) or benzimidazolium (Bzim) acceptor group, is shown in gray. | ||
Pursuant to our continued interest in the triphenylamine biphotonic fluorescent DNA ligands, we set out to modify the third and last parameter of TP structures: the conjugated link between the donor core and the acceptor branches. We report herein on the synthesis and characterization of six new triphenylamine-based fluorophores where the double bond of the original TP structure was replaced with a 2,5-diaryloxazole group (Fig. 1). This new series of compounds was named TP-oxazoles to distinguish it from the previous TP series with a double bond linker.
The 2,5-diaryloxazoles possess attractive photophysical properties such as strong absorption, good Stokes shifts and high fluorescence quantum yields.37–39 Some of them were described as efficient two-photon absorbers.40 These properties have already been reported for applications in biphotonic fluorescence microscopy.41,42 Moreover, even if the TP dyes were proven to be highly photostable,14 the oxazole ring can be expected to be photochemically, but also metabolically, more stable than a vinylic bond which is reactive and prone to isomerization. Two different possible connections of the oxazole group were envisioned affording the TP-Ox5Py and TP-Ox2Py sets containing the two and three-branched derivatives (see oxazole ring numbering and compound naming in Fig. 1). Since the benzimidazolium group was proven to be more effective in the TP series than the pyridinium,14 we also synthesized a TP-Ox5Bzim set (Fig. 1).
The photophysical and DNA-binding properties of the six novel TP-oxazoles were investigated by UV-vis absorption, steady-state fluorescence and circular dichroism spectroscopies and selected dyes were also successfully applied to fixed cell DNA confocal and biphotonic imaging. Depending on the connectivity of the oxazole ring and on the nature of the acceptor group (pyridinium or benzimidazolium), the TP-oxazoles display a surprising variety of photophysical properties in comparison to the TP dyes and constitute a complementary family of versatile DNA probes.
Results and discussion
Synthesis
The preparation of oxazoles 2 was achieved by the van Leusen oxazole ring synthesis in quantitative yields (Scheme 1). The compounds 3 were synthesized by C–H bond activation of oxazoles 2 with 4-bromopyridine hydrochloride using our described protocol.43 They were obtained in moderate yields but two or three carbon–carbon bonds were formed during the reaction. The methylation step was performed with methyl tosylate to avoid the side methylation of the nitrogen on the oxazole rings that could occur with stronger alkylating agents and afforded the final salts TP-3Ox2Py and TP-2Ox2Py in high yields.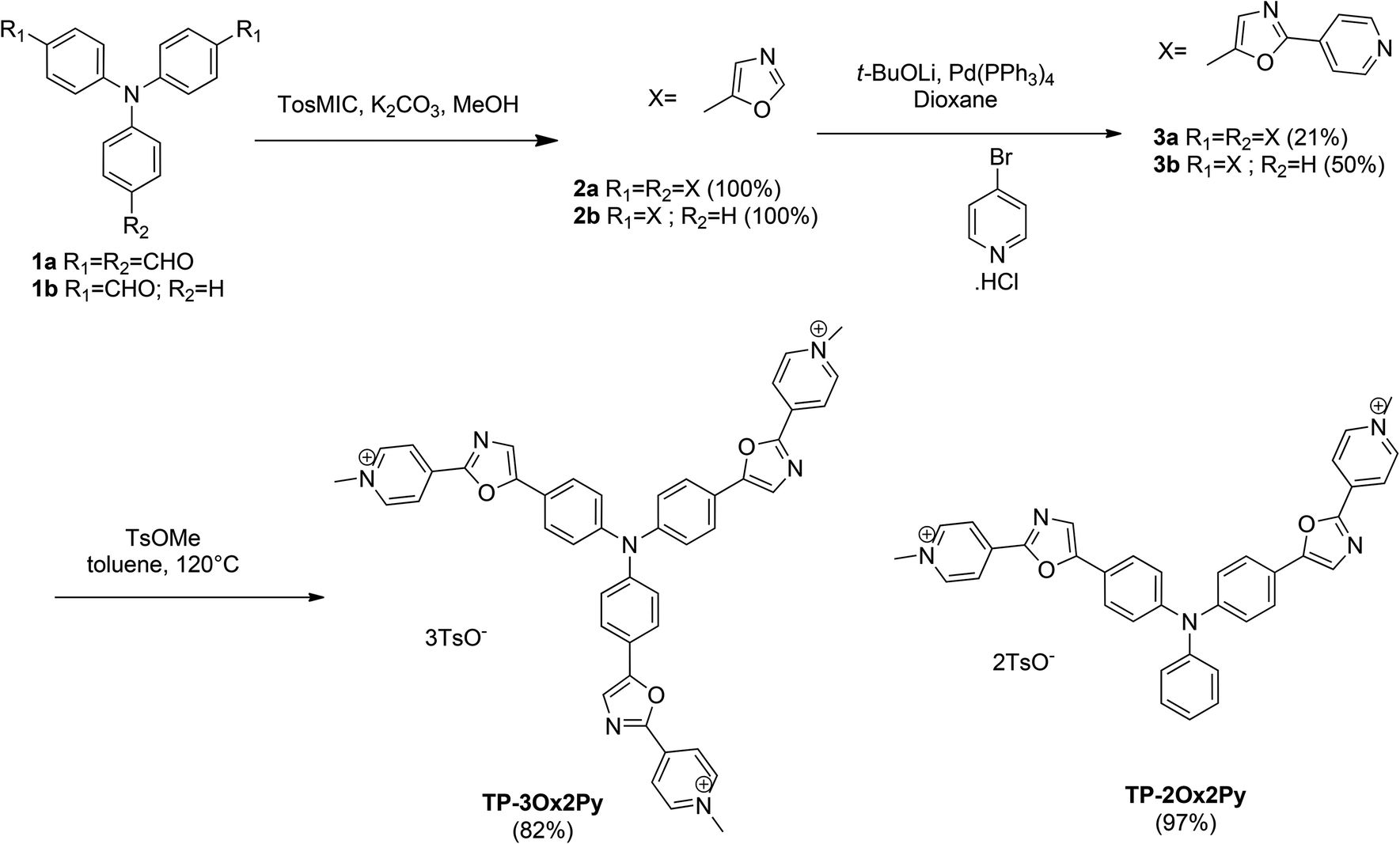 | ||
| Scheme 1 Synthesis of the TP-Ox2Py series. | ||
For the preparation of the regioisomers TP-2Ox5Py and TP-3Ox5Py (Scheme 2), the key reaction was also the direct arylation of oxazole. 5-Pyridyloxazole was synthesized according to the reported Van Leusen procedure and reacted with bis or tris-bromotriphenylamine to afford the compounds 5a or 5b in moderate yields for a two or three-bond forming reaction. The methylation occurred in good yields to afford the desired salts.
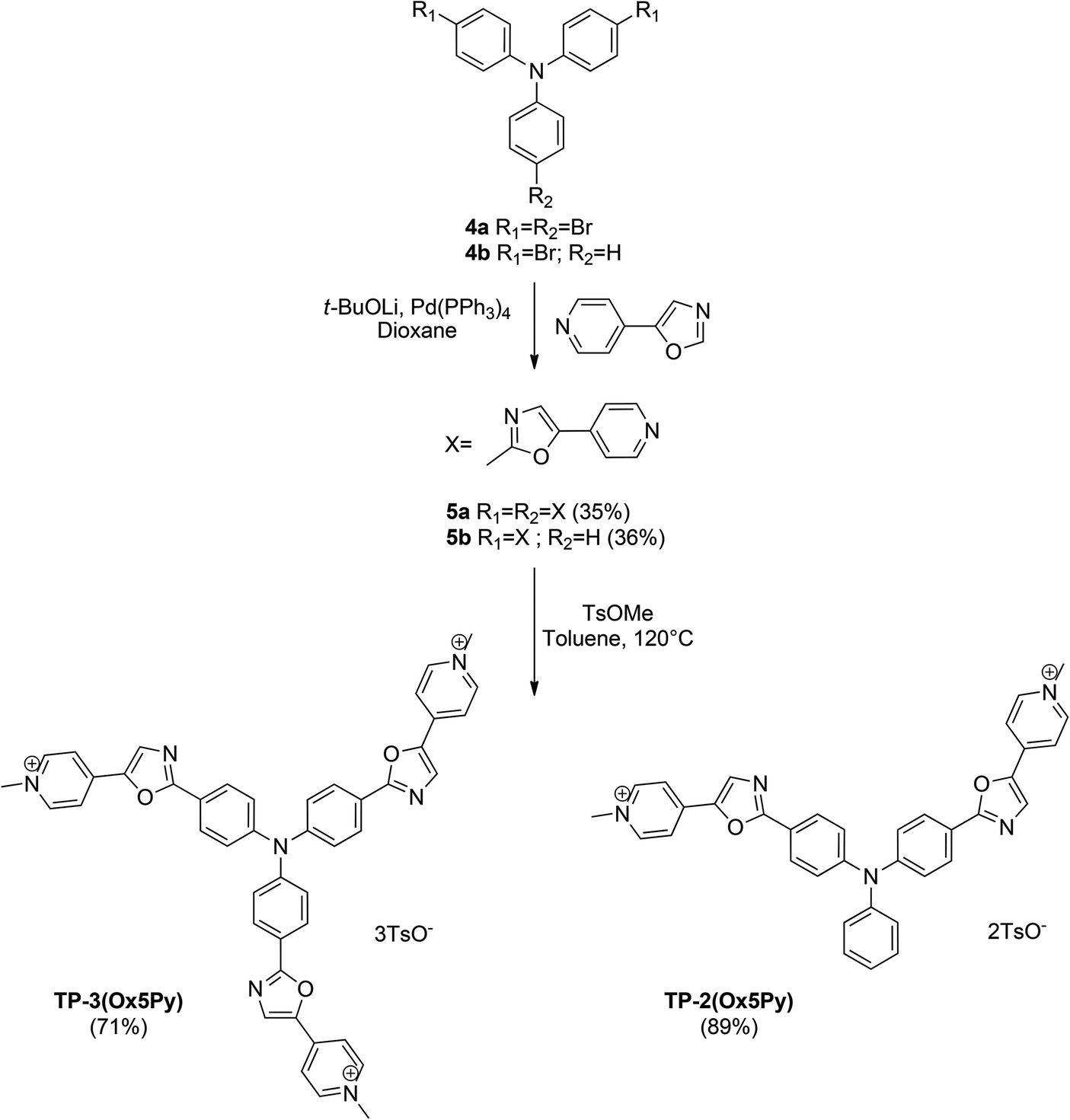 | ||
| Scheme 2 Synthesis of the TP-Ox5Py series. | ||
The same sequence was used to prepare TP-Ox5Bzim (Scheme 3). The 5-(1-methyl-1H-benzo[d]imidazol-2-yl)oxazole was prepared by a Van Leusen reaction starting from 1-methyl-1H-benzo[d]imidazole-2-carbaldehyde. The direct arylation of the 5-(1-methyl-1H-benzo[d]imidazol-2-yl)oxazole afforded the neutral intermediates 6a and 6b in good yield. Unfortunately, the methylation was not complete in both cases. TP-3Ox5Bzim was obtained in mixture with the bismethylated compound in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, TP-2Ox5Bzim was obtained in mixture with the monomethylated compound in the same ratio (see ESI†).
:1 ratio, TP-2Ox5Bzim was obtained in mixture with the monomethylated compound in the same ratio (see ESI†).
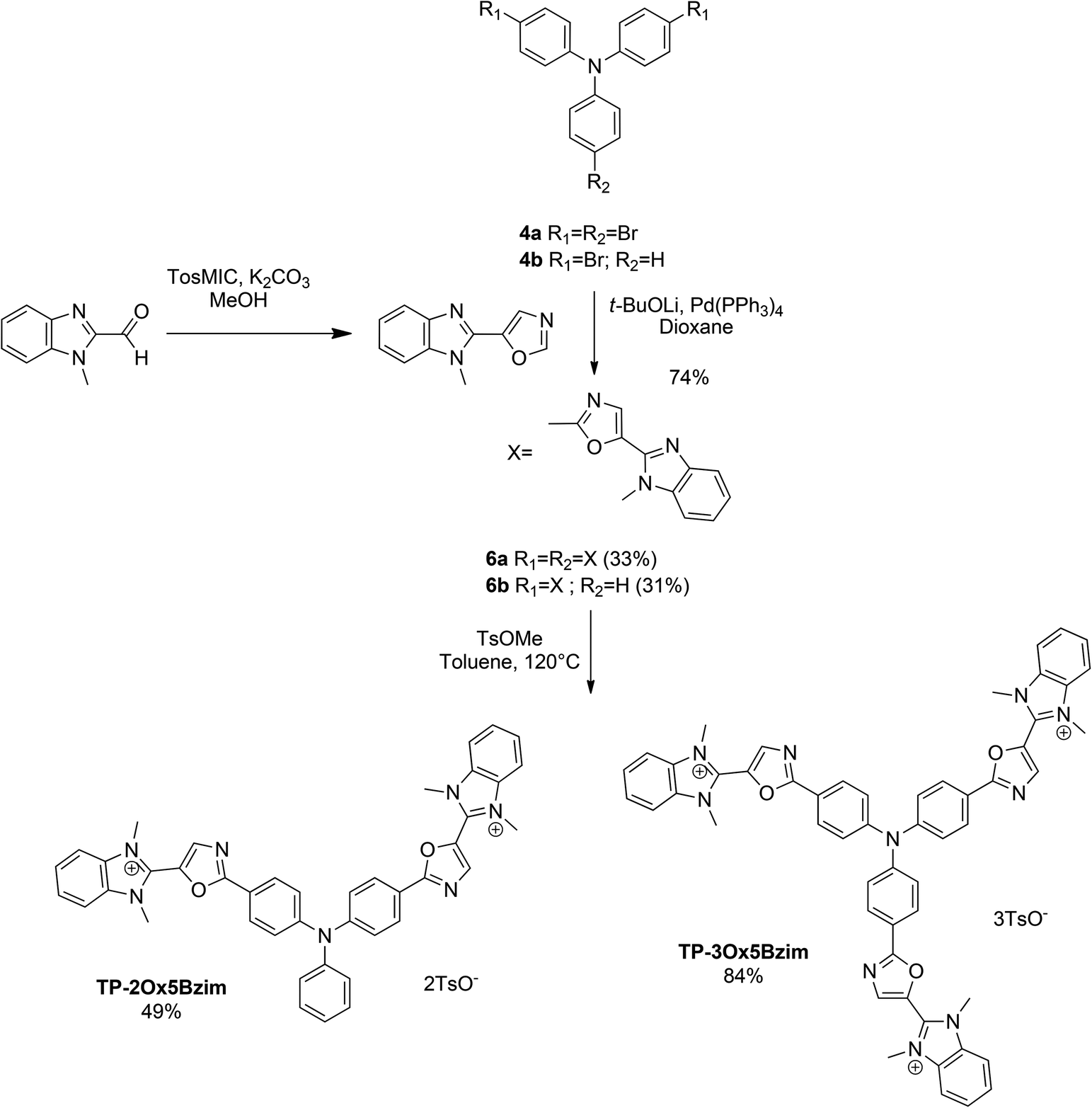 | ||
| Scheme 3 Synthesis of the TP-Ox5Bzim series. | ||
Photophysical properties
The linear and nonlinear optical properties of the TP-oxazole derivatives were measured in the presence of DNA in a sodium cacodylate buffer (pH 7.2) and are summarized in Table 1. The properties of the TP-Py and of TP-Bzim sets are recalled here for comparison. The ligands were studied in drewAT, a 14-mer palindromic sequence (5′-CGCGAAATTTCGCG) used to model AT-rich B-DNA duplexes.| λ abs, nm | λ em, nm | E, L mol−1 cm−1 | Φ F | ε·ΦF | λ 2PA, nm | δ GM | δ·ΦF | |
|---|---|---|---|---|---|---|---|---|
| TP-2(Ox5Py) | 466 | 631 | 21800 |
0.038 | 830 | 850 | 50 | 2 |
| TP-3(Ox5Py) | 444 | 606 | 45500 |
0.031 | 1400 | 820 | 115 | 4 |
| TP-2(Ox2Py) | 505 | — | 15600 |
— | — | — | — | — |
| TP-3(Ox2Py) | 480 | — | 60100 |
— | — | — | — | — |
| TP-2(Ox5Bzim) | 440 | 533 | 17400 |
0.17 | 2960 | nd | nd | nd |
| TP-3(Ox5Bzim) | 426 | 536 | 65500 |
0.28 | 18750 |
nd | nd | nd |
| TP-2Py | 509 | 656 | 31400 |
0.07 | 2200 | 840 | 260 | 18 |
| TP-3Py | 499 | 684 | 51000 |
0.01 | 510 | 840 | 460 | 5 |
| TP-2Bzim | 476 | 585 | 44400 |
0.54 | 24000 |
840 | 1080 | 583 |
| TP-3Bzim | 468 | 591 | 62400 |
0.34 | 21200 |
840 | 250 | 85 |
All compounds exhibit a broad lowest energy absorption band with maxima between 400 and 510 nm and strong molar absorption coefficients between 15000 and 30000 M−1 cm−1 for the two-branched derivatives and 45000 and 60000 M−1 cm−1 for the three-branched ones (Fig. 1 and Table 1). As it is often the case with DNA binders, a hypochromism is observed in presence of DNA (Fig. 2).
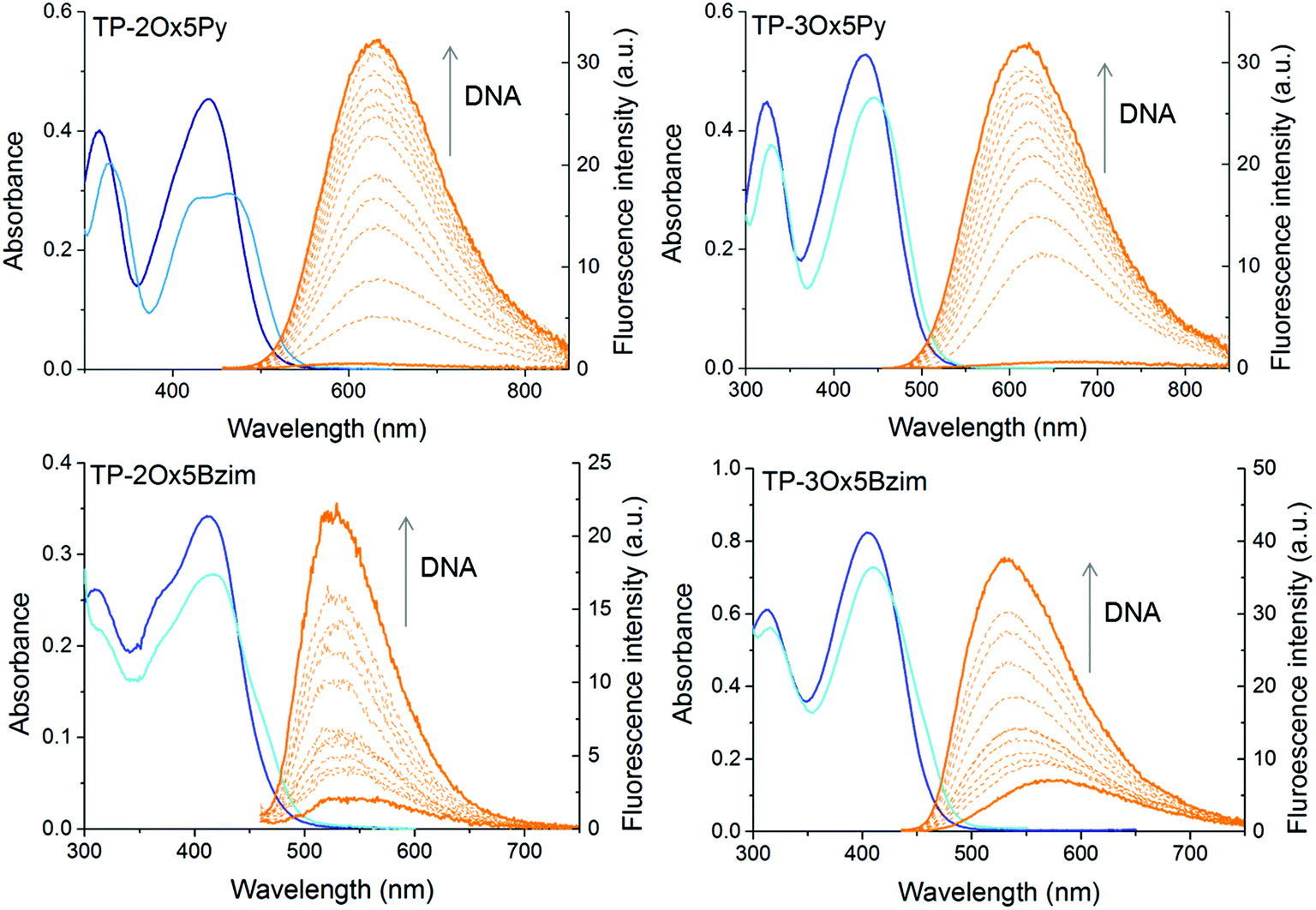 | ||
| Fig. 2 Absorption and emission spectra of the TP-Ox5Py and TP-Ox5Bzim in 10 mM sodium cacodylate pH 7.2 buffer with 400 mM NaCl. Absorbance: [TP-oxazole] = 10 μM free in solution (dark blue) or in the presence of 1 equivalent of drewAT (pale blue). Fluorescence: [TP-Ox5Py] = 1 μM, [drewAT] = 0–10 μM. [TP-3Ox5Bzim] = 0.25 μM, [drewAT] = 0–2.5 μM. | ||
In a general manner, the absorption of the TP-oxazoles is blue-shifted compared to that of their parent compound, pointing to a weaker electronic conjugation between the core and the branches through the oxazole ring. However, this shift depends strongly on the number of branches as well as on the connectivity of the oxazole ring and TP-2Ox2Py absorbs almost at the same wavelength as TP-2Py. As was the case between the TP-Py and the TP-Bzim, the absorption of the TP-Ox5Bzim is blue-shifted compared to that of the TP-Ox5Py but to a slightly lesser extent (18 to 26 nm versus 31 to 33 nm). Regardless of the connecting sense of the oxazole ring, the two-branched derivatives absorb at wavelengths significantly red-shifted of about 20–25 nm compared to their three-branched counterparts. This trend was also observed between TP-2Py and TP-3Py, but to a lesser extent (10 nm).
The TP-Ox5Py and TP-Ox5Bzim retain the fluorescence turn-on property of the TP dyes in DNA (Fig. 2). As was the case for the latter, the TP-Ox5Py are virtually non fluorescent when unbound in aqueous solution (ΦF < 0.005), they have a OFF/ON behaviour. On the contrary, the TP-Ox5Bzim exhibit a significant fluorescence quantum yield in the free state (ΦF = 0.06 for TP-3Ox5Bzim) which is further increased in the presence of DNA up to 0.28. The TP-3Ox5Bzim is much more fluorescent than the TP-Ox5Py already in glycerol (see ESI Table 1†), so a higher quantum yield in DNA was to be expected. However the fact that it is less prone to quenching in aqueous media remains unexpected and unexplained. Although they differ from the TP-Ox5Py only by the connectivity of the oxazole ring, the TP-Ox2Py derivatives surprisingly do not show the same fluorescence restoration in DNA. Like the other TP dyes, they are non fluorescent in the free state and no fluorescence enhancement is observed upon addition of drewAT. The TP-Ox2Py are fluorescent in glycerol (see ESI Table S1†), with a slightly lower but comparable fluorescence quantum yield than the TP-Ox5Py, which means that the absence of fluorescence originates from differences in the DNA binding.
The TP-Ox5Py have slightly lower monophotonic brightnesses (εΦF, Table 1) than the TP-Py, but they remain in the same order of magnitude and they display a higher fluorescence enhancement factor (Fmax/F0, Table 2). Their two-photon absorption maxima of 50 GM at 850 nm and 115 GM at 820 nm for the two and three-branched derivatives respectively are also significantly decreased compared to the parent TP-Py, but still remain very good compared to most of the commonly used fluorophores. On the contrary to the TP dyes, the three-branched derivative has the highest brightness, and TP-3Ox5Py could prove a fluorescent DNA turn-on probe just as efficient as TP-2Py.
| F max/F0 | K a (106 M−1) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F max/F0: ratio of the maximum fluorescence intensity measured at binding saturation and of the initial fluorescence intensity of the free dye. Ka: binding constant. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TP-2(Ox5Py) | 50 | 1.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TP-3(Ox5Py) | 40 | 2.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TP-2Ox5Bzim | 6 | 0.17 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TP-3Ox5Bzim | 9 | 0.21 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TP-2Py | 24 | 1.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TP-3Py | 14 | 4.4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TP-2Bzim | 140 | 12 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
With a strong absorptivity and a large fluorescence quantum yield in DNA, the monophotonic fluorescence brightness of TP-3Ox5Bzim is almost on par with that of TP-2Bzim (Table 1) which was the brightest dye of the TP series. TP-3Ox5Bzim thus represents a promising candidate for DNA imaging and since it also has a decent quantum yield in the free state it could also be used for other applications as a fluorescent probe unquenched in aqueous media.
The switchable fluorescence emission was explained in the case of the TP-Py and TP-Bzim dyes by the hydrophobic interaction with DNA which by sheltering the ligands from water and rigidifying their structure restores their fluorescence emission. It was demonstrated by CD experiments and molecular modeling that the vinyl-TP dyes bind to DNA by inserting the V-shaped motif formed by two of the branches in the minor groove.14 Despite having a very similar structure, the different TP-oxazole derivatives display very diverse photophysical properties in DNA depending on the connectivity of the oxazole ring and on the choice of the electron-poor substituent. To assess whether or not these differences originate from different DNA-binding abilities or lack thereof, we looked at the affinity and binding mode of the dyes to DNA via fluorimetric titrations, circular dichroism and docking experiments.
DNA affinity and binding mode
In the case of the fluorescent dyes TP-Ox5Py and TP-Ox5Bzim, the interaction with DNA can be monitored by recording the increase in fluorescence intensity upon addition of drewAT (Fig. 3). By plotting the fluorescence enhancement factor F/F0versus the concentration of drewAT, we obtained fluorimetric titration curves that were fitted to a one-site binding model function to determine binding constants as we described in a previous paper (Fig. 3).14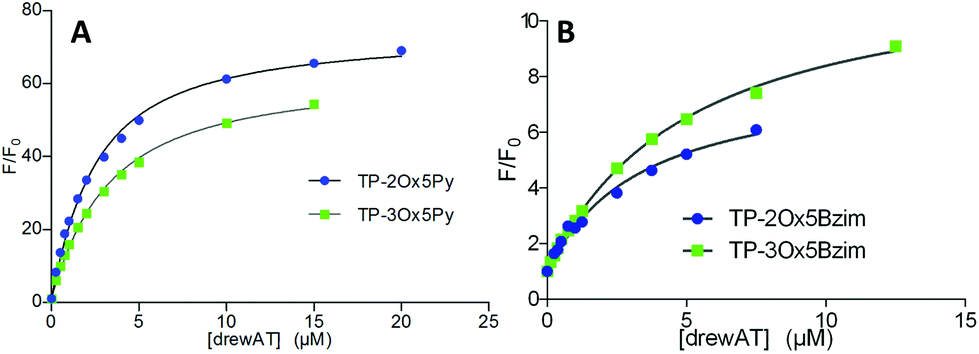 | ||
| Fig. 3 Fluorimetric titrations of (A) the TP-Ox5Py and (B) the TP-Ox5Bzim compounds upon addition of drewAT, fitted to a one-site binding model. [TP-Oxazole] = 0.25 μM in 10 mM sodium cacodylate pH 7.2 buffer with 400 mM NaCl. F: integrated fluorescence intensity for a given concentration of added drewAT. F0: initial integrated fluorescence intensity without drewAT. | ||
The model fitted the experimental data with good accuracy (Fig. 3 and ESI†), showing that, like the TP dyes, the TP-oxazoles bind to drewAT with a 1:1 stoichiometry. The affinity constants were found to be around 106 M−1 for both two and three-branched TP-Ox5Py (Table 2), which is in the same range as the parent TP-Py compounds (Table 2). In the case of the TP-Ox5Bzim, the fluorescence enhancement is much lower due to the fluorescent character of the free dye and the absence of signal saturation suggests a weaker and less specific binding. In the absence of a plateau the curve fitting is less reliable but the calculated affinity constants were found to be one order of magnitude below that of the TP-Py and TP-Ox5Py, which agrees well with the experimental observation.
To monitor the interaction of the non fluorescent TP-Ox2Py with DNA and also to determine the binding mode of all the compounds, we used circular dichroism spectroscopy. After verifying that the achiral dyes alone display no CD signal, we titrated the dyes into a drewAT solution and followed the evolution of the induced CD. For TP-2Ox5Py, a strong induced CD signal was observed in the ligand absorption region, which is a characteristic sign of minor groove binding (Fig. 4). More surprisingly the TP-2Ox2Py displays similar induced CD spectra (Fig. 4) which strongly suggests that this dye interacts with DNA in a similar fashion as the TP-Ox5Py, TP-Py and TP-Bzim despite their different fluorescence properties that may have suggested a lack of binding to DNA. The three-branch compounds TP-3Ox5Py and TP-3Ox5Bzim display the same behavior (see ESI, Fig. 1†).
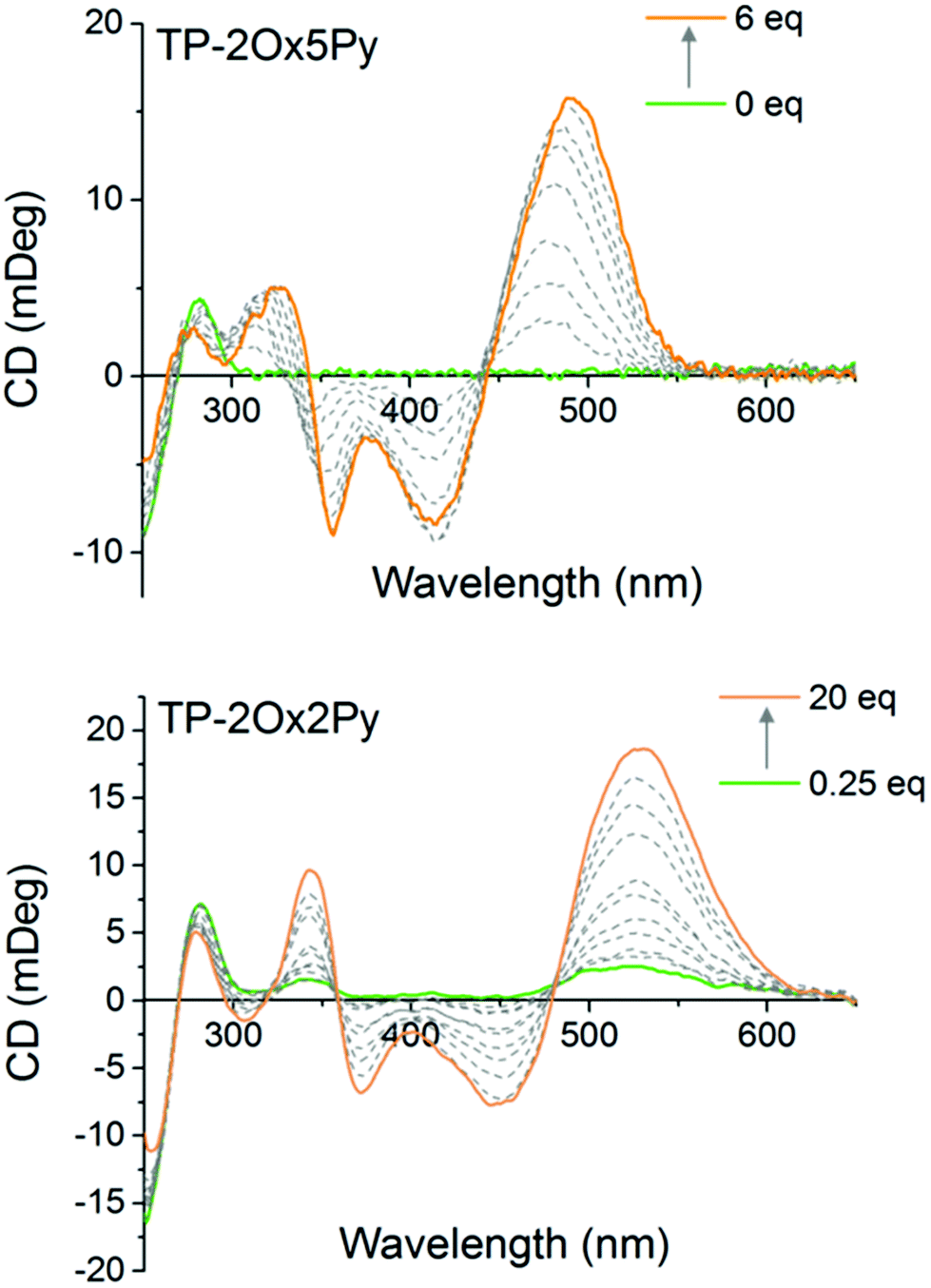 | ||
| Fig. 4 Circular dichroism spectra of drewAT upon addition of TP-2Ox5Py, TP-2Ox2Py ligands. Increasing amounts of dyes (molar equivalents indicated on the graphs) are added to a 2 μM solution of drewAT in 10 mM sodium cacodylate pH 7.2 buffer with 100 mM NaCl. | ||
The CD data together with the fluorimetric titration show that the TP-Ox5Py compounds bind to DNA with similar affinity and in a similar fashion as their parent compounds TP-Py. The TP-Ox5Bzim also bind within the minor-groove of DNA, despite a lower affinity constant and the absence of fluorescence saturation at high dye to DNA ratio which suggests that non-specific binding is also occurring. Although affinity constant could not be measured due to the lack of changes in the fluorescence intensity, the TP-Ox2Py also seems to interact with DNA with the same binding mode. To gain a finer insight into the binding mode of the dyes and explain the difference in binding affinity and photophysical properties, we performed docking studies of the dyes in drewAT as was previously done with the TP dyes (Fig. 5).14
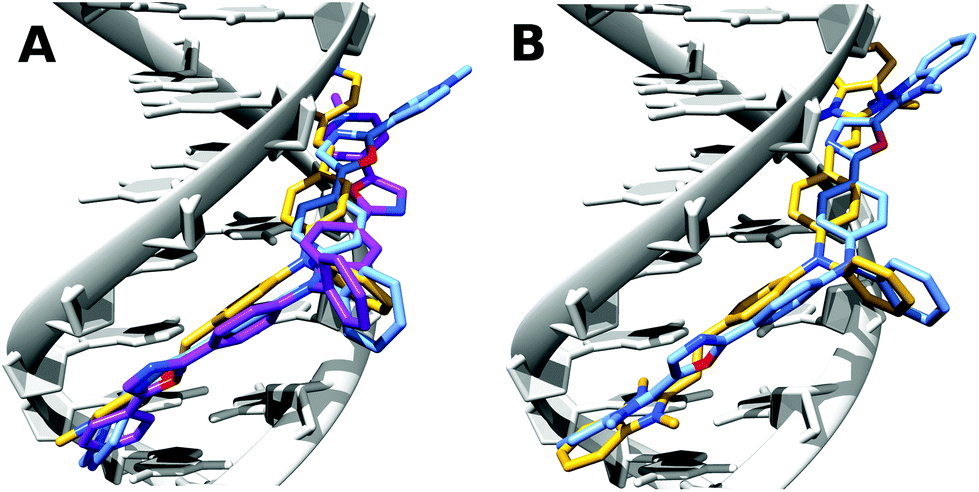 | ||
| Fig. 5 Docking of (A) TP-2Py (yellow), TP-2Ox5Py (purple) and TP-2Ox2Py (pale blue) and (B) TP-2Bzim (yellow) and TP-2Ox5Bzim (pale blue) in drewAT. | ||
As expected from the CD spectra, a good pose was found by the docking program in the DNA minor-groove for these dyes. The TP-2Ox5Py binds similarly as TP-2Py, this explains the very similar properties and binding affinity between the TP-Py and TP-Ox5Py sets. In the case of the TP-2Ox2Py, one of the pyridinium points out of the groove and is then exposed to water. This can explain the absence of restoration of the fluorescence. Moreover, although TP-2Ox5Py adopts a quasi-planar structure, TP-2Ox2Py does not. The angles between the oxazole rings and the pyridiniums are 42°, 36° for TP-2Ox2Py and 5° and 24° for TP-2Ox5Py. This absence of planarity of the structure in DNA can also be an explanation for the non fluorescent character of the TP-Ox2Py series in DNA. For TP-2Ox5Bzim, similar trends can be observed: one benzimidazolium group is out of the groove and the angle between this benzimidazolium and the corresponding oxazole is 41°. This may be a reasonable explanation for the lower affinity for DNA observed for this dye compared to TP-2Bzim.
Cellular imaging
The compounds were first tested in confocal microscopy (Fig. 6 top panel). Live Hela cells were stained with 2 μM dye for 2 hours and washed before fixation and preparation of the microscope slides. The two TP-Ox5Py display a bright and contrasted staining of the nuclear DNA with no RNA binding as attested by the absence of signal in the nucleoli. In particular, the excellent contrast between the residual fluorescence in the cytoplasm and the nucleus exemplifies the high turn-on ratio of these DNA probes. This behavior is similar to that of the TP-Py and TP-Bzim dyes and consistent with the in vitro properties. Live-cell microscopy has shown that the TP dyes are actually trapped in the mitochondria of living cells and only accumulate in the nucleus upon apoptosis or during the cell fixation process.44 The behavior of the TP-Ox5Py dyes is the same (data not shown). Despite a high fluorescence quantum yield and an in vitro DNA binding ability, the staining of TP-3Ox5Bzim is essentially cytoplasmic with only a faint fluorescence signal in the nucleus. This may be due to a competitive binding to a cytoplasmic organelle, since the affinity of the TP-Ox5Bzim was measured to be at least one order of magnitude lower than that of the other TP and TP-Ox5Py dyes. The TP-Ox5Bzim were thus not further investigated in two-photon excited fluorescence microscopy.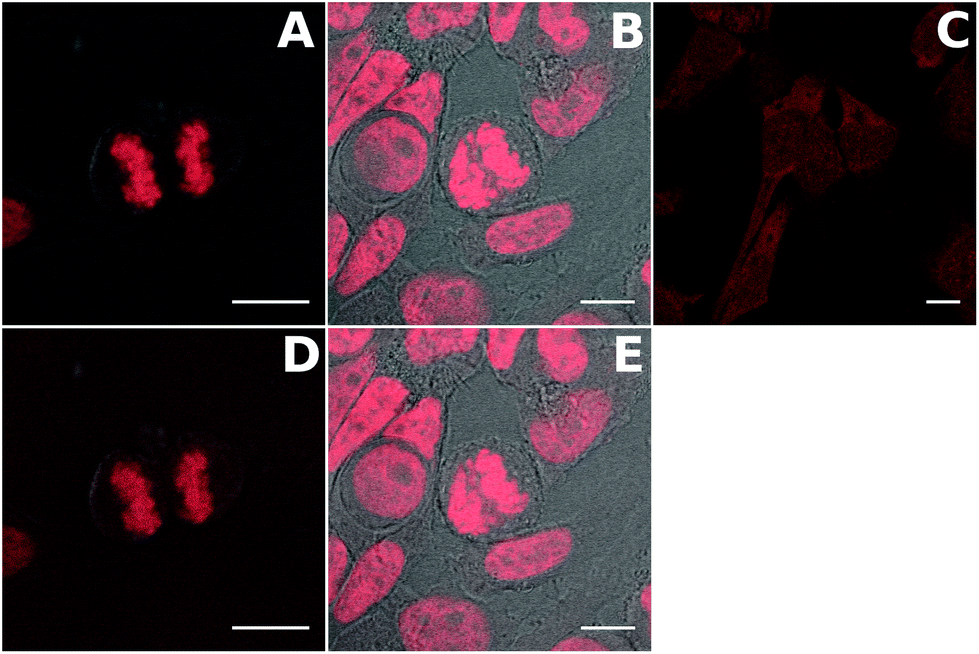 | ||
| Fig. 6 Merged fluorescence and transmission images of fixed Hela cells stained with 2 μM TP-2Ox5Py (A and D) or TP-3Ox5Py (B and E) or TP-3Ox5Bzim (C). Images A, B and C were obtained under one photon excitation (λexc = 488 nm); images D and E under two-photon excitation (λexc = 800 nm). Scale bar is 10 μm. Incubation was performed on live cell and the cells fixed after washing the excess dye. | ||
The TP-Ox5Py were tested in two-photon excited fluorescence microscopy under the same condition (Fig. 6) and gave similar images as in confocal imaging, with bright images. This underlines that despite having much lower two-photon absorption cross-sections than the TP-Bzim set, the TP-Ox5Py remain very good two-photon probes compared to the vast majority of classical fluorescent probe.
Conclusions
A series of six novel triphenylamine-oxazole derivatives with pyridinium or benzimidazolium electron accepting group and with two different possible connecting sense of the oxazole group were synthesized using a direct arylation reaction as a key synthesis step. The substitution of the vinylic bond by an oxazole ring in the TP dye scaffold does not suppress the DNA binding ability and more importantly it enables us to substantially modulate the fluorescence properties of the ligands through minor structure modifications.When the oxazole is tethered to the triphenylamine on position 2 with a pyridinium on position 5, the resulting compounds TP-2Ox5Py and TP-3Ox5Py retain the general properties of the parents TP-Py: they are turn-on fluorescent DNA minor-groove binders with a 50 to 70-fold fluorescence enhancement in DNA, binding affinities around 106 M−1 and fluorescence quantum yield between 3 and 4%. The TP-Ox2Py o the other hand are very poorly fluorescent in aqueous solution, even in the presence of DNA, although CD titration and docking calculations clearly show that they bind to the minor-groove of DNA as well. The TP-Ox2Py is thus a family of non-fluorescent minor-groove binders with strong absorption in the 400–500 nm range which could be used as non-covalent fluorescence acceptor in FRET studies inside nucleic acids systems for instance.
The replacement of the pyridinium group with a benzimidazolium afforded the TP-Ox5Bzim which are fluorescent even in the free form with a quantum yield of 6% for the three-branched compound. In DNA a further increase in the fluorescence is observed affording a very high brightness of 18750 for TP-3Ox5Bzim. TP-3Ox5Bzim has a lower affinity for DNA than its pyridinium counterpart and it does not stain DNA in fixed cell confocal microscopy but its very high brightness makes it a very valuable long-wavelength fluorophore unquenched in aqueous media.
TP-3Ox5Py was successfully applied to cellular imaging of DNA in fluorescence microscopy with comparable performance as the TP-Py. A fine and selective staining of the nucleus is observed both in confocal and two-photon excited fluorescence microscopy although in the latter the lower absorption cross-section results in significantly lower contrast compared to other TP-dyes.
In conclusion, we demonstrated herein that the oxazole appeared as an elegant way of obtaining a series of structurally similar molecules with highly versatile properties: (i) the TP-Ox2Py are non fluorescent DNA ligands with a strong and broad absorption and may be used as fluorescence quencher in DNA (ii) TP-3Ox5Bzim is a bright fluorophore suitable for aqueous media and (iii) the TP-Ox5Py are turn-on DNA probe that may offer a more metabolically stable alternative to the TP dyes for in vivo applications.
Experimental section
Synthesis
All chemicals were purchased from Sigma-Aldrich (USA) or Acros Organics (Belgium) and used as received. Preparative flash chromatographies were carried out with Merck silica gel (Si 60, 35–70 μm). 1H NMR spectra were recorded at 300 MHz on a Bruker Advance 300 spectrometer. Chemical shifts are reported in ppm downfield to TMS (δ = 0.00). High-performance liquid chromatography was carried out on a Waters Alliance equipped with a photodiode array detector using an XterraMS column with a linear gradient of acetonitrile versus water containing 0.05% of TFA ranging from 2 to 30% over 5 min then to 100% over 9 min at a flow rate of 1 mL min−1. Electrospray ionization mass spectrometry (ESI-MS) was performed at the Institut Curie and HRMS were performed at the Small Molecule Mass Spectrometry platform of IMAGIF (ICSN, Gif-sur-Yvette, France).:1) as a yellow solid (66 mg, 0.072 mmol) in 84% yield. 1H NMR (300 MHz, DMSO-d6) δ 8.66 (s, 3H), 8.25 (d, J = 9.0, 6H), 8.16 (m, 6H), 7.74 (m, 6H), 7.44 (d, J = 9.0, 6H), 7.38 (d, J = 9.0, 6H), 7.08 (d, J = 9.0, 6H), 4.27 (s, 18H), 2.25 (s, 9H); 13C NMR (75 MHz, DMSO-d6) 164.1, 149.1, 145.7, 139.1, 138.0, 137.6, 134.5, 132.1, 129.2, 128.9, 128.1, 127.1, 125.5, 120.8, 113.4, 33.7, 20.8; MS (ES+) m/z 293.9; HRMS (ESI) calcd for C54H45N10O3 [M+] 881.3676, found 881.3661.
:1) as a yellow solid (24 mg, 0.0024 mmol) in 49% yield. 1H NMR (300 MHz, DMSO-d6) δ 8.64 (s, 2H), 8.20–8.15 (m, 7H), 7.79–7.67 (m, 6H), 7.45 (d, J = 8.0 Hz, 4H), 7.30–7.25 (m, 8H), 7.09 (d, J = 8.0 Hz, 4H), 4.26 (s, 12H), 2.27 (s, 6H); MS (ES+) m/z 334.6, 840.3 [M + C7H7SO3]; HRMS (ESI) calcd for C49H42N7O5S [M + C7H7SO3] 840.2968, found 840.2972.
Oligonucleotide sequence
DrewAT (5′-CGCG-AAATTTCGCG-3′) were purchased from Eurogentec (Belgium). This autocomplementary strand was heated at 90 °C for 5 min in a 10 mM sodium cacodylate buffer pH 7.2, 100 mM NaCl (approximately duplex concentration 200 μM) and then slowly cooled to room temperature over 12 h. Once prepared, oligonucleotides solutions were stocked at 4 °C. Before use, the concentrations were evaluated by UV (after thermal denaturation 5 min at 90 °C) using the supplier's values for the molar extinction coefficients at 260 nm.Fluorescence and absorption spectroscopies
UV-vis experiments were monitored on a Uvikon XL (Secomam, France) UV spectrophotometer. Fluorescence spectra were recorded on a FluoroMax-3 (Jobin Yvon), at room temperature. Measurements were performed with solutions of OD < 0.1 to avoid re-absorption of the emitted light, and data were corrected with a blank and from the variations of the detector with the emitted wavelength. All experiments were performed in 10 mM sodium cacodylate buffer pH 7.2.Fluorescence quantum yield were measured according to Williams comparative method using quinine bisulfate in 1 N H2SO4 as reference. Absorption and fluorescence spectra were recorded for five solutions of increasing concentrations with an absorbance comprised between 0.01 and 0.1 to avoid reabsorption phenomenon. The fluorescence of the sample and the reference are then plotted versus the absorbance. The data should correlate linearly and after calculating the gradient of the curve, the fluorescence quantum yield is given by eqn (1),
| Φx = Φref (Gradx/Gradref) × (ηx2/ηref2) | (1) |
The variation of the fluorescence signal of a fluorophore solution of fixed concentration (1 μM or 0.25 μM) was monitored upon addition of increasing quantities of an oligonucleotide. A quick UV titration of the fluorophore by the oligonucleotide was first performed to determine the isobestic point when there is one. The excitation wavelength was then chosen so as to minimize the variation of the absorbance upon addition of DNA.
The titration curves were obtained by plotting F/F0versus the concentration of oligonucleotide, where F is the integrated fluorescence intensity of the oligonucleotide-dye complex and F0 the initial integrated fluorescence intensity of the free dye. Data were analyzed using eqn (2) derived from the mass-action law in Graphpad Prism 5.0 software:
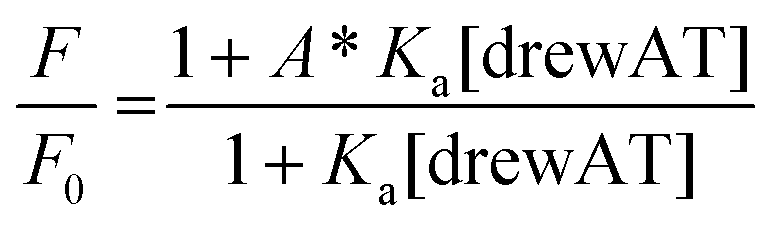 | (2) |
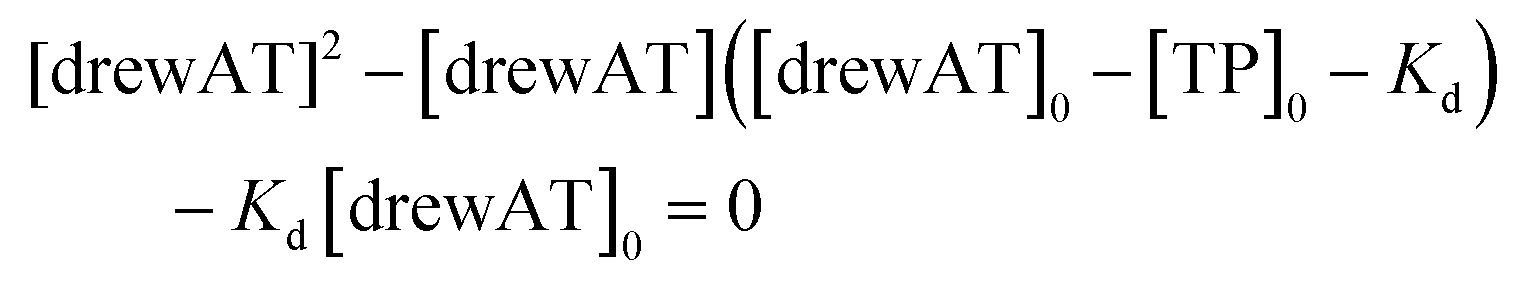 | (3) |
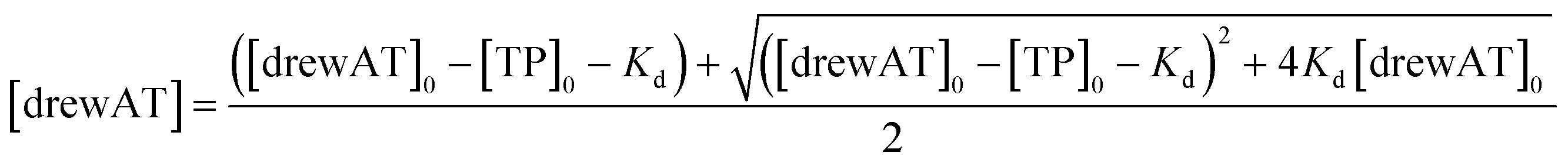 | (4) |
Circular dichroism
The CD spectra were recorded on a Jasco J-710 spectrometer in a 1 cm quartz cuvette thermostated at 22 °C. The DNA concentration was kept constant with increasing concentrations of dyes. Each spectrum is the accumulation of four scans performed with a 1 nm step and a 200 nm min−1 scanning speed.Two-photon absorption cross-section measurements
Two photon induced fluorescence measurements were performed using an inverted microscope set up coupled to a mode locked Ti-sapphire laser (Tsunami, Spectra Physics) delivering 100 fs pulses with a 76 MHz repetition rate over the spectral range covering 740 to 950 nm. The Ti-Saph excitation beam was focused in a cell filled with low concentration solutions using a 40× microscopic objective, the same objective being also used for the two-photon fluorescence signal collection. The signal is then sent either to a channel plate multiplier (Perkin Elmer MP-993-CL) or to a spectrometer coupled to a CCD camera (Andor DU401-BR-DD) for detailed study of the emission spectra. The laser beam was linearly polarized and a set of half-wave plate and polarizer was used to vary the fundamental beam intensity (700 μW at the level of the sample). Excitation spectra were determined from measurements of the whole emitted light using a photomultiplier associated to filters cutting the fundamental beam (SemRock razor edge 785, stopline 785 and 808, FF735 and FF-01-750). The TPIF intensities of the samples were measured relative to a solution of fluorescein in water (pH = 13), the ratio of the fluorescent signals enabling further determination of the 2PA cross section (δ) assuming equal one and two-photon fluorescence quantum yields.9Cellular culture and confocal and two-photon excited fluorescence microscopy
Cells were grown on coverslips at 37 °C in monolayer cultures in complete DMEM (Gibco, Cergy Pontoise, France) with 10% FCS and antibiotics (100 mg per ml streptomycin and 100 mg per ml penicillin) under conditions of 100% humidity, 95% air and 5% CO2 for 24 hours. The cells were then incubated for two hours with the desired compounds (2 μM). After washing twice with PBS, the cells were fixed with 4% paraformaldehyde and washed with PBS twice. The coverslips were then mounted on microscope slides using Prolong Gold antifade reagent (Invitrogen).The confocal and two-photon excited fluorescence microscopy was performed using a confocal laser scanning microscope DMI 6000 with a SP5-AOBS unit (both Leica) equipped with a 63× (NA = 1.4) objective (oil immersion), an argon gas laser for one-photon excitation and a Chameleon Ti:Sappphire laser delivering pulses in the 100 to 200 fs range at an 80 MHz repetition rate with a tunability ranging from 705 to 980 nm for two-photon excitation.
Molecular modeling
Ligand drawing, standardization (protonation state at physiological pH, aromatization, hydrogen treatment), and 3D generation were done with Chemaxon Marvin Beans. Docking was performed with the Surflex-Dock engine of the Tripos SYBYL-X 2.0 suite. The modified GeomX mode was used throughout the calculations with the option Allow DNA movement: six additional starting conformations were generated, pre- and postdock minimizations were performed, a maximum of three final poses were saved, with a minimum of 0.5 RMSD between poses. The spin alignment method was used, with an exhaustive accuracy (density of search 9.0) and 12 number of spins per alignment. A DrewAT-like structure was built using SYBYL-X 2.0 (biopolymer builder module), and the binding site centered on the central AAATTT sequence was determined by protomol probes (CH4 as a steric hydrophobic probe, NH as a H-bond donor probe, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O as a H-bond acceptor probe). The docking results are ranked based on an empirical scoring function including hydrophobic, polar, repulsive, entropic, solvation, and crash terms and expressed in-log10(Kd) units to represent binding affinities.
O as a H-bond acceptor probe). The docking results are ranked based on an empirical scoring function including hydrophobic, polar, repulsive, entropic, solvation, and crash terms and expressed in-log10(Kd) units to represent binding affinities.
Acknowledgements
ANR-07-PCVI-0005-01 (EPATAN) and NanoBioIdf (LabelDNA) are acknowledged for fundings and in particular for post-doctoral fellowship to E.F.-P. This work has benefited from the facilities and expertise of the Small Molecule Mass Spectrometry platform of IMAGIF (Centre de Recherche de Gif – http://www.imagif.cnrs.fr). We are very grateful to Marine SCOAZEC and Marie-Noelle SOLER from the imaging plateform PICT-IBISA@ORSAY (Institut Curie, Orsay, France) for technical assistance in the use of the Leica confocal SP5 system. The authors wish to thank Dr Aude Sourdon for her help in confocal and biphotonic imaging.Notes and references
- W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73–76 CAS.
- W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21, 1369–1377 CrossRef CAS PubMed.
- C. Xu, W. Zipfel, J. B. Shear, R. M. Williams and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10763–10768 CrossRef CAS.
- J. Lecoq, J. Savall, D. Vucinic, B. F. Grewe, H. Kim, J. Z. Li, L. J. Kitch and M. J. Schnitzer, Nat. Neurosci., 2014, 17, 1825–1829 CrossRef CAS PubMed.
- C. D. Andrade, C. O. Yanez, H. Y. Ahn, T. Urakami, M. V. Bondar, M. Komatsu and K. D. Belfield, Bioconjugate Chem., 2011, 22, 2060–2071 CrossRef CAS PubMed.
- B. G. Wang, K. Konig and K. J. Halbhuber, J. Microsc., 2010, 238, 1–20 CrossRef CAS PubMed.
- W. Li, R. G. Nava, A. C. Bribriesco, B. H. Zinselmeyer, J. H. Spahn, A. E. Gelman, A. S. Krupnick, M. J. Miller and D. Kreisel, J. Clin. Invest., 2012, 122, 2499–2508 CAS.
- A. C. Zenclussen, D. N. Olivieri, M. L. Dustin and C. E. Tadokoro, Am. J. Reprod. Immunol., 2013, 69, 281–289 CrossRef CAS PubMed.
- M. A. Albota, C. Xu and W. W. Webb, Appl. Opt., 1998, 37, 7352–7356 CrossRef CAS PubMed.
- S. Yao and K. D. Belfield, Eur. J. Org. Chem., 2012, 2012, 3199–3217 CrossRef CAS.
- D. Kim, H. G. Ryu and K. H. Ahn, Org. Biomol. Chem., 2014, 12, 4550–4566 CAS.
- K. P. Divya, S. Sreejith, P. Ashokkumar, K. Yuzhan, Q. Peng, S. K. Maji, Y. Tong, H. Yu, Y. Zhao, P. Ramamurthy and A. Ajayaghosh, Chem. Sci., 2014, 5, 3469–3474 RSC.
- V. Placide, A. T. Bui, A. Grichine, A. Duperray, D. Pitrat, C. Andraud and O. Maury, Dalton Trans., 2015, 44, 4918–4924 RSC.
- H. M. Kim, B. R. Kim, J. H. Hong, J. S. Park, K. J. Lee and B. R. Cho, Angew. Chem., Int. Ed., 2007, 46, 7445–7448 CrossRef CAS PubMed.
- H. W. Lee, C. H. Heo, D. Sen, H. O. Byun, I. H. Kwak, G. Yoon and H. M. Kim, Anal. Chem., 2014, 86, 10001–10005 CrossRef CAS PubMed.
- H. M. Kim and B. R. Cho, Acc. Chem. Res., 2009, 42, 863–872 CrossRef CAS PubMed.
- A. D'Aleo, A. Bourdolle, S. Brustlein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet and O. Maury, Angew. Chem., Int. Ed., 2012, 51, 6622–6625 CrossRef PubMed.
- E. Macoas, G. Marcelo, S. Pinto, T. Caneque, A. M. Cuadro, J. J. Vaquero and J. M. Martinho, Chem. Commun., 2011, 47, 7374–7376 RSC.
- L. Guo and M. S. Wong, Adv. Mater., 2014, 26, 5400–5428 CrossRef CAS PubMed.
- M. Albota, D. Beljonne, J. L. Bredas, J. E. Ehrlich, J. Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Rockel, M. Rumi, G. Subramaniam, W. W. Webb, X. L. Wu and C. Xu, Science, 1998, 281, 1653–1656 CrossRef CAS PubMed.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- J. Zyss and I. Ledoux, Chem. Rev., 1994, 94, 77–105 CrossRef CAS.
- C. Allain, F. Schmidt, R. Lartia, G. Bordeau, C. Fiorini-Debuisschert, F. Charra, P. Tauc and M. P. Teulade-Fichou, ChemBioChem, 2007, 8, 424–433 CrossRef CAS PubMed.
- C. Katan, F. Terenziani, O. Mongin, M. H. Werts, L. Porres, T. Pons, J. Mertz, S. Tretiak and M. Blanchard-Desce, J. Phys. Chem. A, 2005, 109, 3024–3037 CrossRef CAS PubMed.
- G. S. He, L. S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- L. Porres, O. Mongin, C. Katan, M. Charlot, T. Pons, J. Mertz and M. Blanchard-Desce, Org. Lett., 2004, 6, 47–50 CrossRef CAS PubMed.
- X. He, B. Xu, Y. Liu, Y. Yang and W. Tian, J. Appl. Phys., 2012, 111, 053516 CrossRef.
- X. Jiang, K. M. Karlsson, E. Gabrielsson, E. M. J. Johansson, M. Quintana, M. Karlsson, L. Sun, G. Boschloo and A. Hagfeldt, Adv. Funct. Mater., 2011, 21, 2944–2952 CrossRef CAS.
- Z. Fang, R. D. Webster, M. Samoc and Y.-H. Lai, RSC Adv., 2013, 3, 17914–17917 RSC.
- S. Roquet, A. Cravino, P. Leriche, O. Aleveque, P. Frere and J. Roncali, J. Am. Chem. Soc., 2006, 128, 3459–3466 CrossRef CAS PubMed.
- Z. Yu, Z. Zheng, M. Yang, L. Wang, Y. Tian, J. Wu, H. Zhou, H. Xu and Z. Wu, J. Mater. Chem. C, 2013, 1, 7026–7033 RSC.
- G. Wang, K.-Y. Pu, X. Zhang, K. Li, L. Wang, L. Cai, D. Ding, Y.-H. Lai and B. Liu, Chem. Mater., 2011, 23, 4428–4434 CrossRef CAS.
- V. Parthasarathy, S. Fery-Forgues, E. Campioli, G. Recher, F. Terenziani and M. Blanchard-Desce, Small, 2011, 7, 3219–3229 CrossRef CAS PubMed.
- B. Dumat, G. Bordeau, E. Faurel-Paul, F. Mahuteau-Betzer, N. Saettel, M. Bombled, G. Metge, F. Charra, C. Fiorini-Debuisschert and M. P. Teulade-Fichou, Biochimie, 2011, 93, 1209–1218 CrossRef CAS PubMed.
- B. Dumat, G. Bordeau, E. Faurel-Paul, F. Mahuteau-Betzer, N. Saettel, G. Metge, C. Fiorini-Debuisschert, F. Charra and M. P. Teulade-Fichou, J. Am. Chem. Soc., 2013, 135, 12697–12706 CrossRef CAS PubMed.
- B. Dumat, G. Bordeau, A. I. Aranda, F. Mahuteau-Betzer, Y. El Harfouch, G. Metge, F. Charra, C. Fiorini-Debuisschert and M. P. Teulade-Fichou, Org. Biomol. Chem., 2012, 10, 6054–6061 CAS.
- B. Clapham and A. J. Sutherland, Chem. Commun., 2003, 84–85 RSC.
- Q. Wang and D. S. Lawrence, J. Am. Chem. Soc., 2005, 127, 7684–7685 CrossRef CAS PubMed.
- J. Min, J. W. Lee, Y. H. Ahn and Y. T. Chang, J. Comb. Chem., 2007, 9, 1079–1083 CrossRef CAS PubMed.
- S. Charier, O. Ruel, J. B. Baudin, D. Alcor, J. F. Allemand, A. Meglio, L. Jullien and B. Valeur, Chemistry, 2006, 12, 1097–1113 CrossRef CAS PubMed.
- H. J. Park, C. S. Lim, E. S. Kim, J. H. Han, T. H. Lee, H. J. Chun and B. R. Cho, Angew. Chem., Int. Ed., 2012, 51, 2673–2676 CrossRef CAS PubMed.
- D. L. Silva, L. De Boni, D. S. Correa, S. C. S. Costa, A. A. Hidalgo, S. C. Zilio, S. Canuto and C. R. Mendonca, Opt. Mater., 2012, 34, 1013–1018 CrossRef CAS.
- F. Besselièvre, S. Lebrequier, F. Mahuteau-Betzer and S. Piguel, Synthesis, 2009, 3511–3518 Search PubMed.
- R. Chennoufi, H. Bougherara, N. Gagey-Eilstein, B. Dumat, E. Henry, F. Subra, F. Mahuteau-Betzer, P. Tauc, M. P. Teulade-Fichou and E. Deprez, Chem. Commun., 2015, 51, 14881–14884 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and copies of NMR and HPLC spectra. See DOI: 10.1039/c5ob02225h |
| This journal is © The Royal Society of Chemistry 2016 |