
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First double hydrophilic graft copolymer bearing a poly(2-hydroxylethyl acrylate) backbone synthesized by sequential RAFT polymerization and SET-LRP†
Yinan
Cui‡
a,
Xiuyu
Jiang‡
a,
Chun
Feng
a,
Guangxin
Gu
*b,
Jie
Xu
b and
Xiaoyu
Huang
*a
aKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, People's Republic of China. E-mail: xyhuang@mail.sioc.ac.cn; Fax: +86-21-64166128; Tel: +86-21-54925310
bDepartment of Materials Science, Fudan University, 220 Handan Road, Shanghai 200433, People's Republic of China. E-mail: guangxingu@fudan.edu.cn; Fax: +86-21-65649937; Tel: +86-21-55664033
First published on 7th April 2016
Abstract
Two well-defined double hydrophilic graft copolymers comprising a hydrophilic poly(2-hydroxyethyl acrylate) (PHEA) backbone and hydrophilic poly(N-isopropylacrylamide) (PNIPAM) side chains were synthesized by the combination of reversible addition–fragmentation chain transfer (RAFT) polymerization, single-electron-transfer living radical polymerization (SET-LRP), and a grafting-from strategy. A 2-hydroxyethyl 2-((2-chloropropanoyloxy)methyl)acrylate monomer containing an SET-LRP initiating group (–OCOCH(CH3)Cl) was first RAFT homopolymerized to provide a PHEA-based homopolymer bearing a Cl-containing SET-LRP initiating group in every repeated unit with a narrow molecular weight distribution (Mw/Mn = 1.17). This homopolymer directly initiated SET-LRP of N-isopropylacrylamide at ambient temperature to afford the desired well-defined poly(2-hydroxyethyl acrylate)-graft-poly(N-isopropylacrylamide) graft copolymers (Mw/Mn ≤ 1.33) with hydroxyl in every repeated unit of the backbone without polymeric functionality transformation. The thermo-responsive phase behavior of the obtained double hydrophilic graft copolymers was investigated by UV/vis spectroscopy, 1H NMR, and dynamic light scattering (DLS).
Introduction
Double hydrophilic copolymers (DHCs), which consist of two different hydrophilic segments, have entirely aroused considerable interest because of their environmental friendliness and biocompatibility. In particular, double hydrophilic block copolymers have been widely investigated, which respond to external stimuli1–15 including pH,1–6 temperature,4–15 redox,11 ions,12,15 light,13 and NO,14 and show different phase behaviors16–23 such as self-assembly,16–22 or co-assembly24 in aqueous solution or organic solvents. It apparently turned out that this class of copolymer could be extensively applied in biomedical areas,25–30 for example as a templating agent and carrier for drug delivery systems.However, few studies have focused on double hydrophilic graft copolymers, due to the difficult synthetic process. In order to deepen the understanding of the relationship between the complicated structure and unique properties, our group has made an effort to develop a variety of double hydrophilic graft copolymers in recent years. Feng et al. synthesized a series of double hydrophilic graft copolymers of poly(N-isopropylacrylamide)-b-[poly(ethyl acrylate)-g-poly(2-(diethylamino)ethyl-methacrylate)] (PNIPAM-b-(PEA-g-PDEA)) by the combination of atom transfer radical polymerization (ATRP) and a grafting-from strategy, which could lead to self-assembly forming different aggregates in aqueous media driven by varying pH and/or temperature, and obviously showed “schizophrenic” micellization properties.31 We also reported metallic nano-particles that were stabilized by double hydrophilic graft copolymers,32,33 the size of which could be controlled by adjusting the ratio of metal ion to graft copolymer or varying the length of the side chain. Moreover, Jiang et al. reported a dual-stimuli responsive polymer system, poly(acrylic acid)-g-poly(N-vinylcaprolactam) (PAA-g-PNVCL) in 2013, which possessed responsiveness to pH and temperature simultaneously.34
Generally, when constructing a double hydrophilic polymer system, pendant carboxyls are introduced into the polymeric chain to maintain the hydrophilicity of the corresponding segment, for instance poly(acrylic acid) (PAA)34 or poly(mathacrylic acid) (PMAA)33 have been extensively employed in structuring procedures.35–39 However, the pendent hydroxyls of poly(2-hydroxyethyl methacrylate) (PHEMA) or poly(2-hydroxyethyl acrylate) (PHEA) have hardly been introduced into polymeric chains, though they can be prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization or ATRP. Indeed, to the best of our knowledge, no one has reported the synthesis of a double hydrophilic graft polymer containing PHEMA or PHEA segments with hydroxyl side groups. This is because the pendant hydroxyls of HEA/HEMA are treated with other functionalities to link the side chains so that the hydrophilic hydroxyls are transformed into hydrophobic ester groups.31,32 To construct a new PHEA-based graft copolymer with a hydrophilic PHEA backbone, our group has designed and synthesized a novel trifunctional monomer, 2-hydroxyethyl 2-((2-chloropropanoyloxy)methyl)acrylate (HECPMA), which possessed a polymerizable double bond, a Cl-containing initiating group (–OCOCH(CH3)Cl), and a hydrophilic hydroxyl group simultaneously, and we reported the first example of an amphiphilic graft copolymer bearing a hydrophilic PHEA backbone with hydroxyl in every repeated unit of the backbone in 2014.40
Herein, on the basis of the trifunctional HECPMA monomer developed by our group, we report the synthesis of a well-defined double hydrophilic graft copolymer bearing a PHEA backbone, poly(2-hydroxyethyl acrylate)-g-poly(N-isopropylacrylamide) (PHEA-g-PNIPAM) by successive reversible-deactivation radical polymerization (RDRP). The well-defined PHEA backbone was first formed by RAFT homopolymerization of HECPMA monomer, which possessed a RDRP initiating group (–OCOCH(CH3)Cl) and a pendant hydroxyl in every repeated unit. Single-electron-transfer living radical polymerization (SET-LRP) of N-isopropylacrylamide (NIPAM) was directly initiated by –OCOCH(CH3)Cl to afford the target product of PHEA-g-PNIPAM while the hydroxyl remained inert. This kind of double hydrophilic graft copolymer showed thermo-responsive phase behavior in aqueous media.
Results and discussion
Preparation of RDRP macroinitiator
Since the example of a double hydrophilic graft copolymer consisting of a hydrophilic PHEA/PHEMA backbone with hydroxyl in every repeated unit of the backbone has been never reported, a HECPMA monomer40 possessing a polymerizable double bond, a Cl-containing RDRP initiating group (–OCOCH(CH3)Cl), and a hydroxyl concurrently, which was developed by our group in 2014, was employed in the current case to construct the PHEA-backbone-containing double hydrophilic graft copolymer. A pendant –OCOCH(CH3)Cl initiating group exists in the HECPMA monomer, which means that the most popular “grafting-from” strategy41–49 compared to the other two “grafting-through” and “grafting-onto” approaches can be employed in the present work. This method requires the preparation of a macroinitiator followed by the polymerization of the second monomer for the formation of side chains, initiated by pendant initiating sites on the polymeric backbone of the macroinitiator. Therefore, the HECPMA monomer was first homopolymerized to form the desired macroinitiator with a pendant initiating group in every repeated unit.The –OCOCH(CH3)Cl initiating group can certainly initiate ATRP or SET-LRP of common vinyl monomers, and ATRP/SET-LRP of functional monomers bearing halogen-containing initiating groups provided a hyperbranched polymer, not a linear polymer.50 So, RAFT polymerization,51–55 the most versatile RDRP process because of its mild reaction conditions, variety of monomers with controlled polymerization, tolerance of monomer functionalities, and utilization for the preparation of copolymers with different architectures, may be the right way to polymerize the HECPMA functional monomers containing RDRP initiating groups. In the current case, HECPMA is a kind of acrylate monomer, for which CDB was often chosen as the chain transfer agent (CTA).56 Thus, the RAFT homopolymerization of the HECPMA 1 monomer was performed in DMF at 80 °C using AIBN as an initiator and CDB as the CTA (Scheme 1). The crude product was a pink powder due to the presence of a dithiobenzoate end functionality. Although the content of the dithiobenzoate moiety was quite low, it may influence the subsequent RDRP graft polymerization and the observation during the subsequent polymerization may be interfered with by the pink color of the dithiobenzoate moiety. The dithiobenzoate end group was then removed by AIBN (20 eq.)57 and a white powder was obtained after the reaction, which demonstrated the absence of the dithiobenzoate residue. The complete removal of the dithiobenzoate end group was also confirmed by the disappearance of the characteristic peak of a dithiobenzoate end group (510 nm) in the UV/vis spectrum after the treatment with AIBN57 compared to that before the treatment.
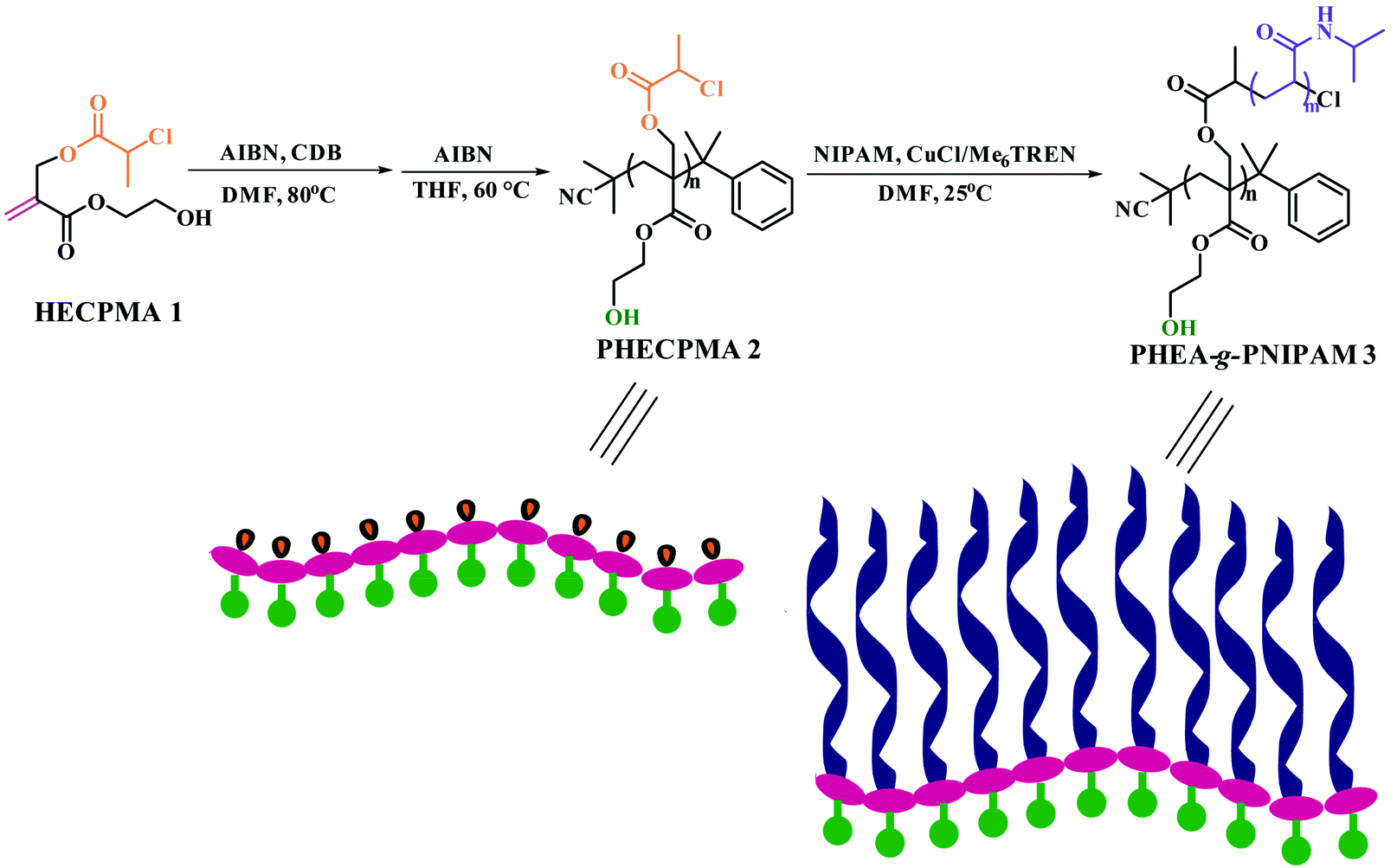 | ||
| Scheme 1 Synthesis of PHEA-g-PNIPAM well-defined double hydrophilic graft copolymer via sequential RAFT polymerization and SET-LRP. | ||
The white powder was first characterized by GPC and a unimodal and symmetric eluent peak with a narrow molecular weight distribution of 1.17 appeared as shown in Fig. 1, which affirmed the well-defined structure of the obtained product from the RAFT homopolymerization. The chemical structure of the white powder was examined by FT-IR, 1H NMR, and 13C NMR. The FT-IR spectrum of the homopolymer is shown in Fig. 2A and the typical stretching vibration absorption signal of the double bond of the HECPMA monomer was not found to appear at 1636 cm−1, which illustrates the successful polymerization of HECPMA monomer. This point was also verified by the disappearance of the resonance signals of the double bond (5.00–7.00 ppm) in the 1H NMR spectrum (Fig. S1A†), and those at 129.0 and 134.4 ppm in the 13C NMR spectrum (Fig. S1B†). The presence of a polyacrylate skeleton after the RAFT homopolymerization of the HECPMA monomers was demonstrated by the resonance signals at 1.97 (CH2CCO2) ppm in the 1H NMR spectrum (Fig. S1A†), and at 44.4 (CH2CCO2) and 64.7 (CH2CCO2) ppm in the 13C NMR spectrum (Fig. S1B†). The RAFT mechanism of the polymerization was evidenced by the weak resonance signals (7.00–7.50 ppm) in the 1H NMR spectrum (Fig. S1A†) and weak peaks at 126.1 and 128.3 ppm in the 13C NMR spectrum (Fig. S1B†), which were attributed to the phenyl end groups originating from the CTA of CDB. Furthermore, we can see the stretching vibration absorption of carbonyl at 1735 cm−1 and the broad peak at 3435 cm−1 corresponding to the stretching vibration absorption of hydroxyl in Fig. 2A. In particular, it should be noted that the peak at 4.62 ppm (Fig. S1A†) belongs to the resonance signal of 1 proton of CH3CHCl, which evidenced that the RDRP initiating groups remained inert during RAFT polymerization. The proton resonance signal at 4.87 ppm corresponds to the hydroxyl of CH2CH2OH. The resonance signal of the 2 protons of CH2CCH2O was found to be located at 4.07 ppm after polymerization (Fig. S1A†), and was located at 4.93 ppm in the HECPMA monomer due to the presence of a double bond. Now, it can be concluded from all the aforementioned evidence that RAFT homopolymerization of the HECPMA 1 monomer was successfully performed and provided a well-defined PHECPMA 2 homopolymer bearing a –OCOCH(CH3)Cl RDRP initiating group in every repeated unit.
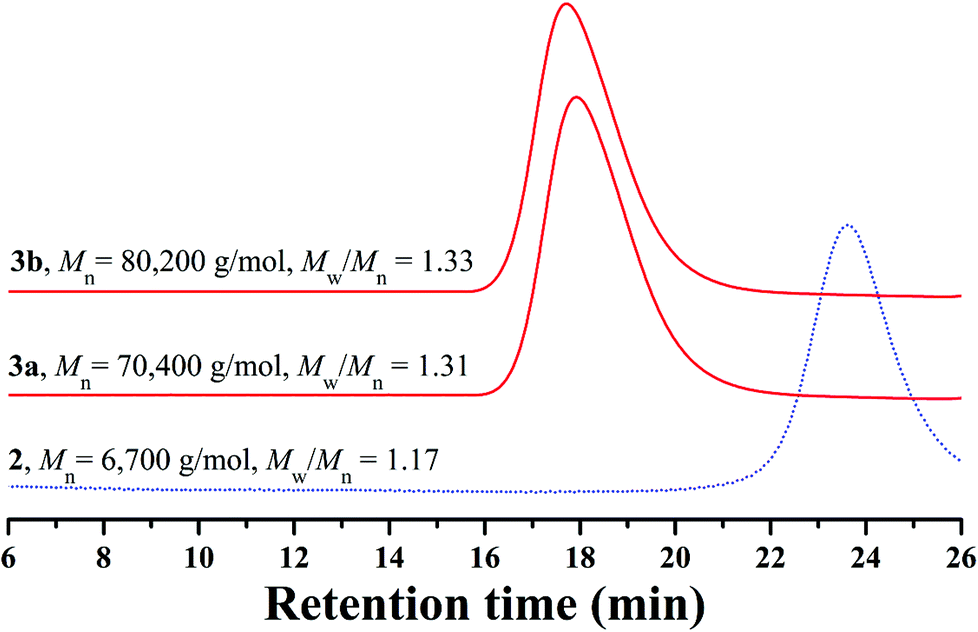 | ||
| Fig. 1 GPC traces of PHECPMA 2 and PHEA-g-PNIPAM 3 in LiBr-added DMF. | ||
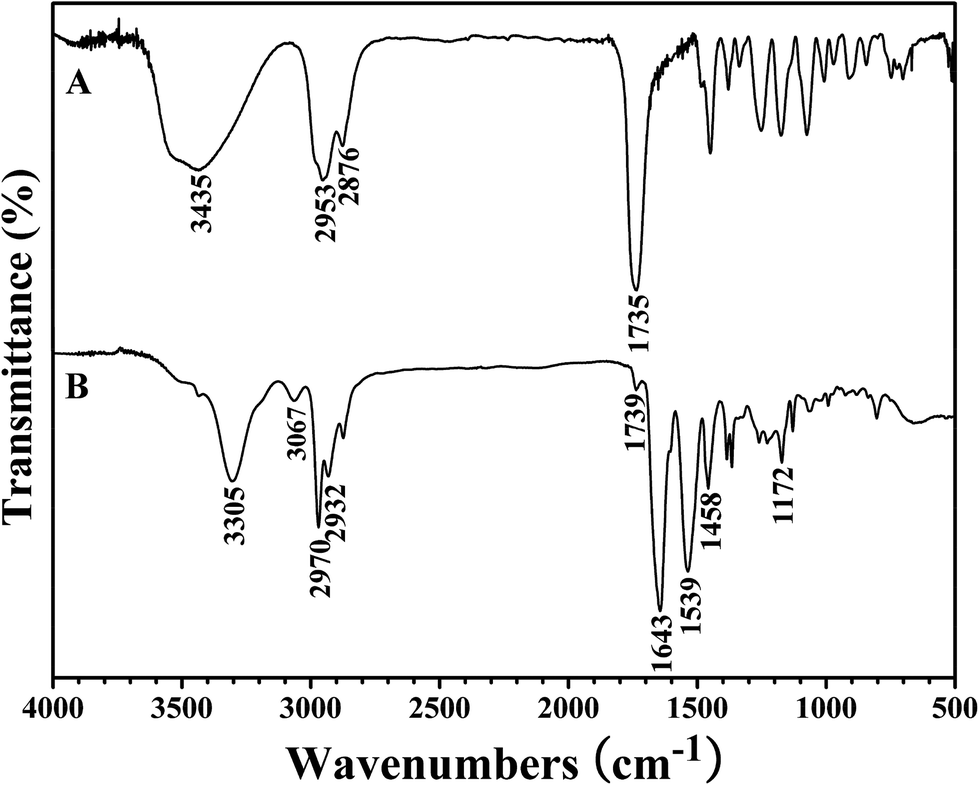 | ||
| Fig. 2 FT-IR spectra of PHECPMA 2 (A) and PHEA-g-PNIPAM 3 (B). | ||
Though linear poly(methyl methacrylate) standards were employed to calibrate the GPC system, the molecular weight of the PHECPMA 2 homopolymer (6700 g mol−1) obtained from GPC was not very accurate because of the relatively strong polarity of the pendant hydroxyl groups in the homopolymer. Thus, GPC/MALS was employed in the current case to determine the absolute molecular weight of the PHECPMA 2 homopolymer and the value was 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 460 g mol−1. The number of HECPMA repeated units in the PHECPMA 2 homopolymer was then estimated from the data of absolute molecular weight according to eqn (1) (187 and 236.5 are the molecular weights of the CTA moiety and HECPMA monomer, respectively). The result showed that every PHECPMA 2 chain possessed 56.1 Cl-containing RDRP initiating groups.
460 g mol−1. The number of HECPMA repeated units in the PHECPMA 2 homopolymer was then estimated from the data of absolute molecular weight according to eqn (1) (187 and 236.5 are the molecular weights of the CTA moiety and HECPMA monomer, respectively). The result showed that every PHECPMA 2 chain possessed 56.1 Cl-containing RDRP initiating groups.
| nHECPMA = (Mn,GPC/MALS − 187)/236.5 | (1) |
Synthesis of the PHEA-g-PNIPAM well-defined graft copolymer
PHEA-g-PNIPAM well-defined graft copolymer was designed to be constructed by RDRP of NIPAM initiated by the pendant –OCOCH(CH3)Cl initiating groups of the PHECPMA 2 homopolymer via the grafting-from strategy. Herein, RDRP of NIPAM with a feeding ratio ([NIPAM]:[Cl group]) of 100:1 was performed in DMF at ambient temperature using the PHECPMA 2 homopolymer as a macroinitiator and CuCl/Me6TREN as a catalytic system. Two PHEA-g-PNIPAM graft copolymers with different molecular weights were obtained by extending the polymerization time (1.0–1.5 h) as summarized in Table 1. It can be seen from Table 1 that the molecular weights of both graft copolymers (≥70400 g mol−1) obtained from conventional GPC are much higher than that of the PHECPMA 2 macroinitiator (6700 g mol−1) obtained from conventional GPC, evidencing the occurrence of the RDRP of NIPAM. Moreover, both graft copolymers presented unimodal and symmetric eluent peaks with relatively narrow molecular weight distributions (Mw/Mn ≤ 1.33) in Fig. 1, which showed that the polymerization of NIPAM proceeded as a controlled process and intermolecular coupling could be neglected.45
| Entry | [NIPAM]:[Cl group] |
Time (h) | M n,GPC (g mol−1) | M w/Mnb | N% | N NIPAM | n NIPAM | M n,EA (g mol−1) |
|---|---|---|---|---|---|---|---|---|
|
a Initiated by PHECPMA 2 macroinitiator (Mn,GPC = 6700 g mol−1, Mn,GPC/MALS = 13460 g mol−1, Mw/Mn = 1.17, 56.1 Cl-containing ATRP initiating groups) in DMF. [Cl group]:[CuCl]:[Me6TREN] = 1:1:1, polymerization temperature: 25 °C.
b Measured by GPC in DMF at 35 °C.
c The total number of NIPAM repeated units obtained from elemental analysis.
d The number of NIPAM repeated units per side chain obtained from elemental analysis.
e Calculated from elemental analysis.
|
||||||||
| 3a | 100:1 |
1.0 | 70400 |
1.31 | 11.485 | 1540.4 | 27.4 | 187800 |
| 3b | 100:1 |
1.5 | 80200 |
1.33 | 11.685 | 2023.5 | 36.1 | 242400 |
Note that DMF, a polar solvent with strong polarity, was used as the solvent for the RDRP of NIPAM as DMF is a good solvent for both PHECPMA and PNIPAM polymers. However, CuX has been reported to disproportionate into Cu(0) and CuX2 readily in polar solvents including DMF in the presence of Me6TREN ligand.58–62 Therefore, a control experiment was conducted to clarify the mechanism of the RDRP of NIPAM in the current case. Two different mixtures (2 mL DMF + 9.9 mg CuCl (0.1 mmol) + 27 μL Me6TREN (0.1 mmol), or 2 mL DMF + 13.45 mg CuCl2 (0.1 mmol) + 27 μL Me6TREN (0.1 mmol)) were added separately to two Schlenk flasks under N2, followed by three cycles of freezing–pumping–thawing. Both solutions were stirred at 25 °C for 30 min and the solution containing CuCl2 turned blue without any precipitation at the bottom of the flask (inset of Fig. 3). But, the solution containing CuCl turned green and a clear black sediment appeared at the bottom of the flask (inset of Fig. 3). In addition, the maximum UV absorption wavelength of the solution containing CuCl was located at 925 nm (blue line in Fig. 3) close to that of the solution containing CuCl2 (green line in Fig. 3), which meant that this absorption might originate from the Cu(II) complex. Both facts strongly supported that Cu(I) disproportionated into Cu(0) and Cu(II) species in DMF in the presence of the Me6TREN ligand and the black precipitate should be Cu(0) because of the limited solubility of Cu(0). Previous studies have demonstrated that the mechanism of SET-LRP invokes the reversible activation of dormant polymer chains via a Cu(0)-mediated outer-sphere electron transfer process involving heterolytic C–X cleavage.58,62,63 This mechanism shows that the disproportionation of Cu(I)X generates a Cu(0) activator and Cu(II)X2 deactivator in situ and the extent of Cu(I)X disproportionation is mainly dependent on the selection of ligand and solvent.64–73 Thus, it can be concluded from the above-mentioned results that the mechanism of the RDRP of NIPAM should be that of SET-LRP. Indeed, SET-LRP has emerged as a powerful tool for the rapid synthesis of various polymers with excellent control of molecular weight, molecular weight dispersity, high chain end fidelity, and ultrahigh molecular weight in polar and nonpolar solvents.59,62,64,65,67
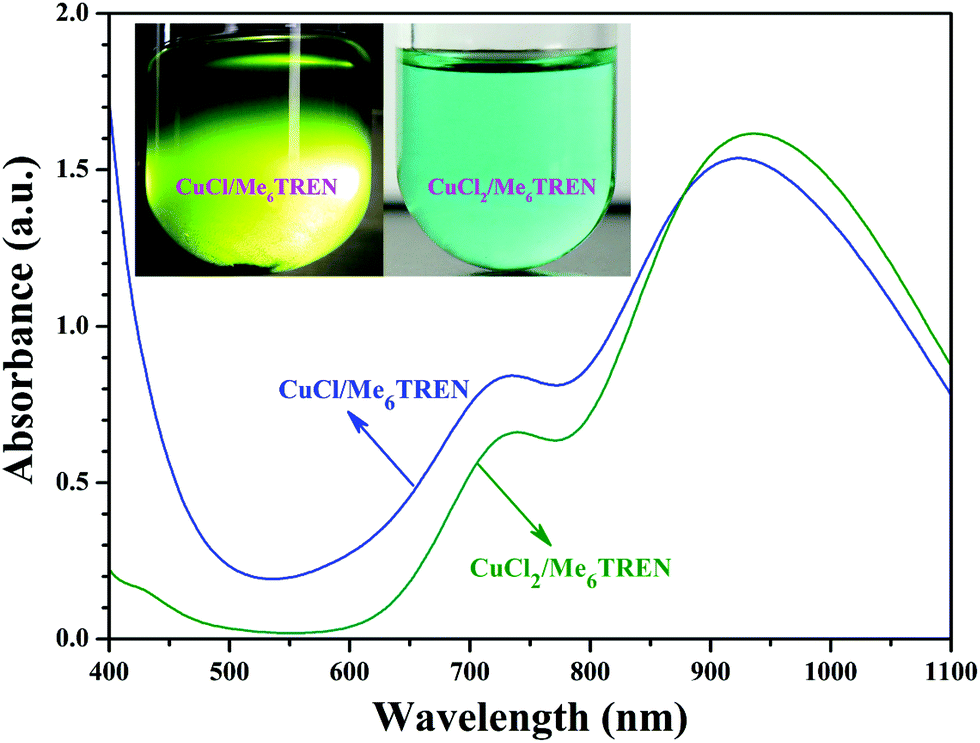 | ||
| Fig. 3 UV/vis spectra of DMF solutions containing CuCl/Me6TREN and CuCl2/Me6TREN, the insets are digital images of the solutions. | ||
SET-LRP of NIPAM initiated by the PHECPMA 2 macroinitiator was performed at relatively low conversions (<52%) of NIPAM to suppress the intermolecular coupling reactions.41,74 The polymerization kinetics were investigated as depicted in Fig. 4. It was found that the conversion of NIPAM increased linearly with the extension of polymerization time. These first order polymerization kinetics clearly showed a constant number of propagating species during the polymerization, which is a typical character of SET-LRP.62
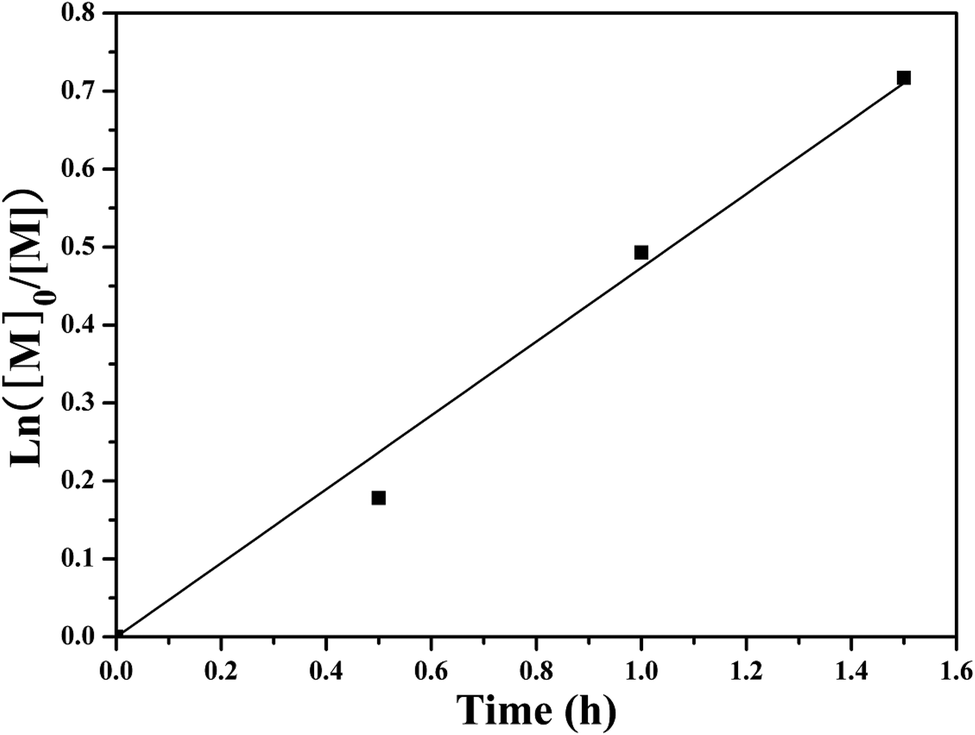 | ||
| Fig. 4 Kinetics plot for SET-LRP graft copolymerization of NIPAM in DMF. | ||
PHEA-g-PNIPAM 3 graft copolymer was characterized by FT-IR and 1H NMR. In comparison with the FT-IR spectrum of the PHECPMA 2 macroinitiator (Fig. 2A), new signals originating from the PNIPAM side chains appeared in Fig. 2B after the SET-LRP of NIPAM. The peaks located at 3305 and 1643 cm−1 were attributed to the characteristic stretching vibration absorption of –N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the amide group in the NIPAM repeated unit, respectively, while the band at 1539 cm−1 corresponded to the bending vibration absorption of –N–H of the amide group. The signals of the ester carbonyl (1739 cm−1) and hydroxyl (3438 cm−1) groups still appeared in Fig. 2B, showing the existence of the PHEA backbone; however, both signals became weaker due to the introduction of the PNIPAM side chains. 1H NMR spectrum of the PHEA-g-PNIPAM 3 graft copolymer is shown in Fig. 5. The strong peak located at 1.16 ppm (‘a’) was attributed to the 6 protons of NCH(CH3)2 in the PNIPAM side chains and the broad peak at 2.09 (‘c’) ppm originated from the 1 proton of CH2CH in the NIPAM repeated unit. The weak resonance signal at 3.49 (‘g’) ppm corresponded to the 2 protons of CO2CH2CH2OH in the HEA repeated unit, indicating the presence of the PHEA backbone. In brief, FT-IR and 1H NMR measurements confirmed the co-existence of the PHEA backbone and PNIPAM side chains in the PHEA-g-PNIPAM 3 graft copolymer.
O of the amide group in the NIPAM repeated unit, respectively, while the band at 1539 cm−1 corresponded to the bending vibration absorption of –N–H of the amide group. The signals of the ester carbonyl (1739 cm−1) and hydroxyl (3438 cm−1) groups still appeared in Fig. 2B, showing the existence of the PHEA backbone; however, both signals became weaker due to the introduction of the PNIPAM side chains. 1H NMR spectrum of the PHEA-g-PNIPAM 3 graft copolymer is shown in Fig. 5. The strong peak located at 1.16 ppm (‘a’) was attributed to the 6 protons of NCH(CH3)2 in the PNIPAM side chains and the broad peak at 2.09 (‘c’) ppm originated from the 1 proton of CH2CH in the NIPAM repeated unit. The weak resonance signal at 3.49 (‘g’) ppm corresponded to the 2 protons of CO2CH2CH2OH in the HEA repeated unit, indicating the presence of the PHEA backbone. In brief, FT-IR and 1H NMR measurements confirmed the co-existence of the PHEA backbone and PNIPAM side chains in the PHEA-g-PNIPAM 3 graft copolymer.
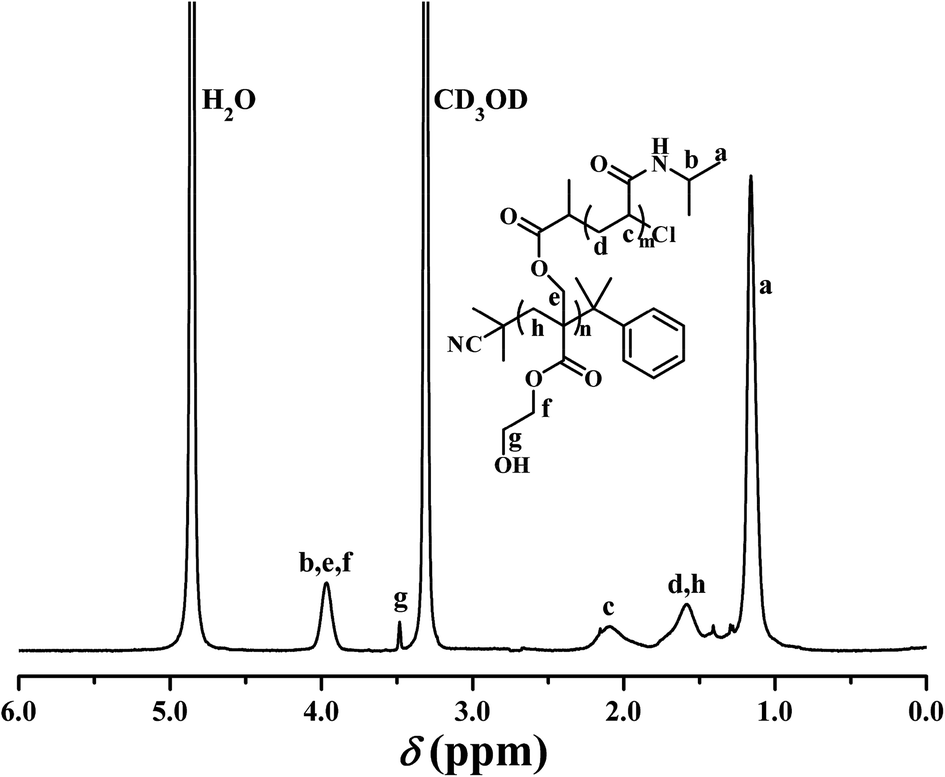 | ||
| Fig. 5 1H NMR spectrum of PHEA-g-PNIPAM 3 in CD3OD. | ||
In particular, it should be pointed out that no proton resonance signal was found around 4.50 ppm in Fig. 5 while the resonance signal of the 1 proton of –OCOCH(CH3)Cl appeared at 4.62 ppm in Fig. S1A.† This fact distinctly demonstrates that the initiation efficiency for the SET-LRP of NIPAM was 100% in the present work, i.e. PHEA-g-PNIPAM 3 possessed 56.1 PNIPAM side chains.
It has been proved that the molecular weight of the graft copolymer measured by conventional GPC is very different from the ‘real’ value.75,76 ‘Absolute’ molecular weights of the PHEA-g-PNIPAM 3 graft copolymers were obtained from elemental analysis since only the PNIPAM side chains possessed the N element. From the data of the absolute molecular weight of the PHECPMA 2 macroinitiator (Mn = 13460 g mol−1) and the nitrogen content of PHEA-g-PNIPAM 3 graft copolymers, the total number of NIPAM repeated units (NNIPAM) and the length of the PNIPAM side chain (nNIPAM) can be calculated according to eqn (2) and (3) (13460 and 113.16 are the molecular weights of the PHECPMA 2 macroinitiator and NIPAM, respectively; 56.1 is the number of –OCOCH(CH3)Cl initiating sites on the backbone), respectively, and the results are summarized in Table 1. Indeed, the ‘absolute’ molecular weights obtained from elemental analysis were much higher than those obtained from conventional GPC.
| N% = 14NNIPAM/(113.16 NNIPAM + 13460) | (2) |
| nNIPAM = NNIPAM/56.1 | (3) |
Thus, all this evidence strongly supported the PHEA-g-PNIPAM 3 graft copolymer possession of a well-defined structure: a poly(2-hydroxyethyl acrylate) backbone (56.1 repeated units) and 56.1 PNIPAM side chains (length: 27.4 or 36.1 NIPAM repeated units).
Thermo-responsive phase behavior of the PHEA-g-PNIPAM graft copolymer
It is well known that PHEA is a kind of water-soluble polymer because the pendant hydroxyl groups have hydrophilicity while PNIPAM is a kind of thermo-responsive polymer, which undergoes an abrupt phase transition upon heating at its lower critical solution temperature (LCST) around 32 °C.77 PNIPAM with this LCST is readily soluble in cold water, but undergoes a phase transition to an insoluble state when heated above a certain temperature at which an abrupt change in conformation, solubility, and hydrophilic–hydrophobic balance of the polymer occurs.78 PNIPAM contains a large number of hydrophilic amide groups, but also contains hydrophobic groups. Hydrogen bonding between the hydrophilic segments of the polymeric chain and water molecules is dominant at temperatures lower than the LCST. When the temperature is raised, a partial displacement of water from the polymer coil weakens these hydrogen bonds and increases interactions between the hydrophobic segments of the polymer.79 Therefore, the transition from a hydrophilic state to hydrophobic state occurs at a certain temperature, the LCST, and the polymer phase separates. Considering the double hydrophilic nature of the PHEA-g-PNIPAM 3 graft copolymer comprising a hydrophilic PHEA backbone and densely grafted hydrophilic thermo-responsive PNIPAM side chains, it can be expected that the PHEA-g-PNIPAM 3 graft copolymer will exhibit thermo-responsive phase behavior in aqueous solution.As shown in the insets of Fig. 6, a colorless and homogeneous aqueous solution of PHEA-g-PNIPAM 3 graft copolymer at 25 °C (below the LCST) turned turbid at 50 °C (above the LCST). This transition was reversible upon cooling and the solution became transparent again. The phase transition temperature of the PHEA-g-PNIPAM 3 graft polymers could be determined from the temperature dependence of their UV/vis transmittance. Fig. 6 shows the UV/vis transmittance of the aqueous solutions of the PHEA-g-PNIPAM 3 graft copolymer with different concentrations as a function of temperature in a heating process. For copolymer 3a with shorter PNIPAM side chains, the transmittance of the aqueous solutions at 27.5 °C was 87% (0.5 mg mL−1), 83% (1 mg mL−1), and 77% (2 mg mL−1); all three samples displayed a sharp and reversible transition at the LCST. The transmittance of the aqueous solutions decreased sharply with the rising of the temperature above the LCST. The transmittance of the aqueous solutions at 70 °C was 74.8% (0.5 mg mL−1), 50.8% (1 mg mL−1), and 18.3% (2 mg mL−1). This fact pointed out that the PNIPAM side chains became hydrophobic upon raising the temperature, and therefore the PHEA-g-PNIPAM 3 graft copolymer possessed thermal responsiveness.
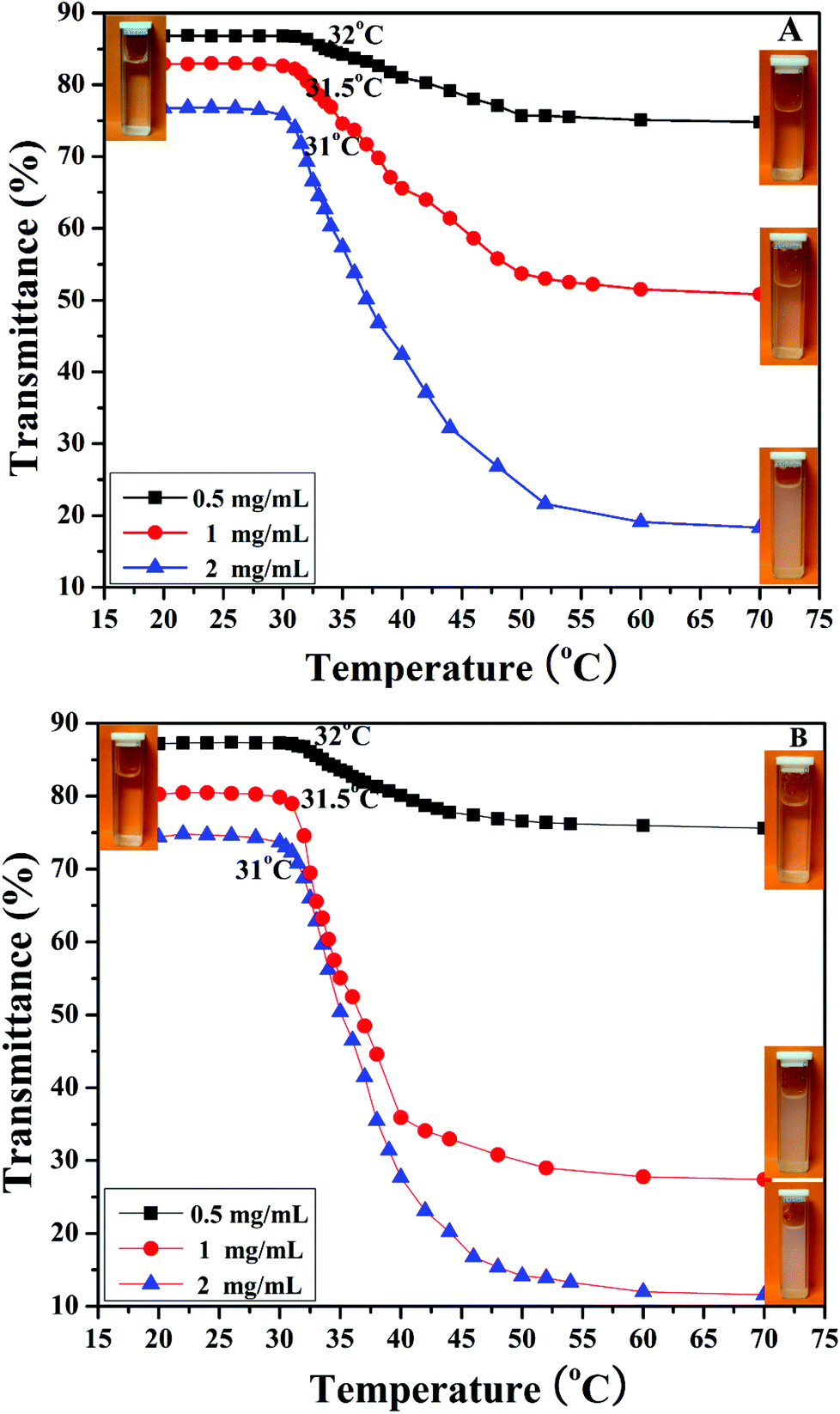 | ||
| Fig. 6 Temperature-dependent transmittance for aqueous solutions of PHEA-g-PNIPAM 3a (A) and 3b (B) graft copolymers with different concentrations. | ||
The values of the LCSTs of the aqueous solutions of the PHEA-g-PNIPAM 3a graft copolymer were 32 °C (0.5 mg mL−1), 31.5 °C (1 mg mL−1), and 31 °C (2 mg mL−1), which were the same as those of the PHEA-g-PNIPAM 3b graft copolymer with longer PNIPAM side chains. These results indicated that the LCST of the PHEA-g-PNIPAM 3 graft copolymer was dependent on the concentration of the copolymer, not the length of the PNIPAM side chain, which is similar to reports in the previous literature.80–82
The thermo-sensitivity of the PHEA-g-PNIPAM 3 graft copolymer was also checked by temperature-dependent 1H NMR. 1H NMR is a versatile technique that reflects the change in the solubility of the copolymer because the proton resonance signals of the desolvated and associated segments become strongly attenuated or even vanish, whereas the proton resonance signals of the water-soluble segments remain unchanged. To deeply understand the phase transition behavior of the PHEA-g-PNIPAM 3 graft copolymer, temperature-dependent 1H NMR spectra of copolymer 3a in D2O ranging from 20 °C to 40 °C were collected and are shown in Fig. 7. It can be seen that the corresponding signals of both the PHEA and PNIPAM segments appeared below 30 °C, indicating the complete dissolution of the whole graft copolymer. However, a drastic drop in the peak intensity of the corresponding protons of the PNIPAM side chains (peaks ‘a’, ‘b’, ‘c’, and ‘d’) was detected when the temperature was raised above 32 °C and these peaks almost completely disappeared at 40 °C because the intramolecular hydrogen bonding between the PNIPAM segment induced the collapse of the PNIPAM chains and the PNIPAM segment became water-insoluble.
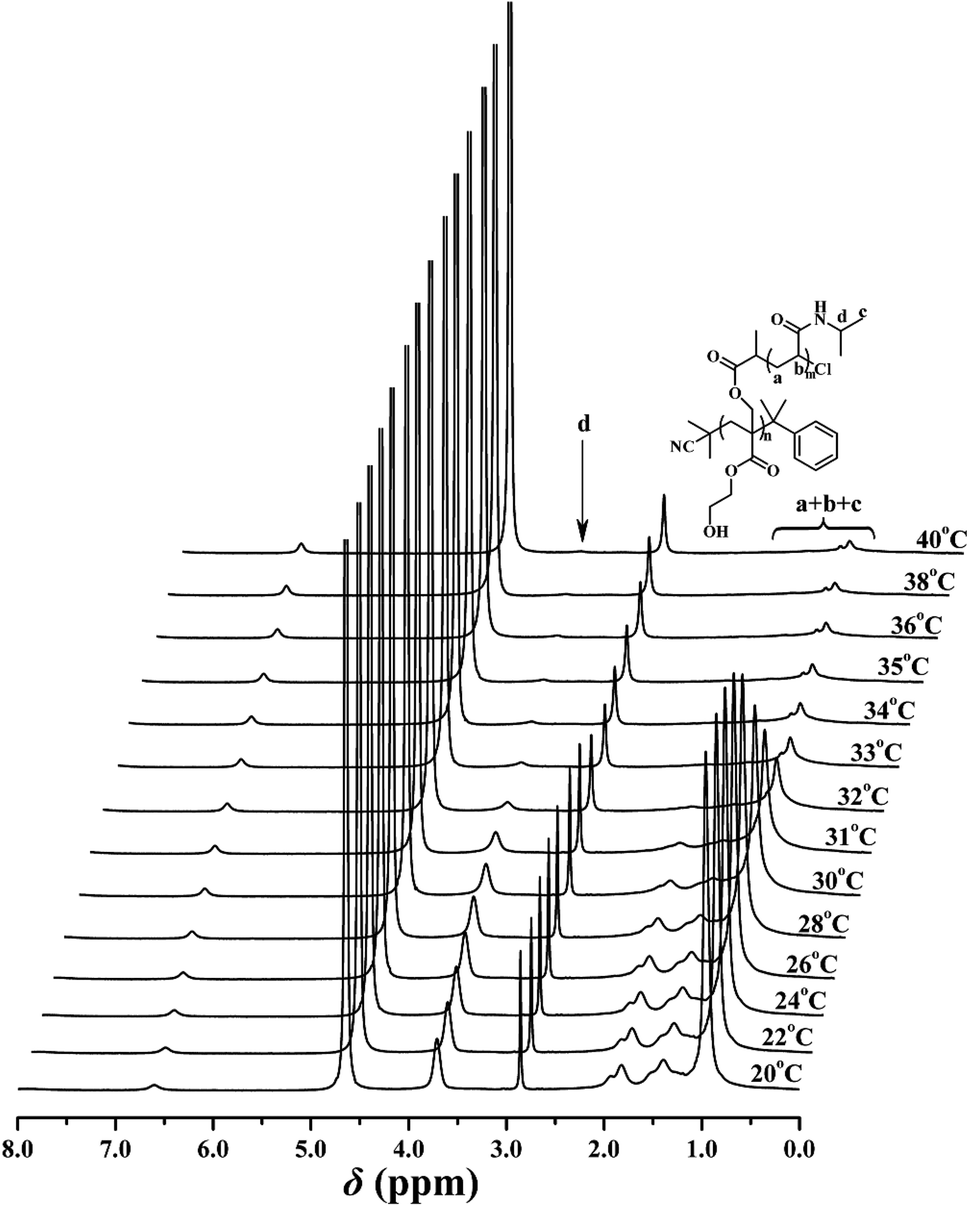 | ||
| Fig. 7 Temperature-dependent 1H NMR spectra of PHEA-g-PNIPAM 3a in D2O. | ||
Further insight into the thermo-responsive behavior of the PHEA-g-PNIPAM 3 graft copolymer was investigated by DLS. The hydrodynamic diameter (Dh) of the PHEA-g-PNIPAM 3b graft copolymer was obviously influenced by the temperature as shown in Fig. 8. When the temperature was just 20 °C, copolymer 3b dissolved molecularly in aqueous media with low Dhs around 20 nm (Fig. 8). However, the Dh of the PHEA-g-PNIPAM 3 graft copolymer increased sharply around 31 °C upon the increase of the temperature and the Dh of copolymer 3b increased to about 390 nm at 40 °C (Fig. 8). This result is in good accordance with those of UV/vis and 1H NMR.
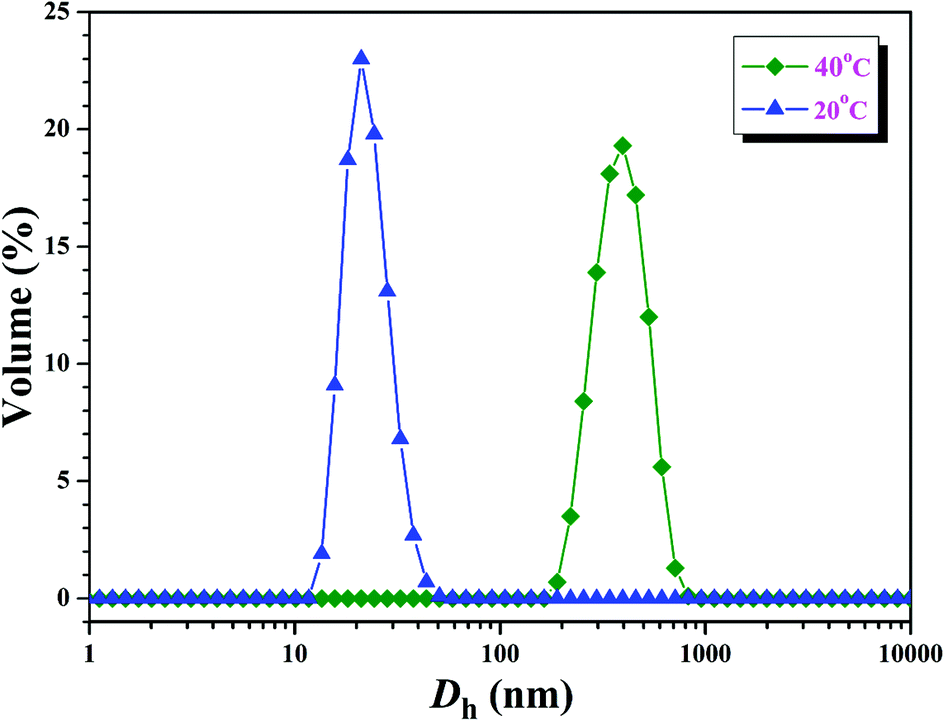 | ||
| Fig. 8 Hydrodynamic diameter distributions of the PHEA-g-PNIPAM 3b graft copolymer at different temperatures with a concentration of 1 mg mL−1. | ||
To summarize, the above-mentioned results have shown that the PHEA-g-PNIPAM 3 graft copolymer was sensitive to temperature: it dissolved molecularly below 30 °C, and it displayed a sharp transition from a double hydrophilic to amphiphilic nature at the LCST in aqueous solution.
Conclusions
We have presented a detailed synthesis of PHEA-g-PNIPAM well-defined double hydrophilic graft copolymers with relatively narrow molecular weight distributions (Mw/Mn ≤ 1.33) through successive RDRP via the grafting-from strategy, using HECPMA trifunctional monomer as a starting material. Both the syntheses of the backbone and side chains are controllable and the whole synthesis process avoided post-polymerization functionality transformation because HECPMA monomer containing a RDRP initiating group could be copolymerized with another suitable monomer without the RDRP initiating group providing well-defined backbones containing a certain amount of initiating sites, and furthermore affording well-defined graft copolymers with a tunable density of side chains. Moreover, the mechanism of the graft copolymerization of NIPAM was proved to be that of SET-LRP. The graft copolymer consisted of a hydrophilic PHEA backbone and hydrophilic thermo-sensitive PNIPAM side chains, thus PHEA-g-PNIPAM graft copolymer possessed a double hydrophilic nature. To the best of our knowledge, this is the first example of a double hydrophilic graft copolymer bearing a hydrophilic PHEA backbone. UV/vis, 1H NMR, and DLS results showed that aqueous solutions of PHEA-g-PNIPAM graft copolymers responded sharply to temperature and their LCSTs were dependent on the concentration of the solution. The development of the HECPMA trifunctional monomer will make a great contribution to the controlled synthesis of well-defined graft copolymers because PNIPAM hydrophilic side chains used in the current case can be easily extended to various hydrophilic polymers to generate a great deal of double hydrophilic graft copolymers with different stimuli-responsiveness.Acknowledgements
The authors are thankful for the financial support from National Natural Science Foundation of China (21274162 and 21474127) and Shanghai Scientific and Technological Innovation Project (14520720100, 14QA1404500, and 13ZR1464800).Notes and references
- S. Lee, K. Saito, H. R. Lee, M. J. Lee, Y. J. Shibasaki, Y. Oishi and B. S. Kim, Biomacromolecules, 2012, 13, 1190–1196 CrossRef CAS PubMed.
- E. Molina, J. Warnant, M. Mathonnat, M. Bathfield, M. In, D. Laurencin, C. Jerome, P. Lacroix-Desmazes, N. Marcotte and C. Gerardin, Langmuir, 2015, 31, 12839–12844 CrossRef CAS PubMed.
- S. Kumar, R. Acharya, U. Chatterji and P. De, Langmuir, 2013, 29, 15375–15385 CrossRef CAS PubMed.
- G. Yang, Z. Yang, C. G. Mu, X. D. Fan, W. Tian and Q. Wang, Polym. Chem., 2015, 6, 3382–3386 RSC.
- M. Bathfield, J. Warnant, C. Gerardin and P. Lacroix-Desmazes, Polym. Chem., 2015, 6, 1339–1349 RSC.
- Y. C. Tang, L. Liu, J. H. Wu and J. X. Duan, J. Colloid Interface Sci., 2013, 397, 24–31 CrossRef CAS PubMed.
- Y. Y. Jiang, G. H. Liu, X. R. Wang, J. M. Hu, G. Y. Zhang and S. Y. Liu, Macromolecules, 2015, 48, 764–774 CrossRef CAS.
- R. I. S. Romao, M. Beija, M. T. Charreyre, J. P. S. Farinha, A. M. P. S. G. Silva and J. M. G. Martinho, Langmuir, 2010, 26, 1807–1815 CrossRef CAS PubMed.
- S. Petrova, C. G. Venturini, A. Jager, E. Jager, M. Hruby, E. Pavlova and P. Stepanek, RSC Adv., 2015, 5, 62844–62854 RSC.
- P. Pramanik and S. Ghosh, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2444–2451 CrossRef CAS.
- N. Chan, S. Y. An, N. Yee and J. K. Oh, Macromol. Rapid Commun., 2014, 35, 752–757 CrossRef CAS PubMed.
- M. Khimani, S. Yusa, A. Nagae, R. Enomoto, V. K. Aswal, E. Kesselman, D. Danino and P. Bahadur, Eur. Polym. J., 2015, 69, 96–109 CrossRef CAS.
- Y. H. Wu, H. M. Hu, J. M. Hu, T. Liu, G. Y. Zhang and S. Y. Liu, Langmuir, 2013, 29, 3711–3720 CrossRef CAS PubMed.
- J. M. Hu, M. R. Whittaker, S. H. Yu, J. F. Quinn and T. P. Davis, Macromolecules, 2015, 48, 3817–3824 CrossRef CAS.
- J. B. Li, J. Liang, W. L. Wu, S. J. Zhang, K. Zhang and H. Y. Zhou, New J. Chem., 2014, 38, 2508–2513 RSC.
- S. M. Brosnan, H. Schlaad and M. Antonietti, Angew. Chem., Int. Ed., 2015, 54, 9715–9718 CrossRef CAS PubMed.
- M. Hartlieb, D. Pretzel, M. Wagner, S. Hoeppener, P. Bellstedt, M. Gorlach, C. Englert, K. Kempe and U. S. Schubert, J. Mater. Chem. B, 2015, 3, 1748–1759 RSC.
- N. Morimoto, T. Wazawa, Y. Inoue and M. Suzuki, RSC Adv., 2015, 5, 14851–14857 RSC.
- A. Matuszewska, M. Uchman, A. Adamczyk-Wozniak, A. Sporzynski, S. Pispas, L. Kovacik and M. Stepanek, Biomacromolecules, 2015, 16, 3731–3739 CrossRef CAS PubMed.
- N. Morimoto, K. Muramatsu, T. Wazawa, Y. Inoue and M. Suzuki, Macromol. Rapid Commun., 2014, 35, 103–108 CrossRef CAS PubMed.
- Q. Jin, G. Y. Liu and J. Ji, Chin. J. Chem., 2014, 32, 73–77 Search PubMed.
- A. Wibowo, K. Osada, H. Matsuda, Y. Anraku, H. Hirose, A. Kishimura and K. Kataoka, Macromolecules, 2014, 47, 3086–3092 CrossRef CAS.
- J. P. Wu, Z. Wang, Y. H. Yin, R. Jiang and B. H. Li, Macromolecules, 2015, 48, 8897–8906 CrossRef CAS.
- M. Uchman, S. Pispas, L. Kovacik and M. Stepanek, Macromolecules, 2014, 47, 7081–7090 CrossRef CAS.
- E. Y. Zeynep, D. Antoine, C. Brice, B. Frank and J. Christine, J. Mater. Chem. B, 2015, 3, 7227–7236 RSC.
- G. Layrac, M. Destarac, C. Gérardin and D. Tichit, Langmuir, 2014, 30, 9663–9671 CrossRef CAS PubMed.
- X. L. Hu, Y. Li, T. Liu, G. Y. Zhang and S. Y. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 15551–15560 CAS.
- V. Dordovic, M. Uchman, M. Reza, J. Ruokolainen, A. Zhigunov, O. I. Ivankov and P. Matejicek, RSC Adv., 2016, 6, 9884–9892 RSC.
- E. Seo, S. J. Ko, S. H. Min, J. Y. Kim and B. S. Kim, Chem. Mater., 2015, 27, 4789–4798 CrossRef CAS.
- N. Mebarek, R. Vicente, A. Aubert-Pouessel, J. Quentin, A. L. Mausset-Bonnefont, J. M. Devoisselle, C. Jorgensen, S. Bégu and P. Louis-Plence, Eur. J. Pharm. Biopharm., 2015, 92, 216–227 CrossRef CAS PubMed.
- C. Feng, Z. Shen, L. N. Gu, S. Zhang, L. T. Li, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5638–5651 CrossRef CAS.
- C. Feng, L. N. Gu, D. Yang, J. H. Hu, G. L. Lu and X. Y. Huang, Polymer, 2009, 50, 3990–3996 CrossRef CAS.
- L. N. Gu, Z. Shen, C. Feng, Y. G. Li, G. L. Lu, X. Y. Huang, G. W. Wang and J. L. Huang, J. Mater. Chem., 2008, 18, 4332–4340 RSC.
- X. Y. Jiang, C. Feng, G. L. Lu, Y. J. Li and X. Y. Huang, Polym. Chem., 2013, 4, 3876–3884 RSC.
- J. F. Gohy, Adv. Polym. Sci., 2005, 190, 65–136 CrossRef CAS.
- K. Yu and A. Eisenberg, Macromolecules, 1998, 31, 3509–3518 CrossRef CAS.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- C. M. Schilli, M. F. Zhang, E. Rizzardo, S. H. Thang, Y. K. Chong, K. Edwards, G. Karlsson and A. H. E. Muller, Macromolecules, 2004, 37, 7861–7866 CrossRef CAS.
- H. I. Lee, J. Pietrasik, S. S. Sheiko and K. Matyjaszewski, Prog. Polym.Sci., 2010, 35, 24–44 CrossRef CAS.
- X. Y. Jiang, G. L. Lu, C. Feng and X. Y. Huang, Polym. Chem., 2014, 5, 4915–4925 RSC.
- G. L. Cheng, A. Boker, M. F. Zhang, G. Krausch and A. H. E. Muller, Macromolecules, 2001, 34, 6883–6888 CrossRef CAS.
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759–785 CrossRef CAS.
- G. Cavallaro, M. Licciardi, M. D. Stefano, G. Pitarresi and G. Giammona, Macromolecules, 2009, 42, 3247–3257 CrossRef CAS.
- Y. Q. Zhang, Z. Shen, D. Yang, C. Feng, J. H. Hu, G. L. Lu and X. Y. Huang, Macromolecules, 2010, 43, 117–125 CrossRef CAS.
- C. Feng, Y. J. Li, D. Yang, J. H. Hu, X. H. Zhang and X. Y. Huang, Chem. Soc. Rev., 2011, 40, 1282–1295 RSC.
- X. Y. Jiang, Y. J. Li, G. L. Lu and X. Y. Huang, Polym. Chem., 2013, 4, 1402–1411 RSC.
- C. Feng, G. L. Lu, G. Sun, X. W. Liu and X. Y. Huang, Polym. Chem., 2014, 5, 6027–6038 RSC.
- H. Liu, S. Zhang, C. Feng, Y. J. Li, G. L. Lu and X. Y. Huang, Polym. Chem., 2015, 6, 4309–4318 RSC.
- B. B. Xu, G. X. Gu, C. Feng, X. Jiang, J. H. Hu, G. L. Lu, S. Zhang and X. Y. Huang, Polym. Chem., 2016, 7, 613–624 RSC.
- K. Matyjaszewski, S. G. Gaynor and A. H. E. Muller, Macromolecules, 1997, 30, 7034–7041 CrossRef CAS.
- S. Pal, S. G. Roy and P. De, Polym. Chem., 2014, 5, 1275–1284 RSC.
- A. P. Bapat, J. G. Ray, D. A. Savin, E. A. Hoff, D. L. Patton and B. S. Sumerlin, Polym. Chem., 2012, 3, 3112–3120 RSC.
- M. Lejars, A. Margaillan and C. Bressy, Polym. Chem., 2013, 4, 3282–3292 RSC.
- M. Kaupp, T. Tischer, A. F. Hirschbiel, A. P. Vogt, U. Geckle, V. Trouillet, T. Hofe, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2013, 46, 6858–6872 CrossRef CAS.
- Y. Y. Zhang, Z. Z. Cheng, X. R. Chen, W. Zhang, J. H. Wu, J. Zhu and X. L. Zhu, Macromolecules, 2007, 40, 4809–4817 CrossRef CAS.
- S. H. Wang, Q. X. Shen, M. H. Nawaz and W. A. Zhang, Polym. Chem., 2013, 4, 2151–2157 RSC.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- M. J. Monteiro, T. Guliashvili and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1835–1847 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2745–2754 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- T. Guliashvili and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1607–1618 CrossRef CAS.
- G. Lligadas, B. M. Rosen, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8360–8364 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Macromolecules, 2009, 42, 2379–2386 CrossRef CAS.
- N. H. Nguyen, H. J. Sun, M. E. Levere, S. Fleischmann and V. Percec, Polym. Chem., 2013, 4, 1328–1332 RSC.
- M. E. Levere, N. H. Nguyen, X. F. Leng and V. Percec, Polym. Chem., 2013, 4, 1635–1647 RSC.
- S. R. Samanta, H. J. Sun, A. Anastasaki, D. M. Haddleton and V. Percec, Polym. Chem., 2014, 5, 89–95 RSC.
- S. R. Samanta and V. Percec, Polym. Chem., 2014, 5, 169–174 RSC.
- S. R. Samanta, R. L. Cai and V. Percec, Polym. Chem., 2014, 5, 5479–5491 RSC.
- S. R. Samanta, V. Nikolaou, S. Keller, M. J. Monteiro, D. A. Wilson, D. M. Haddleton and V. Percec, Polym. Chem., 2015, 6, 2084–2097 RSC.
- S. R. Samanta, R. L. Cai and V. Percec, Polym. Chem., 2015, 6, 3259–3270 RSC.
- M. Gavrilov, T. J. Zerk, P. V. Bernhardt, V. Percec and M. J. Monteiro, Polym. Chem., 2016, 7, 933–939 RSC.
- M. F. Zhang, T. Breiner, H. Mori and A. H. E. Muller, Polymer, 2003, 44, 1449–1458 CrossRef CAS.
- Z. F. Jia, Q. Fu and J. L. Huang, Macromolecules, 2006, 39, 5190–5193 CrossRef CAS.
- L. N. Gu, Z. Shen, S. Zhang, G. L. Lu, X. H. Zhang and X. Y. Huang, Macromolecules, 2007, 40, 4486–4493 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- J. Wang, P. Gao, L. Ye, A. Y. Zhang and Z. G. Feng, Polym. Chem., 2011, 2, 931–940 RSC.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS PubMed.
- M. Gao, X. R. Jia, G. C. Kuang, Y. Li, D. H. Liang and Y. Wei, Macromolecules, 2009, 42, 4273–4281 CrossRef CAS.
- J. Xu, J. Ye and S. Y. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- J. Ramos, A. Imaz and J. Forcada, Polym. Chem., 2012, 3, 852–856 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00489j |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |