
A review of metal oxynitrides for photocatalysis
Manan
Ahmed
* and
Guo
Xinxin
Department of Environmental Engineering, Faculty of Engineering and Green Technology, Universiti Tunku Abdul Rahman, Perak, Malaysia. E-mail: manan_ahmed@live.com
First published on 26th January 2016
Abstract
Photocatalytic hydrogen (H2) production represents a very promising but challenging contribution to a clean, sustainable and renewable energy system. Photocatalytic water splitting into hydrogen and oxygen is a method to directly convert solar energy into storable chemical energy, and has received considerable attention for use in large scale solar energy utilization because of its great potential for low cost and clean energy production. Developing efficient and cost-effective photocatalysts for water splitting is a growing need, and semiconductor photocatalysts have recently attracted more attention due to their stability and simplicity. Over the past few decades, various metal oxide photocatalysts for water splitting have been developed and their photocatalytic application studied under UV irradiation. To harness solar energy efficiently, a recent main concern has been the development of visible-light (λ > 400 nm) active photocatalysts for water splitting. Metal oxynitrides are emerging materials that may exhibit the properties of both oxides and nitride. Metal oxynitride photocatalysts are of significant interest in the field of photocatalytic water splitting as the observed small band gaps lead to activity in the visible range. Titanium, tantalum, niobium, gallium and zirconium form important photocatalysts which show promise in visible light-driven photoreactions. Along with perovskite structures, development of double complex perovskite oxynitrides that are active under visible light are also reviewed. This review article provides a broad overview of the development of water-splitting photocatalysts for generating hydrogen, summarizing the current state of work with a focus on the recent progress in visible-light induced overall water splitting on oxynitride photocatalysts.
 Manan Ahmed | Manan Ahmed received his degree in Industrial Chemistry from GC University Lahore, Pakistan. Currently he is a Master of Engineering Science candidate at Universiti Tunku Abdul Rahman, under the supervision of Dr Guo Xinxin. His main research focuses on the design, synthesis and analysis of photocatalysts, inorganic metal complexes and analysis of atmospheric contamination. |
 Guo Xinxin | Dr Guo Xinxin was born in Jilin, China. After completing a bachelor's degree in Environmental Chemistry at Jilin University, she went to the UK for further studies and obtained a master's degree in Environmental Sciences from the University of Nottingham, UK, in 2004, and then she did her PhD in the University of East Anglia involving the study of aerosol chemistry of phenols in the atmosphere. In 2010, she joined the Faculty of Engineering and Green Technology, Universiti Tunku Abdul Rahman, Malaysia. She currently holds the position of Assistant Professor at the Department of Environmental Engineering. Her research interests include indoor and outdoor air pollution, chemistry of atmospheric aerosols, and chemistry of organic acids and metal–organic complexes in the atmosphere, but now she is concentrating on atmospheric surfactants, their characteristics, concentration, composition around the lake ecosystems and photocatalytic water splitting. |
1. Introduction
Providing clean and renewable energy is arguably the most important challenge facing humanity in the 21st century. The world's energy demand is projected to more than double by mid-century and more than triple by 2100.1 One of the most important goals of our modern society is to construct a sustainable environment. Growing environmental concerns related to the extensive use of non-sustainable fossil fuels (oil, natural gas and coal) and a constantly increasing energy demand will force mankind, sooner or later, to tap into clean and sustainable sources of energy.Capturing solar energy by splitting water into hydrogen and oxygen has long been considered a favorable idea, in 1874 Jules Verne, recognizing the finite supply of coal and potential of hydrogen derived from water electrolysis, made the comment that “water will be the coal of the future”.2 Water electrolysis by means of solar light mimics photosynthesis by converting water into H2 and O2 using inorganic photo-semiconductors (photocatalyst) that catalyzed the water-splitting reaction. Hydrogen is a clean energy carrier because the chemical energy stored in the H–H bond is easily released when it combines with oxygen, yielding only water vapor as the reaction by-product. Hydrogen (H2) is widely considered to be the future clean energy carrier in many applications, such as environmentally friendly vehicles, domestic heating, and stationary power generation. Currently, large-scale hydrogen production is performed by reforming or gasifying fossil fuels.3
Photocatalysis has long been studied and is expected to make a great contribution to both environmental treatment (emission cleaning and water purification) and renewable energy. Over the past few decades, the number of applications based on photocatalysis increased sharply; while a wide range of materials systems have been developed.4 Photocatalytic H2 production from water is one of the most promising ways to realize a hydrogen economy for three reasons. (1) This technology is based on photon (or solar) energy, which is a clean, permanent source of energy, and mainly water, which is a renewable resource; (2) it is an environmentally safe technology without undesirable by-products and pollutants; and (3) the photochemical conversion of solar energy into a storable form of energy, i.e. hydrogen allows to deal with the intermittent character and seasonal variation of the solar influx.
An essential prerequisite for the photocatalyst is its resistance to reaction at the solid/liquid interface, which can comprise its properties. However, it has proven difficult to find an ideal photocatalyst, which meets all the requirements (chemical stability, corrosion resistance, visible light harvesting and suitable band edges) that would render photocatalytic H2 production a viable alternative. Fortunately, nano science and nanotechnology have boosted the modification of existing photocatalysts and the discovery and development of new candidate materials.5 The rapidly increasing number of scientific publications constitutes clear bibliographical evidence for the significance of this hot topic. Since 2004, the number of publications on nano photocatalytic H2 production has increased by a factor of about 1.5 times every year. Many papers studied the impact of different nanostructures and nanomaterials on the performance of photocatalysts, since their energy conversion efficiency is principally determined by nano-scale properties.
Transition-metal oxynitrides are an important class of emerging materials that, in optimal cases, may combine the advantages of oxides and nitrides. Transition-metal oxynitrides are well suited for catalytic applications as they have good electrical conductivities and corrosion resistance. Their activities in hydro-denitrogenation and hydro-desulfurization resemble those of noble metals.6 Generally, oxynitride stabilities in air and moisture are greater than those of the pure nitrides, but with smaller band gaps than those of comparable oxides. This leads to useful electronic and/or optical properties, such as N-doping of TiO2 to tune the band gap from the ultraviolet to the visible region for photocatalysis. Many useful oxynitrides of high valence (d0 electron configuration) transition metals adopt the AMX3 (X = N/O) perovskite-type crystal structure; CaTaO2N and LaTaON2 solid solutions are non-toxic, red-yellow pigments; BaTaO2N has a high dielectric constant and acts as a photocatalyst in the decomposition of water, and EuNbO2N and EuWON2 are ferromagnetic and show colossal magneto resistances.7
We choose oxynitrides as a promising candidate to implement photocatalysis in this review because most of the existing photocatalysts are oxides.8 However, the band gap energies of metal oxides (>3 eV) are usually too large to absorb visible light and most of the material is active under UV light irradiation. This is mainly due to a too low valance band (VB) energy which is derived from the 2p orbitals of the oxygen atoms.9 Therefore, the photocatalyst that functions in the visible light region (λ = 400–800 nm) must be developed for the particle use for photocatalytic water splitting. A requirement for the visible light-induced photocatalyst is that the optimum band-gap energy should be less than 3 eV.10 To solve this problem, non-oxides such as nitrides and sulfides have been proposed, since their valance band (VB) position is usually higher in energy. Unfortunately, nitrides usually suffer from poor aqueous stability and cannot maintain photocatalytic activity in water over a long period of time.11 By changing the N/O ratio for a constant cationic composition it is possible to modulate the oxidation state of the cations modifying the physical properties. When introducing nitrogen into an oxidic network the physical and chemical properties may change even if this takes place at a doping level. Nitrogen is less electronegative than oxygen so the optical gap between the anion-based valence band and the cation-based conduction band decreases as oxide is substituted by nitride. Nitrogen is more polarizable than oxygen; the metal–ligand bond in metal oxynitride is more covalent and this increases the nephelauxetic effect (expansion of the electron cloud), and then decreases the electronic repulsion and the energy of the d orbitals.12 Indeed, the band gap values published for oxynitrides fall into the interval of 1.6–3.3 eV. The band gap overlap with the solar spectrum makes this class of materials interesting for applications as visible-light driven photocatalysts.13
The review presents the progress in the study of photocatalysts for overall water splitting, with a focus on recent advances in visible-light water splitting on metal oxynitride photocatalysts.
2. Mechanism of photocatalytic water splitting or hydrogen production
Photocatalytic water splitting is not a catalytic process according to the traditional definition of catalysis because it is an uphill reaction. It can also be called photo-induced water splitting on a semiconductor. Photocatalytic water splitting involves a sequence of multiple competing steps, including light harvesting, charge generation, separation, transportation, recombination and surface reaction.14 Generally, inorganic-based semiconductors are used for photocatalysis and their electronic structure plays a key role in photocatalytic water splitting. An ideal semiconductor/photocatalyst should have a band gap, Eg, that constitutes the energy difference between the conduction band (CB) and the valence band (VB) with sufficient energy to drive the water splitting reaction. Solid materials have an electronic structure with some empty and some filled electronic states. If there is an energy gap between these two sets of electronic states, the material is classified as a semiconductor or insulator. The filled and empty energy states are called the valance and conduction band, and the energy gap between the valance and conduction band is called the band gap.The basic working principle of photocatalysis is simple and similar to electrolysis. In the ground state, all electrons exist in the valance band (VB) and under visible light irradiation the semiconductor (photocatalyst) absorbs a photon to excite an electron from the valence band to the conduction band: resulting in an excited state and the formation of an electron (e−)–hole (h+) pair. The lifetime of an e−–h+ pair is only a few nanoseconds, but this may be enough to promote redox reaction. The photo-generated electrons and holes cause redox reactions similarly to electrolysis. These photo-generated electrons and hole may recombine in the bulk or on the surface of a photocatalyst. The two protons, which are required to generate hydrogen gas, can use the electrons that are excited in the photocatalyst. The hole in the valence band can be filled with an electron produced by oxygen generation (Fig. 1).15
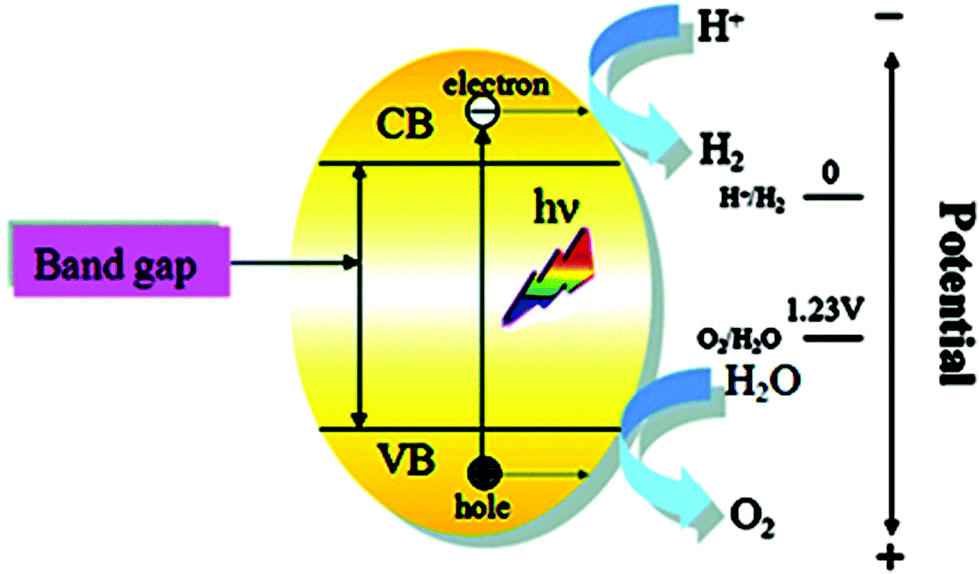 | ||
| Fig. 1 Mechanism of photocatalytic water splitting (reproduced from ref. 15 with permission from the Royal Society of Chemistry). | ||
One requirement for photocatalytic water splitting is that the conduction band (CB) and valence band (VB) for a semiconductor material should straddle the redox potentials for H+/H2 and H2O/O2. For H2 production to occur, the conduction band-edge must be more negative than the reduction potential of H+ to H2 (EH+/H2 = 0 V vs. NHE at pH = 0), while the valance band top-edge should be more positive than the oxidation potential of H2O to O2 (EO2/H2O = 1.23 vs. NHE at pH = 0) for O2 formation from water to occur.16 Thermodynamically, the theoretical minimum photon energy required for splitting water into hydrogen and oxygen corresponding to a 2-electron transfer step is 1.23 eV. This photon energy 1.23 eV is equivalent to a 1000 nm wavelength and it is located in the near infrared region. However, Murphy et al. determined that there are fundamental thermodynamic limits to the photo conversion efficiency, which depend on the band-gap of the semiconductor being used, and on the spectrum of the incident radiation. These limiting efficiencies cannot be achieved, due to losses such as the incomplete absorption of the incident radiation in the semiconductor, reflection of the incident radiation from the surface of the semiconductor and other surfaces. The actual minimum required band gap for the photocatalytic overall water splitting is estimated to be approximately 2 eV.17 So that the visible light (400–800 nm in wavelength) that represents half of the solar energy spectrum can be thermodynamically used for water splitting. The challenge is to obtain an efficient photocatalyst that has an absorption edge in a longer wavelength and the ability to split water.18 However, in practice, a slightly higher photon energy is necessary due to the presence of over potentials, and a photon energy of 1.7 eV (730 nm) is thought to be a realistic goal.
3. Method of syntheses of oxynitrides
The methods of syntheses of nitride-based materials are less straightforward than for oxides because of their lower stability. To synthesize oxynitrides from oxides a suitable nitriding agent is required. The most common method for the syntheses of ternary oxynitrides is the thermal treatment at high temperature (between 1300 °C and 1600 °C) containing a stoichiometric mixture of metal oxides, carbonates and/or nitrides under a high-pressure N2(g), or a mixture N2/H2 (typically 90/10 or 95/5% v/v) if reducing conditions are required.19 The use of high pressure is expected to be a general and very convenient approach in oxynitrides synthesis since it suppresses the decomposition to oxides and nitrogen gas and it should be useful for the stabilization of new phases at moderate temperatures. High pressure is also expected to stabilize the oxynitride structure with high coordination numbers for cations and anions, as for instance in perovskites.12 However, very few direct solid-state syntheses of oxynitrides at high pressures have been reported.Ammonolysis of a precursor oxide has been well documented as a simple experimental technique and inexpensive method for the syntheses of a wide variety of oxynitride products. Ammonolysis of oxides under flowing NH3 gas has enabled many new oxynitrides to be synthesized in which ammonia behaves as a nitriding agent and additionally induces a change in the oxidation state of the involved transition metals. Due to the high bonding energy of the N to N triple bond, the application of N2 in syntheses usually requires high activation energy.20 The corresponding reaction between a suitable solid precursor and gaseous ammonia is often referred to as “thermal ammonolysis”.
Due to the large difference in the free energies of formation between the binary rare earth oxides and nitrides, the ammonolytic syntheses of rare earth quaternary oxynitrides usually start with ternary oxides (A2B2O7, ABO4 with a B-site cation being in its stable oxidation state) or oxide–carbonate mixtures (ACO3–BxOy), as the most often used precursors for the perovskite-type oxynitrides synthesis.21,22 Nitridation reaction are usually carried out in an alumina boat comprising the oxide precursor powder placed inside an alumina tube through which ammonia gas flows normally at a rate up to 30–40 L h−1 at 800 °C. The reaction between a precursor and ammonia is usually carried out at atmospheric pressure in the temperature region of T = 600–1000 °C, depending on the precursor and system used, with a heating rate of about 10 °C min−1. The ammonia flow rate depends on the reaction temperature; higher the temperature, higher the rate in order to minimize the dissociation of NH3 into dinitrogen and dihydrogen before reaching the product.22 The key parameters for the ammonolysis reaction are temperature, nitrogen flow rate; reaction time and sample placement in the reaction tube and the gas composition (NH3, N2 and H2) around the sample should be considered to optimize the purity of the oxynitride phase.23 Ammonia acts as a reducing and an oxidizing (nitriding) agent, this dual behavior nitriding and reducing is essential for the ammonolysis reaction.
Ammonia decomposition reactions on oxide surfaces are not well studied. However, it is suggested that ammonia dissociates at the surface, forming active nitriding species (N, NH, NH2) in a native state, and molecular hydrogen.24 The highly reactive hydrogen combines with the oxygen atoms from the oxides and eliminates them as water vapor; the formation of water vapor provides the thermodynamic driving force in the reaction, while nitrogen is introduced into the lattice through substitution. Several types of metallic compounds can be nitrides with the help of flowing ammonia gas by using adequate temperatures. At a sufficiently high temperature range of 450–1200 °C the use of ammonia gas at atmospheric pressure is equivalent to using N2(g)/H2(g) at high pressure.25
| Oxide + NH3(g) → Oxynitride + H2O(v)↑ |
Thin films of functional oxynitrides have been synthesized using physical and chemical methods. Pulsed laser deposition (PLD) was employed for the synthesis of thin films of perovskites with dielectric properties as BaTaO2N26 and reactive radio frequency magnetron sputtering has been used for the preparation of thin films of LaTiO2N with photo-catalytic applications.27 Nanostructured meso-porous nitrogen doped TiO2 films have been prepared by combining sol–gel, templating and ultrafast evaporation-induced self-assembly followed by controlled treatment in NH3 gas.28
4. Oxynitride photocatalyst for visible light utilization
Research in this field of photocatalysis was initiated by the pioneering work conducted by Honda and Fujishima in 1972.29 Since the discovery of the Honda–Fujishima effect, a number of experiments have been conducted to establish new stable photocatalytic powder systems which were mostly based on metal oxides and oxynitrides to stoichiometrically split water into H2 and O2.30 Many types of semiconductors over the past 40 years, with over 130 materials including oxides, nitrides, sulfides, carbides, and phosphides, have been reported to act as efficient particle photocatalysts for hydrogen evolution via water splitting. Unfortunately, most of the photocatalysts are active only under UV light, and materials active under visible light are quite limited. More recently non-oxide materials such as sulfides, oxynitrides and oxysulfides have been found to be active for H2 and O2 evaluation under visible light irradiation (Table 1). Until now, the quantum efficiency for overall water splitting over visible light driven particle photocatalysts only achieves low values and is still the current ‘bottleneck’ of hydrogen production from solar light.| Photocatalysts | |
|---|---|
| Titanium oxide and titanate oxide | Metal oxynitrides |
| TiO2–Cr–Sb | TaON |
| TiO2–N | Ti3O3N2 |
| SrTiO3–Cr–Ta | Zr3O3N2 |
| SrTiO3–Cr–Sb | Metal sulfides |
| La2Ti2O7–Cr | CdS |
| La2Ti2O7–Fe | CdS–CdO–ZnO |
| Other transition metal oxides | CdS–Zn |
| BiVO4 | ZnS–Cu |
| Ag3VO4 | ZnS–Ni |
| SnNb2O6 | ZnS–Pb |
| Nb2O5 | (AgIn)xZn2(1−x)S2 |
| Ta2O5 | Sm2Ti2S2O5 |
| BiTaO4 | (CuIn)xZn2(1−x)S2 |
| ZnO | (CuAg In)xZn2(1−x)S2 |
| Bi2O3 | Na14In17Cu3S35 |
| WO3 | Metal perovskite oxynitride |
| Ga2O3 | LaTaON2 |
| Mixed metal oxynitrides | LaMgxTa1−xO1+3xN2−3x |
| (Ga1−xZnx)(N1−xOx) | CaTaO2N |
| (Zn1+xGe)(N2Ox) | SrTaO2N |
| Metal nitride | BaTaO2N |
| Ta3N5 | Sr2Nb2O7−xNx |
| Metal free nitride | LaTiO2N |
| C3N4 | CaNbO2N |
| SrNbO2N | |
| BaNbO2N | |
| Y2Ta2O5N2 | |
Solar spectrum comprises 4–5% of ultraviolet light whereas approximately 40% of solar photons are in the visible region. Most of the reported photocatalysts are metal oxides. A major drawback of pure metal oxides is the large band gap that restricts the utilization of visible light which is the main component of the solar spectrum and it can only be activated upon irradiation of UV light (λ ≤ 387 nm for anatase), limiting the practical efficiency of solar application.31 Therefore current research focus is to develop a stable visible light responsive photocatalyst capable of achieving overall water splitting. Only a handful of bulk oxynitride materials of d0 and d10 electronic configuration transition metal cations are the primary usable metal components for a water splitting photocatalyst under visible light because their orbitals can form a suitable conduction band. The valence bands of these oxynitride materials are populated by hybridized N2p and O2p orbitals while conduction is mostly composed of the empty d orbitals of the corresponding metal ions, resulting in more negative valence band levels and smaller band gaps compared to those of conventional oxide semiconductors, allowing visible-light induced H2 production from water.16,32–34 In this case, it is expected that the conduction band levels are not significantly affected because the cationic components are not changed.35 Since N3− has a similar ionic radius to that of O2− partial or full substitution of N3− for all or part of the O2− in the oxide is possible in some cases.
The photocatalyst materials for water splitting are usually in the form of particles or powders suspended in aqueous solution in which each particle acts as a micro-photoelectrochemical cell that performs both oxidation and the reduction reactions of water on its surface.
This eliminates the need for a conducting substrate, enabling the use of conventional synthesis routes, which are therefore much simpler and less expensive to develop and use photoelectrochemical cells. However, particulate photocatalytic systems have disadvantages when compared to photo electrochemical cells with regard to the separation of charge carriers, which is not as efficient as with a photoelectrode system, and there are difficulties associated with the effective separation of the stoichiometric mixture of oxygen and hydrogen to avoid the backward reaction.3
4.1. Tantalum oxynitrides
Numerous typical metal oxides with d10-electronic configurations have been demonstrated to be usable as photocatalysts for efficient overall water splitting. Metal oxide photocatalysts have attracted extensive research attention for the purposes of solar energy conversion and environmental remediation. Overall water splitting using a photocatalyst is an attractive solution for the supply of clean and recyclable hydrogen energy. Although a large number of photocatalysts have been proposed to date, most function only in the ultraviolet (wavelength <400 nm) region (e.g., TiO2) due to the inherently large band gap of metal oxides.36 Metal oxynitrides are candidates for visible-light responsive photocatalysts, and promising results have been reported for TaON which have suitable conduction and valence band positions to produce hydrogen and oxygen from water. It contains nitrogen that forms the top of the valence band, and it makes its band gap energy smaller than those of metal oxides. As a result, it absorbs visible light more effectively and has better photocatalytic ability. Domen et al. demonstrated that TaON absorbed the visible light up to ca. 530 nm, with the valence band at 2.23 eV versus NHE, and exhibited a high quantum efficiency of 34% for water photooxidation (oxygen photoevolution) in the presence of a sacrificial oxidizing reagent such as Ag+.37 They also reported that tantalum nitride (Ta3N5) exhibited high photocatalytic activity for water photo-oxidation as well, though its valence band was mainly composed of N2p orbitals and located at 1.6 V versus NHE.38,39Tantalum oxynitride is one of the most attractive materials not only for the half reaction, which generates hydrogen or oxygen from two water molecules, but also for the full reaction, which generates hydrogen and oxygen molecules from water simultaneously in the same reactor.40 Its crystal structure is usually monoclinic, isostructural to baddeleyite ZrO2 (β-TaON), but there is also monoclinic VO2 (B) (γ-TaON) that is metastable consisting of edge-sharing octahedra of tantalum in layers that are connected through the Ta(ON)6 octahedral corners41 and from neutron diffraction studies; oxygen and nitrogen are ordered in the two anion sites of the baddeleyite structure. However, tantalum is hepta-coordinate and there are two crystallographically independent sites for anions where N and O are in order42 (Fig. 2). This order can be rationalized in many oxynitrides, mix anionic compound and predicted by using Pauling's second crystal rule.
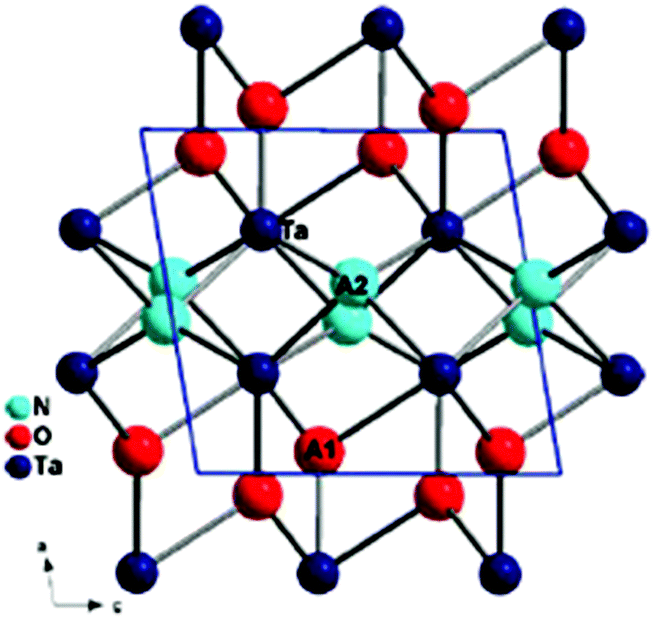 | ||
| Fig. 2 Crystal structure of TaON showing the coordination of two available anion sites occupied respectively by O and N (reproduced from ref. 12 with permission from the Royal Society of Chemistry). | ||
From the band structure of β-TaON, as calculated by density functional theory (DFT), the bottom of the conduction band is based on empty Ta5d orbitals similar to Ta2O5, whereas the top of the valence band consists of a hybridization of N2p and O2p orbitals in which the N2p contribution is larger than that of O2p.43 The potential energy of the hybridized orbital is higher than that of an O2p orbital in an oxide, resulting in smaller band gap energy sufficient to absorb visible light (Fig. 3). However, problems will arise on the stability and efficiency of the TaON photocatalyst because it is expected that the metal–N bonds in N-doped metal oxynitride are weak compared with the metal–O bonds in metal oxides and that photogentrated holes are trapped at higher N2p induced levels at the surface, resulting in a large decrease in the oxidation power compared with the O2p valance band holes. In addition, N-doping will introduce more or less distortion in the metal oxide crystal structures leading to their destabilization.
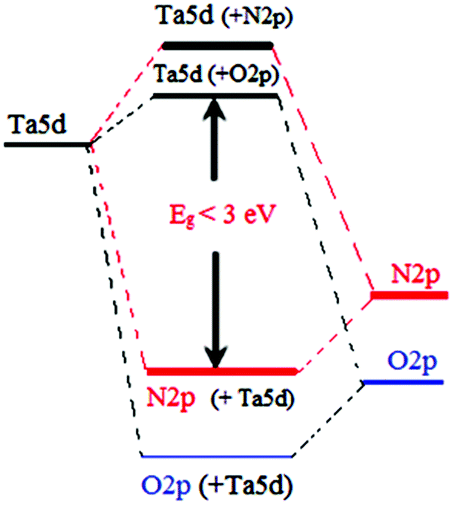 | ||
| Fig. 3 Schematic band structure of tantalum oxynitride (reproduced from ref. 46 open access). | ||
This problem was studied by Nakamura et al. and they reported the mechanism of water photooxidation at the surface of TaON photocatalysis using Fe3+ and clarified how water photoxidation proceeds at the metal oxynitride surface. Their experiments have indicated that the TaON surface is oxidized under visible-light irradiation, indicating that the oxygen photo-evolution on TaON actually occurs on a thin Ta–oxide over-layer.44
The synthesis of β-TaON is usually done by ammonolysis of commercial Ta2O5 powder at 800–850 °C, using either low flow rates of NH3 (typically 20–50 cm3 min−1) or some PH2O (bubbling NH3 in water) to prevent the formation of a more stable phase, the nitride Ta3N5. As the nitridation reaction precedes the O2− anions in the precursor Ta2O5 are gradually replaced by N3− anions from the NH3. It can also be prepared by treating Ta2O5 under nitrogen gas saturated with NH4OH. Nanoparticles of β-TaON are prepared by a Ca-assisted urea route and efficient photo-anodes have been prepared by electrophoretic deposition.45
4.2. Gallium–zinc oxynitride
The most active photocatalyst under visible light irradiation is a gallium–zinc oxynitride solid solution of GaN and ZnO also known as (Ga1−xZnx)(N1−xOx), one of the materials that meets all the previously stated conditions with highest photocatalytic activity for over all water splitting.32 This photocatalyst has an absorption edge at ∼500 nm and a quantum yield of 5.2% for 410 nm light, as shown in Fig. 4. The figure shows that the absorption edges of (Ga1−xZnx)(N1−xOx) are located at longer wavelengths than those of GaN or ZnO, but wavelengths shift to shorter with increasing nitridation. The band gap energy of the (Ga1−xZnx)(N1−xOx) is roughly estimated to be 2.6–2.8 eV based on the onsets of the diffuse reflectance spectra, which is substantially smaller than that for either GaN (3.4 eV) or ZnO (3.2 eV). | ||
| Fig. 4 Diffuse reflection UV-visible spectra of (Ga1−xZnx)(N1−xOx) with varying x = 6–8. GaN and ZnO have absorption edges in the UV region (<400 nm), but the solid solution has absorption in the visible region (reproduced from ref. 32 open access). | ||
GaN is a well-known material with a bandgap of 3.4 eV and has been studied extensively for application in light-emitting diodes and laser diodes.47 GaN has also been examined as a photoelectrode, and it has been confirmed to have the potential for overall water splitting under UV irradiation.48 The bandgap of ZnO is 3.2–3.4 eV, and it has been used for many applications such as in light-emitting diodes and gas sensors.49,50 GaN and ZnO are well known materials with wurtzite crystal structures with almost the same lattice parameters and band gaps of ca. 3.4 and 3.2 eV, respectively, providing response only in the UV region (Fig. 5). However, the solid solution of the oxide and nitride, (Ga1−xZnx)(N1−xOx) with d10 electronic configuration, which retains the wurtzite structure, exhibits a band gap of 2.6–2.8 eV, suitable for response in the visible region. DFT calculation indicates that the small band gap is attributed to the repulsion of N2p–Zn3d (p–d) electrons in the upper valance band, thus defining the valance band maximum of (Ga1−xZnx)(N1−xOx).51 Therefore, the visible light response of the solid solution originates from the contribution of the Zn3d atomic orbital to the valance band formation, where the bonding between Zn and N atoms is formed as a result of the formation of the solid solution.
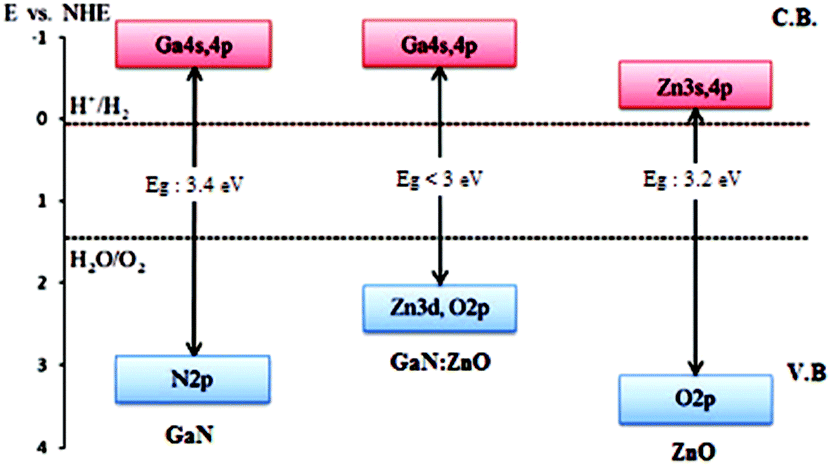 | ||
Fig. 5 Schematic illustration of the band structure of GaN, ZnO and GaN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ZnO solid solution (reproduced from ref. 55 with permission from the Royal Society of Chemistry). :ZnO solid solution (reproduced from ref. 55 with permission from the Royal Society of Chemistry). | ||
The photocatalytic activity of (Ga1−xZnx)(N1−xOx) for water splitting under visible light (at wavelengths longer than 400 nm) is greatly enhanced by loading the catalyst surface with nanoparticles of a mixed oxide of rhodium and chromium.52 Typically, nanoparticles of co-catalysts deposited on the surface of photocatalysts, as the hydrogen production sites, are used to enhance the rate of water reduction half-reaction. The main role of co-catalysts is to give away the photo excited electrons from the bulk of the photocatalyst and bring them to the water–photocatalyst interface. Noble metals or transition-metal oxides (e.g., Cr, Pt or Rh) are often used as co-catalysts to facilitate the water reduction half-reaction. Such co-catalysts are typically applied as nanoparticles to the photocatalyst surface by different methods including in situ photo deposition. In situ photo deposition allows the co-catalysts to be located selectively at the reaction sites without the need for an activation treatment.53 The rate of hydrogen evolution under visible light irradiation (λ > 400 nm) for GaN:ZnO solid solution was initially reported to be about 180 μmol h−1 g−1 using RuO2 as the co-catalyst.54
The GaN:ZnO solid solution photocatalyst is typically synthesized by nitridation of a mixture of Ga2O3 and ZnO (Zn:Ga = 1:1) under dry ammonia flow at high temperatures around 1223 k for 5–15 hours via the solid-state reaction process, which is the traditional method.56 Elemental analyses by inductively coupled plasma optical emission spectroscopy (ICP-OES) have revealed that the ratios of Ga to N and Zn to O in the as-prepared material are close to unity and that the N and O concentrations increase with the Ga and Zn concentrations, respectively.57
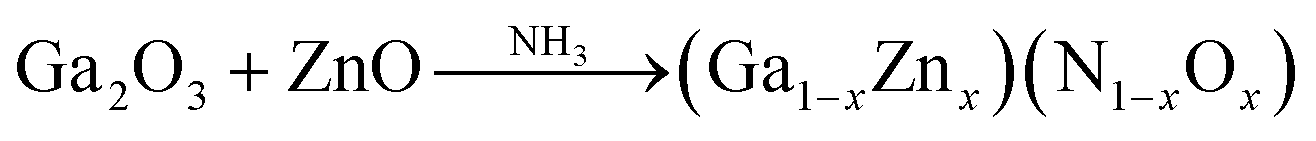
Although the photocatalyst prepared through the solid state reaction process demonstrates the highest activity for overall water splitting, long synthesis time and high temperatures are considered as drawbacks of this synthesis technique. The produced materials have a relatively low surface area, bulk defects and low Zn content (usually with Zn/(Zn + Ga) ratios of less than 15%). This composition range is far from the optimum needed to minimize the band gap. Moreover, the Zn/Ga ratio of the solid solution is very low, even with low synthesis times (<0.28 at 5 h) due to the volatilization of ZnO at high synthesis temperatures.56 Various attempts have been made to increase the Zn content without losing the crystallinity of the synthesized photocatalyst, including the application of different precursors in different amounts, different annealing times, as well as pre- and post-treatment techniques.
Madea et al., examined in detail the photocatalytic activities for water splitting on GaN:ZnO solid solutions for various compositions and with various loading amounts of the RuO2 cocatalyst. Among these, (Ga1−xZnx)(N1−xOx) with x = 0.12 loaded with a 5 wt% RuO2 showed the highest activity.54 H2 and O2 evolved steadily and stoichiometrically upon light irradiation, and the reaction continued even under visible light irradiation. The optical absorption edge and band gap for the solid solution with this composition were 482 nm and 2.68 eV, respectively. This was the first reproducible example of visible-light induced overall water splitting on a single photocatalyst system.55 It is proposed that the modification of (Ga1−xZnx)(N1−xOx) with Rh and Cr mixed–oxide nanoparticles increased the quantum efficiency for overall water splitting to 2–3% at 420–440 nm, which is approximately 10 times greater than that achieved using the RuO2-loaded catalyst. The function of the Rh–Cr–O mixed oxide nanoparticles is to trap the excited electron and holes to harness them for the photocatalytic splitting of water. The Rh3+ are the H2 evolution sites and Cr3+ oxide shells prevent the O2/H2 backward reaction to to form water and the bulk GaZnOx or their contact points are the possible O2 evolution sites. Interestingly, modification of (Ga1−xZnx)(N1−xOx) with either RhO2 or Cr oxide alone did not provide a significant increase in photocatalytic activity.57,58
4.3. Gallium–zinc–indium oxynitrides
A solid solution of (Ga1−xZnx)(N1−xOx), with the d10 electronic configuration has been reported to act as a photocatalyst for overall water splitting under visible light irradiation.32 It has been shown that the absorption edge of (Ga1−xZnx)(N1−xOx) depends on the zinc and oxygen concentration and the highest photocatalytic activity is obtained in a narrow compositional range near x = 0.18. The absorption edge band for this composition occurs at around ∼500 nm. For efficient use of solar energy, compounds exhibiting an absorption band at longer wavelengths are crucial.9,59Kamata et al. reported the new photocatalytic material Ga–Zn–In–O–N mixed oxynitride with d10 electronic configuration and a wide absorption band at visible wavelengths. One of the approaches to prepare new materials is to extend the absorption edge of (Ga1−xZnx)(N1−xOx) by forming a solid solution with indium nitride (InN), having the same wurtzite structure as (Ga1−xZnx)(N1−xOx). Ga–Zn–In–O–N mixed metal oxynitrides have a more suitable characteristic as InN have a narrow band gap (<1 eV) as compared to (Ga1−xZnx)(N1−xOx) (2.6–2.8 eV). It exhibits an absorption band near 600 nm and demonstrates a potential for overall water splitting under visible light irradiation.60
4.4. Titanium and zirconium oxynitrides
Wu et al. reported the theoretical aspect of the two new Ti3O3N2 and Zr3O3N2 photocatalysts. Both of these oxynitride materials are not yet reported and predicted for future work. Both of these compounds can be generated from the crystal structure of Ta3N5 and their crystal structures are shown in Fig. 6. The instability energy for Ti3O3N2 is ΔH 31.0 meV per atom and 1.0 meV per atom for Zr3O3N2. This suggests that both materials are expected to be synthesizable. The band edge position suggests that Ti3O3N2 is particularly interesting as its conduction and valance bands bracket the water redox levels and band gap predicted as 2.37 eV, is small enough for visible light absorption (Fig. 7). Comparing the band properties of Ti3O3N2 with TaON, the best oxynitride photocatalyst so far, it is found that both of them have their conduction and valance bands associated with the water redox levels, but the band gap of Ti3O3N2 is expected to be smaller than the band gap of TaON (2.83 eV in calculation and 2.4 eV in experiment). Therefore, Ti3O3N2 has a potential to exhibit better photocatalytic performance than TaON.11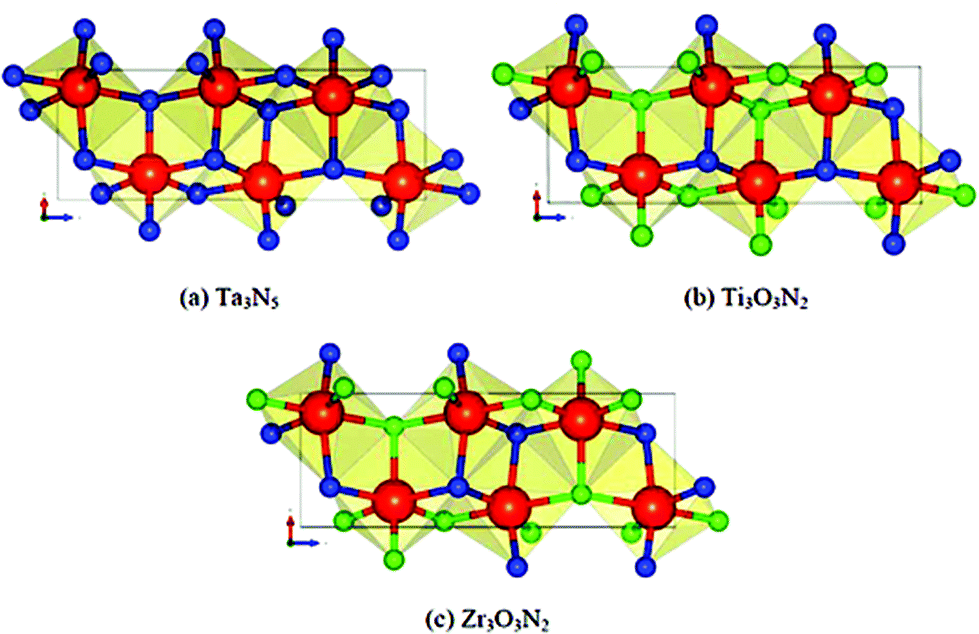 | ||
| Fig. 6 Comparison of crystal structure. Blue atoms are N, green atoms are O and red atoms are (a) Ta, (b) Ti, and (c) Zr (reproduced from ref. 11 with permission from the Royal Society of Chemistry). | ||
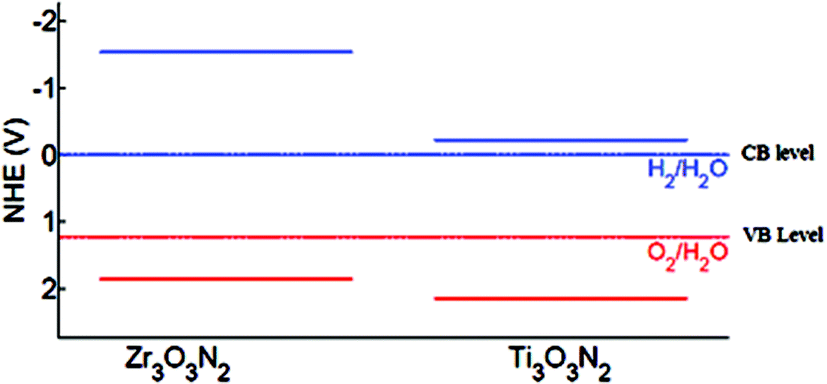 | ||
| Fig. 7 Band gap level of Zr3O3N2 and Ti3O3N2 with normal hydrogen electrode reference (reproduced from ref. 11 with permission from the Royal Society of Chemistry). | ||
Zr3O3N2 also has its conduction and valance bands associated with the water redox level but it is predicted that the band gap of Zr3O3N2 is large (3.40 eV). However, Fig. 7 suggests that the large band gap of Zr3O3N2 is mainly due to its wide conduction band (CB) level and predicted to be a good photocatalyst under UV irradiation without the presence of a metal co-catalyst because the conduction band (CB) has sufficient potential for H2 formation by reduction of water [E0(H2O/H2) = 0 eV at pH 0] and may also be visible light driven with some CB engineering.61 Shifting the CB downwards is relatively easy which can be achieved with cation doping. If its CB can be shifted to be slightly higher than the H2/H2O level while retaining its valance band position, the band gap will be reduced to 2.0 eV, making it a promising candidate for visible light driven photocatalysts.11
4.5. Perovskite oxynitrides
Perovskite-type materials are known for numerous kinds of applications because their crystal structure is very flexible towards substitutions since it is possible to control the properties of these materials by suitably modifying their composition,62 which may induce interesting physical properties such as colossal magneto-resistance, superconductivity, photocatalytic activity and visible light photo absorption.63 Oxynitride perovskites with the general formula ABO2−xN1+x were first reported by R. Marchand and co-workers and further investigated by several groups.64 Oxynitrides perovskites are generally derived from the corresponding oxides by introducing nitrogen into the anionic network. The resulting increase of negative charge is compensated by (i) cationic substitution at the A site (ii) changes in the oxidation state of the transition metal and (iii) reduction of the number of anions.62 Most substitutions concern the A-site and the transition metals in the B-site of the perovskite lattice, but substitution of the oxygen sites with the nitrogen content up to a maximum of 2.16 atoms per formula offers a powerful way to alter the electronic structure of the valence band.65 The conventional method for the synthesis of oxynitrides is ammonolysis. Synthesis of a perovskite type compound (ABO2N) is generally performed by reactions in NH3(g) between 600 °C and 1100 °C, starting with their oxide precursors (A2B2O7) or mixtures of oxides and oxysalts e.g. carbonates. The synthesis of the precursors was carried out by either soft-chemistry methods or conventional solid–solid reactions.66 The use of precursors is needed for rare earths as the reactivity of R2O3 (R = La, lanthanide) in NH3 is fairly low. However binary rare-earth oxides R2O3 usually do not react with transition metal oxides in NH3 at moderate temperatures so that the ammonolytic synthesis of rare-earth transition-metal oxynitrides requires the use of ternary oxides or very reactive, amorphous precursors. Other convenient methods are solid state reactions in N2 at higher temperatures (ca. 1500 °C), starting with the mixtures of oxynitrides and oxides. High pressure also stabilizes structures with high coordination numbers such as perovskites. However, very few methods of direct solid-state syntheses of oxynitrides at high pressures have been reported.67| A2B2O7 + 2NH3 → 2ABO2N + 3H2O |
Photocatalytic oxynitride-perovskites are a comparatively new research topic. The first report on the photocatalytic water splitting into H2 and O2 with an oxynitride photocatalyst appeared in 2002. Study of Kasahara et al. commented on the photocatalytic properties of LaTiO2N and La0.75Ca0.25TiO2.75N0.25 prepared by thermal ammonolysis of the citric route and that they produced corresponding oxide precursors. LaTaON2 has a perovskite structure with La3+ and Ta5+ at the A- and B-sites, respectively (Fig. 8).68 The tantalum, niobium and titanium oxynitride perovskites are promising candidates for the photocatalytic splitting of water under illumination with visible light. The band gaps and band edge positions of CaTaO2N, SrTaO2N, LaTaON2 and BaNbO2N are suitable for both water oxidation and reduction. They have smaller band gaps than TiO2 (1.5–2.5 eV) and are stable in aqueous solutions, and the presence of tantalum offers the promise of high efficiency because many of the highest quantum efficiency photocatalysts contain tantalum.69 For efficient conversion of solar energy, band gaps smaller than 2 eV (equivalent to an absorption edge of 620 nm) are convenient, and the band gaps of this group of compounds range from 1.8 eV (for LaTaON2) to 2.5 eV (for CaTaO2N). The tantalum perovskites are unable to perform overall water splitting. The LaTaON2 evolve H2 from water (620 nm) but they are not active in O2 production and show efficiencies in water reduction up to 20 percent.70
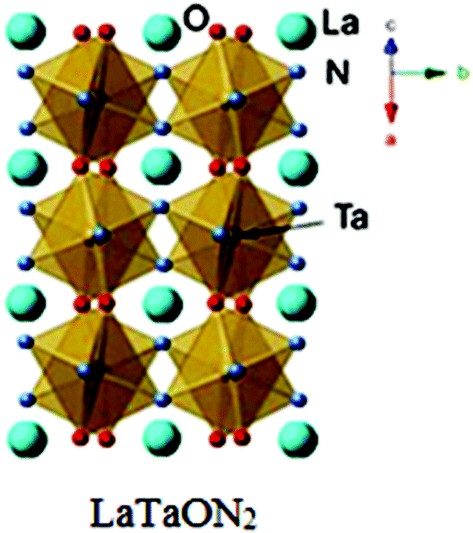 | ||
| Fig. 8 LaTaON2 perovskite structure (reproduced from ref. 71 open access source). | ||
However overall water splitting with visible light up to 600 nm has been recently reported for the double tantalum/magnesium perovskite with formula LaMgxTa1−xO1+3xN2−3x (x > 1/3) and it can be synthesized by thermal ammonolysis of the corresponding oxides and/or were synthesized by the controlled mixing of (1–3/2x) LaTaON2 and 3/2x LaMg2/3Ta1/3O3 (Fig. 9).71,72 The oxide precursors are prepared via a molecular route called the citric acid method. This method produces an oxide precursor wherein each component can be intimately mixed, and this precursor is suitable for the production of multicomponent crystalline oxynitrides.73
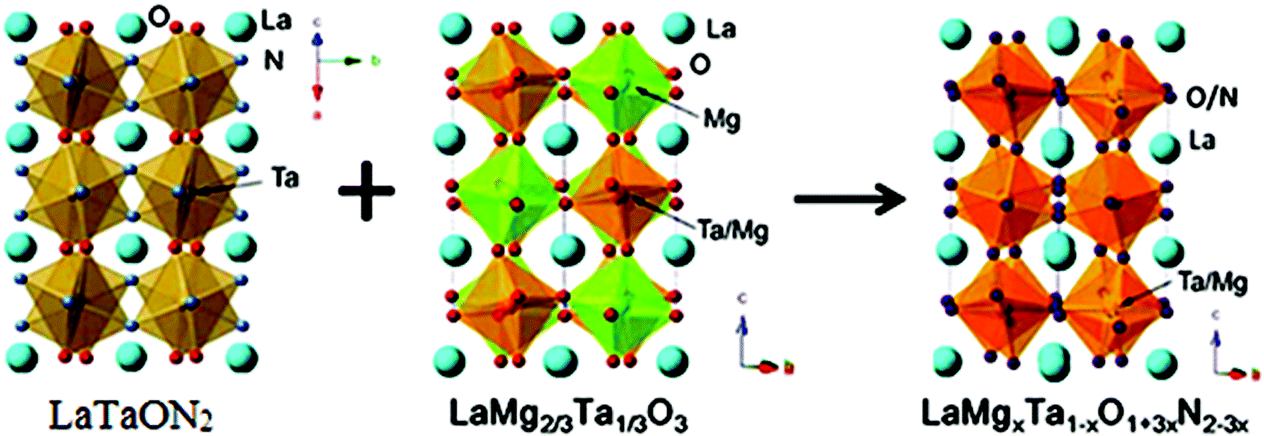 | ||
| Fig. 9 Synthesis and crystal structure of LaMgxTa1−xO1+3xN2−3x (reproduced from ref. 72 open access). | ||
The similar lattice constants of the precursors enabled the formation of uniform solid solutions with tunable band structures. UV/Vis diffuse reflectance spectra show that the wavelength of the absorption edge of LaMgxTa1−xO1+3xN2−3x is blue-shifted from 640 to 525 nm when the molar ratio of Mg2+ in the solid solution is increased from x = 0 to 0.6 (Fig. 10).71,72 The increase in band-gap energy was mainly attributed to the shift in the level of the valence band maximum as a result of the change in the O and N content and should be at least advantageous for O2 evolution. For these oxynitride solid solutions, the lower part of the conduction band mainly consists of Ta5d orbitals, while the upper part of the valence band consists mainly of N2p orbitals with a small contribution from O2p orbitals. The increase in the Mg content decreases the N/O ratio, which shifts the VBM downward.74 The concept for the band engineering by compositional tuning is schematically illustrated in Fig. 11. The LaMg1/3Ta2/3O2N solid solution (x = 1/3) with a band-gap energy of 2.08 eV was identified as the most suitable one for overall water splitting.75
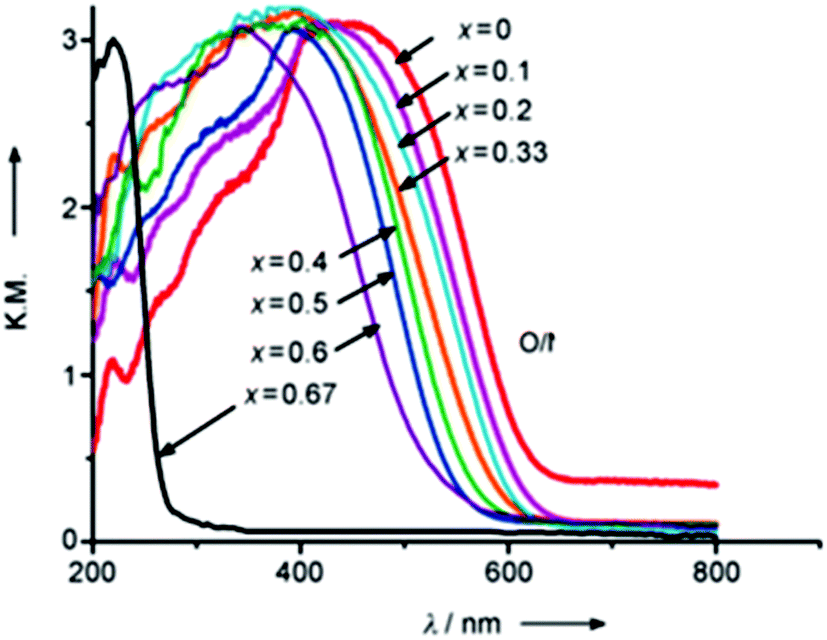 | ||
| Fig. 10 UV/Vis diffuse reflectance spectra of the LaMgxTa1−xO1+3xN2−3x photocatalyst (reproduced from ref. 72 open access). | ||
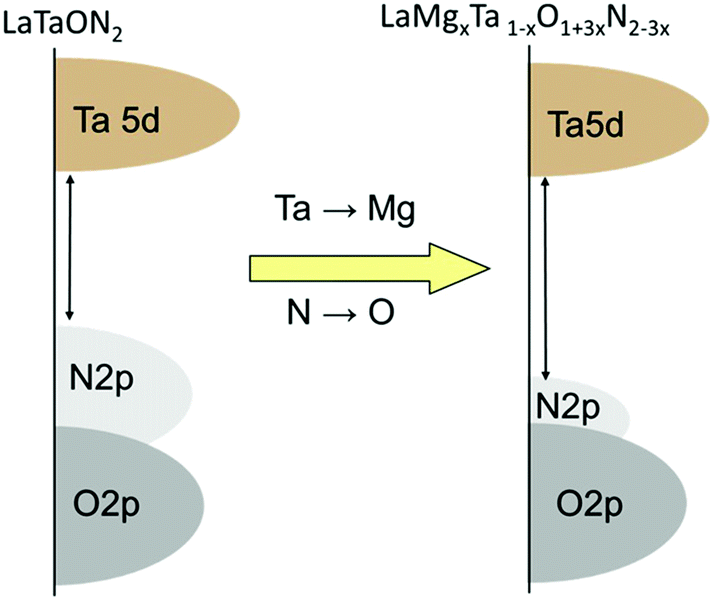 | ||
| Fig. 11 Schematic of band engineering by compositional tuning (reproduced from ref. 72 open access). | ||
Zhang et al. reported the n-type semiconductor LaTiO2N oxynitride (LTON) material with a perovskite structure and a band gap of 2.1 eV (∼600 nm absorption edge). LTON is favorable for water splitting due to low energy band positions and also prepared by relatively cheap material. In the previous work Kasahara et al. found that by using this material, water splitting for H2 and O2 production in the presence of a sacrificial reagent is theoretically and experimentally feasible under visible light irradiation. Although a quantum efficiency of ∼5% for water oxidation has been achieved using IrO2-loaded LTON. The quantum efficacy can be further improved if a precious co-catalyst, IrO2-loaded LTON, was replaced with a more earth abundant metal species.
The LaTiO2N CoOx-modified photocatalyst was synthesized by the flux (FX) and polymerized complex (PC) method using oxide precursors (La2Ti2O7) under a flow of NH3 at 1223 K. The obtained nitride powders are referred to as FX-LTON and PC-LTON, respectively. Cobalt oxide (CoOx) was deposited on LaTiO2N samples as a co-catalyst by an impregnation method from aqueous Co(NO3)2 solution, followed by NH3 treatment at 973 K and calcination at 473 K in air.
The CoOx co-catalyst was most efficient as compared to IrO2 which is well known as one of the most efficient water oxidation co-catalysts and also resulted in marked enhancement in the water oxidation activity. The optimized CoOx–LTON was found with a high efficiency of 27.1 ± 2.6% at 440 nm which significantly exceeds the value reported for the pervious particulate photo-catalyst with a 600 nm absorption edge. Further they noticed that FX-LTON gave a higher performance than PC-LTON. The significantly enhanced quantum efficiency can be attributed to improvements in both the photocatalyst and co-catalyst components.68,76
BaNbO2N was synthesized by nitriding the corresponding oxide precursor Ba5Nb4O15 under an NH3 flow. The oxide precursor, Ba5Nb4O15 was prepared by calcining a mixture of BaCO3 and Nb2O5, with NaCl added as a flux at 1173 K. After a slow cool-down to room temperature, the oxide sample was rinsed with copious amounts of distilled water to eliminate the NaCl flux. BaCO3 was added to Ba5Nb4O15 before nitridation to suppress the possible generation of niobium oxynitride.
Each of the niobium compounds is found to be iso-structural with its tantalum analogue. Interestingly, the niobium compounds consistently have a unit cell volume that is larger than its tantalum analogue by 0.7–1.2%. In the diffuse reflectance spectra (DRS) of the nitridation products, the onset of light absorption characteristics of the band gap excitation of BaNbO2N was observed at around 740 nm (Fig. 12). Additional light absorption was observed in the longer wavelength region, which could be attributed to defects such as reduced B-site cations (Nb) and anion vacancies. Hisatomi et al. also demonstrated that BaNbO2N can be activated for photocatalytic water splitting in the presence of sacrificial reagents by modifying the starting material for nitridation and loading appropriate co-catalysts. BaNbO2N generates oxygen from an aqueous AgNO3 solution under illumination up to 740 nm, the longest wavelength ever reported for (oxy) nitride photocatalysts.77–79
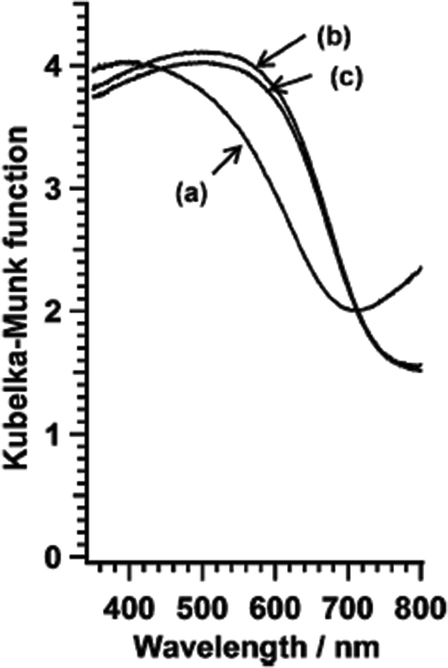 | ||
| Fig. 12 Diffuse reflectance spectra of BaNbO2N oxynitride, nitride from BaCO3 and Nb2O5 (a) 100 (b) 300 and (c) 500 mL min−1 NH3 (reproduced from ref. 76 with permission from the Royal Society of Chemistry). | ||
5. Effect of electrolyte on activity of photocatalysis
Most of the research in photocatalytic water splitting that has been reported is materials based and has been extensively studied as a potential method to supply hydrogen from sunlight and water. Only a few have investigated the effects of the electrolytic environment on photocatalytic efficiency.80–83 Many researchers investigated photocatalyst water splitting using a variety of electrolyte including H2SO4, KNO3, NaCl, and sodium acetate, H3PO4, NaClO4 and KOH.84–88 However, there has been a slight comparison between different electrolytes on photocatalytic activity. From the prospective of particle application, the ability to produce hydrogen from natural seawater would be highly desirable as seawater is the most abundant water resource in the world. However, seawater (acts as an electrolyte) contains a number of inorganic anions and cations and the effect of such ions on the activity of water splitting reaction remains to be investigated in more detail. The effects of electrolyte (identity and concentration) in overall water splitting reaction have to rigorously and quantitatively be taken into account because they may cause reaction switching and additional adsorption onto the active sites, which may enhance or decrease the overall catalytic efficiency. Some studies reported that electrolyte reduces the efficiency of a photocatalyst but some reported improved effects and some researchers found that electrolytes at high pH are more effective in promoting photocatalytic water splitting.89,90Ji and co-workers investigated the overall water splitting in seawater using NiOx-loaded La2Ti2O7 under ultraviolet irradiation (λ > 200 nm). They reported that water splitting can be achieved in seawater, although at half the rate of activity as compared to that in pure water. It was concluded that the suppression of activity in seawater is mainly due to the presence of magnesium cations in the reactant solution. Their study also investigated the visible light photocatalytic activity of a platinum loaded CdS/TiO2 composite catalyst for hydrogen production in the presence of SO32− and S2− as sacrificial electron donors and a Fe2O3 thin-film electrode for anodic photo-current generation.91
Medea et al. investigated the effects of various electrolytes on the photocatalytic activity of (Ga1−xZnx)(N1−xOx) for overall water splitting under visible light irradiation (λ > 400 nm) with respect to the physicochemical properties of (Ga1−xZnx) (N1−xOx), the co-catalyst and the pH of the reactant solution. The highest activity for overall water splitting achieved by this photocatalytic system, has been obtained using (Ga1−xZnx) (N1−xOx) modified Rh2−yCryO3-loaded co-catalyst in aqueous H2SO4 solution at pH 4.5. They also noticed that the addition of an appropriate amount of electrolyte such as NaCl or Na2SO4 to the reactant solution enhances the overall water splitting reaction. Addition of electrolytes, particularly NaCl and Na2SO4, to the reactant solution might facilitate surface redox reactions and/or compensate for the detrimental effect of surface defects that can act as recombination centers between photogenerated electrons and holes, thereby enhancing activity.92 However the same photocatalyst was also demonstrated to split artificial seawater under visible light irradiation, approximately half of the activity achieved as compared to pure water. Similar positive effects on photocatalytic activity for overall water splitting by electrolyte addition have been observed for UV active metal-oxide photocatalysts, although the mechanism for such an effect has not been reported.93
6. Conclusion
Photocatalysts are expected to be a future trend, since nano-sized photocatalysts have shown much better performance than their bulk counterparts. In the literature, a lot of research has been reported over the past few decades, in which an attempt is made to develop a photocatalyst with the highest activity. Development of a large number of photocatalysts and screening them for their activity is the general applied method but a more fundamental approach is to improve simple screening. In general, light absorption, electron–hole separation, hole transportation, and surface reaction are the key features in research and design, requiring materials of optimized composition, favorable structure, and morphology. Therefore we tried to provide more insight into the subject of photocatalysis for future.The first step in the synthesis of a suitable photocatalyst is to tune the band gap in such a way that it lies between 1.6 and <3 eV. The valence band should be lower in energy than the oxidation potential of oxygen and subsequently the conduction band should be higher than the reduction potential of hydrogen. Because the reaction proceeds via half reactions using ions, it is preferable that the photocatalyst splits water heterolytically into a proton and a hydroxyl anion and must reduce generation of water from molecular oxygen and hydrogen. This can be facilitated by the use of suitable metal oxide precursors. Most of the photocatalysts are limited for half reactions so a co-catalyst is necessary to generate hydrogen gas. In addition the designed photocatalyst must exhibit the same properties as a semiconductor.
Recently there has been significant progress in the development of metal oxynitrides, and studies on a potential visible-light photocatalyst with appreciable activities have been carried out. This development has laid a good foundation for future work in this area. The improvement of new technologies needs collaboration with a strong theoretical background for a better understanding of the hydrogen production mechanism, in order to come up with a low-cost and environmentally friendly water-splitting process for hydrogen production.
References
- B. Adeli and F. Taghipour, ECS J. Solid State Sci. Technol., 2013, 2, 118–126 CrossRef.
- F. E. Osterloh and B. A. Parkinson, MRS Bull., 2011, 36, 17–22 CrossRef CAS.
- R. M. N. Yerga, M. C. A. Galvan, F. del Valle, J. A. V. de la Mano and J. L. G. Fierro, ChemSusChem, 2009, 2, 471–485 CrossRef PubMed.
- P. Kanhere and Z. Chen, Molecules, 2014, 19, 19995–20022 CrossRef CAS PubMed.
- J. Zhu and M. Zach, Curr. Opin. Colloid Interface Sci., 2009, 14, 260–269 CrossRef CAS.
- B. Cao, G. M. Veith, R. E. Diaz, J. Liu, E. A. Stach, R. R. Adzic and P. G. Khalifah, Angew. Chem., Int. Ed., 2013, 125, 10953–10957 CrossRef.
- M. Yang, J. Ore-Sole, J. A. Rodgers, A. B. Jorge, A. Fuertes and J. P. Attifield, Nat. Chem., 2011, 3, 47–52 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem., 2007, 111, 7851–7861 CAS.
- S. M. Ji, P. H. Borse, H. G. Kim, D. W. Hwang, J. S. Jang, S. W. Bae and J. S. Lee, Phys. Chem. Chem. Phys., 2005, 7, 1315–1321 RSC.
- Y. Wu, P. Lazic, G. Hautier, K. Persson and G. Ceder, Energy Environ. Sci., 2013, 6, 157–168 CAS.
- A. Fuertes, Dalton Trans., 2010, 39, 5942–5948 RSC.
- D. Logvinovich, Doctoral Dissertation, Solid State Chemistry and Catalysis, 2008 Search PubMed.
- Z. F. Huang, L. Pan, J. J. Zou, X. Zhang and L. Wang, Nanoscale, 2014, 6, 14044–14063 RSC.
- L. Yang, H. Zhou, T. Fan and D. Zhanga, Phys. Chem. Chem. Phys., 2014, 16, 6810–6826 RSC.
- J. Zhu and M. Zäch, Curr. Opin. Colloid Interface Sci., 2009, 14, 260–269 CrossRef CAS.
- A. B. Murphy, P. R. F. Barnesa, L. K. Randeniyaa, I. C. Plumba, I. E. Greyb, M. D. Horneb and J. A. Glasscocka, Int. J. Hydrogen Energy, 2006, 31, 1999–2017 CrossRef CAS.
- Y. Sakata, Y. Matsuda, T. Yanagida, K. Hirata, H. Imamura and K. Teramura, Catal. Lett., 2008, 125, 22–26 CrossRef CAS.
- T. Kurushima, G. Gundiah, Y. Shimomura, M. Mikami, N. Kijima and A. K. Cheetham, J. Electrochem. Soc., 2010, 157, 64–68 CrossRef.
- R. Marchanda, Y. Laurenta, J. Guyadera, P. L'Haridona and P. Verdiera, J. Eur. Ceram. Soc., 1991, 8, 197–213 CrossRef.
- S. H. Elder, F. J. Di Salvo, L. Topor and A. Navrotsky, Chem. Mater., 1993, 5, 1545–1553 CrossRef CAS.
- G. Tessier and R. Marchand, J. Solid State Chem., 2000, 171, 143–151 CrossRef.
- M. R. Brophy, S. M. Pilgrim and W. A. Schulze, J. Am. Ceram. Soc., 2011, 94, 4263–4268 CrossRef CAS.
- A. Hellwig and A. Hendry, J. Mater. Sci., 1994, 29, 4686–4693 CrossRef CAS.
- S. J. Clarke, B. P. Guinot, C. W. Michie, M. J. C. Calmont and M. J. Rosseinsky, Chem. Mater., 2002, 14, 288–294 CrossRef CAS.
- Y. Kim, W. Si, P. M. Woodward, E. Sutter, S. Park and Y. Vogt, Chem. Mater., 2007, 19, 618–623 CrossRef CAS.
- C. Le Paven-Thivet, A. Ishikawa, A. Ziani, L. Le Gendre, M. Yoshida, J. Kubota, F. Tessier and K. Domen, J. Phys. Chem. C, 2009, 113, 6156–6162 CAS.
- E. Martınez-Ferrero, Y. Sakatani, C. Boissiere, D. Grosso, A. Fuertes, J. Fraxedas and C. Sanchez, Adv. Funct. Mater., 2007, 17, 3348–3354 CrossRef.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- Y. Moriya, T. Takata and K. Domen, Coord. Chem. Rev., 2013, 257, 1957–1969 CrossRef CAS.
- M. Pelaez, et al. , Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- J. Kubota and K. Domen, Electrochem. Soc. Interface, 2013, 22, 57–62 CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- K. Maeda, H. Hashiguchi, H. Masuda, R. Abe and K. Domen, J. Phys. Chem. C, 2008, 112, 3447–3452 CAS.
- C. M. Fang, E. Orhan, G. A. De Wijs, H. T. Hintzen, R. A. De Groot, R. Marchand, J. Y. Saillard and G. De With, J. Mater. Chem., 2001, 11, 1248–1252 RSC.
- M. Yashima, Y. Lee and K. Domen, Activity Report on Neutron Scattering Research: Experimental Reports, 15, 2008.
- M. Hara, E. Chiba, A. Ishikawa, T. Takata, J. N. Kondo and K. J. Domen, Phys. Chem. B, 2003, 107, 13441–13445 CrossRef CAS.
- G. Hitoki, A. Ishikawa, T. Takata, J. N. Kondo, M. Hara and K. Domen, Chem. Lett., 2002, 31, 736–737 CrossRef.
- A. Ishikawa, T. Takata, J. N. Kondo, M. Hara and K. Domen, J. Phys. Chem. B, 2004, 108, 11049–11053 CrossRef CAS.
- K. Ueda, H. Kato, M. Kobayashi, M. Hara and M. Kakihana, J. Mater. Chem. A, 2013, 1, 3667–3674 CAS.
- P. Li, W. Fan, Y. Li, H. Sun, X. Cheng, X. Zhao and M. Jiang, Inorg. Chem., 2010, 49, 6917–6924 CrossRef CAS PubMed.
- D. Armytage and B. E. F. Fender, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 809–812 CrossRef CAS.
- K. Maeda, D. Lu and K. Domen, Angew. Chem., Int. Ed., 2013, 52, 6488–6491 CrossRef CAS PubMed.
- R. Nakamura, T. Tanaka and Y. Nakato, J. Phys. Chem. B, 2005, 109, 8920–8927 CrossRef CAS PubMed.
- A. Fuertes, Mater. Horiz., 2015, 2, 453–461 RSC.
- H. N. Kim, “Photocatalytic Water Splitting Reactions Based on Tantalum Oxynitrides” Literature Seminar, October 15, 2013.
- F. A. Ponce and D. P. Bour, Nature, 1997, 386, 351–359 CrossRef CAS.
- I. M. Huygens, K. Strubbe and W. P. Gomes, J. Electrochem. Soc., 2000, 147, 1797–1802 CrossRef CAS.
- M. J. Chen, J. R. Yang and M. Shiojiri, Semicond. Sci. Technol., 2012, 27, 074005 CrossRef.
- Y. S. Choi, J. W. Kang, D. K. Hwang and S. J. Park, IEEE Trans. Electron Devices, 2010, 57, 26–41 CrossRef CAS.
- S. P. Phivilay, C. A. Roberts, A. A. Puretzky, K. Domen and I. E. Wachs, J. Phys. Chem. Lett., 2013, 4, 3719–3724 CrossRef CAS.
- Y. Lee, H. Terashima, Y. Shimodaira, K. Teramura, M. Hara, H. Kobayashi, K. Domen and M. Yashima, J. Phys. Chem. C, 2007, 111, 1042–1048 CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 7554–7560 CAS.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed.
- Z. Wang, Y. Liu, B. Huang, Y. Dai, Z. Lou, G. Wang, X. Zhanga and X. Qina, Phys. Chem. Chem. Phys., 2014, 16, 2758–2774 RSC.
- G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Commun., 2002, 1698–1699 RSC.
- K. Maeda, K. Teramura, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, Pure Appl. Chem., 2006, 78, 2267–2276 CAS.
- Z. Zou and H. Arakawa, J. Photochem. Photobiol., A, 2003, 158, 145–150 CrossRef CAS.
- K. Maeda, et al. , J. Phys. Chem. B, 2005, 109, 20504–20510 CrossRef CAS PubMed.
- K. Kamata, K. Maeda, D. Lu, Y. Kako and K. Domen, Chem. Phys. Lett., 2009, 470, 90–94 CrossRef CAS.
- T. Mishima, M. Matsuda and M. Miyake, Appl. Catal., A, 2007, 324, 77–82 CrossRef CAS.
- A. Rachel, S. G. Ebbinghaus, M. Gungerich, P. J. Klar, J. Hanss, A. Weidenkaff and A. Reller, Thermochim. Acta, 2005, 438, 134–143 CrossRef CAS.
- D. Oka, Y. Hirose, H. Kamisaka, T. Fukumura, K. Sasa, S. Ishii, H. Matsuzaki, Y. Sato, Y. Ikuhara and T. Hasegawa, Sci. Rep., 2014, 4, 4987 CAS.
- R. Marchand, C. R. Acad. Sci., 1976, 282, 329–348 CAS.
- A. E. Maeglia, E. H. Otala, T. Hisatomib, S. Yoona, C. M. Leroyb, N. Schäublea, Y. Lua, M. Grätzelb and A. Weidenkaff, Energy Procedia, 2012, 22, 61–66 CrossRef.
- F. Tessier and R. Marchand, J. Solid State Chem., 2003, 171, 143–151 CrossRef CAS.
- M. Yang, J. A. Rodgers, L. C. Middler, J. Oro-Sole, A. B. Jorge, A. Fuertes and J. P. Attfield, Inorg. Chem., 2009, 48, 11498–11500 CrossRef CAS PubMed.
- A. Kasahara, K. Nukumizu, G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Phys. Chem. A, 2002, 106, 6750–6753 CrossRef CAS.
- S. Balaz, H. S. Porter, M. P. Woodward and J. L. Brillson, Chem. Mater., 2013, 25, 3337–3343 CrossRef CAS.
- D. Yamasita, T. Takata, M. Hara, J. N. Kondo and K. Domen, Solid State Ionics, 2004, 172, 591–595 CrossRef CAS.
- C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., Int. Ed., 2015, 54, 2955–2959 CrossRef CAS PubMed.
- T. Takata, C. Pan and K. Domen, Sci. Technol. Adv. Mater., 2015, 16, 033506 CrossRef.
- C. Marcilly, P. Courty and B. Delmon, J. Am. Ceram. Soc., 1970, 53, 56–57 CrossRef CAS.
- Y. I. Kim and P. M. Woodward, J. Solid State Chem., 2007, 180, 3224–3233 CrossRef CAS.
- J. Zhang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 7230–7232 CrossRef CAS PubMed.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348–8351 CrossRef CAS PubMed.
- T. Hisatomi, C. Katayama, Y. Moriya, T. Minegishi, M. Katayama, H. Nishiyama, T. Yamadad and K. Domen, Energy Environ. Sci., 2013, 6, 3595–3599 CAS.
- T. Hisatomi, C. Katayama, K. Teramura, T. Takata, Y. Moriya, T. Minegishi, M. Katayama, H. Nishiyama, T. Yamada and K. Domen, ChemSusChem, 2014, 7, 2016–2021 CrossRef CAS PubMed.
- B. Siritanaratkul, K. Maeda, T. Hisatomi and K. Domen, ChemSusChem, 2011, 4, 74–78 CrossRef CAS PubMed.
- H. L. Wang, T. Deutsch and J. A. Turner, J. Electrochem. Soc., 2008, 155, F91–F96 CrossRef CAS.
- E. L. Miller, B. Marsen, D. Paluselli and R. Rocheleau, Electrochem. Solid-State Lett., 2005, 8, A247–A249 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- E. Bae and W. Choi, J. Phys. Chem. B, 2006, 110, 14792–14799 CrossRef CAS PubMed.
- W. B. Ingler and S. U. M. Khan, Electrochem. Solid-State Lett., 2006, 9, G144–G146 CrossRef CAS.
- A. Belghazi, S. Bohm, D. A. Worsley and H. N. McMurray, Z. Phys. Chem., 2005, 219, 1539–1546 CrossRef CAS.
- T. F. Jaramillo, S. H. Baeck, A. Kleiman-Shwarsctein, K. S. Choi, G. D. Stucky and E. W. McFarland, J. Comb. Chem., 2005, 7, 264–271 CrossRef CAS PubMed.
- Y. F. Su, T. C. Chou, T. R. Ling and C. C. Sun, J. Electrochem. Soc., 2004, 151, A1375–A1382 CrossRef CAS.
- E. Thimsen, N. Rastgar and P. Biswas, J. Phys. Chem. C, 2008, 112, 4134–4140 CAS.
- S. Q. Zhang, H. J. Zhao, D. L. Jiang and R. John, Anal. Chim. Acta, 2004, 514, 89–97 CrossRef CAS.
- C. B. Almquist and P. Biswas, Chem. Eng. Sci., 2001, 56, 3421–3430 CrossRef CAS.
- S. M. Ji, H. Jun, J. S. Jang, H. C. Son, P. H. Borse and J. S. Lee, J. Photochem. Photobiol., A, 2007, 189, 141–144 CrossRef CAS.
- K. Maeda, H. Masuda and K. Domen, Catal. Today, 2009, 147, 173–178 CrossRef CAS.
- K. Sayama and H. Arakawa, J. Photochem. Photobiol., A, 1996, 94, 67–76 CrossRef CAS.
| This journal is © the Partner Organisations 2016 |