
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThioether-terminated nickel(II) coordination clusters with {Ni6} horseshoe- and {Ni8} rollercoaster-shaped cores†
Sebastian
Schmitz
a,
Jan
van Leusen
a,
Arkady
Ellern
b,
Paul
Kögerler
 *ac and
Kirill Yu.
Monakhov
*a
*ac and
Kirill Yu.
Monakhov
*a
aInstitut für Anorganische Chemie, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: paul.koegerler@ac.rwth-aachen.de; kirill.monakhov@ac.rwth-aachen.de
bChemistry Department, Iowa State University, 1711 Gilman Hall, Ames, IA 50011, USA
cJülich-Aachen Research Alliance (JARA-FIT) and Peter Grünberg Institute (PGI-6), Forschungszentrum Jülich, 52425 Jülich, Germany
First published on 19th January 2016
Abstract
We report two polynuclear nickel(II) compounds whose supramolecular structures are controlled by small inorganic templating anions and π-conjugated Schiff-base ligands (L·SMe3− and HL·SMe2−) with peripheral, structurally exposed methylthioether groups. The central component of the compound [Ni6(L·SMe)3(CO3)(MeOH)6(THF)2]Cl·2MeOH (1, monoclinic space group P2/c) displays a horseshoe-shaped {Ni6} core templated by μ6-CO32− moiety due to the fixation of atmospheric carbon dioxide. According to thermogravimetric analysis, compound 1 retains the structural integrity of its carbonate-bonded metal core [Ni6(L·SMe)3(CO3)]+ up to ca. 390 °C. In the crystal lattice, compound 1 features an one-dimensional hydrogen-bonded chain structure [{CO3 ⊂ Ni6}⋯Cl⋯{CO3 ⊂ Ni6}]∞. The structure of the compound [Ni8(HL·SMe)2(L·SMe)2(OH)4(MeCN)4(H2O)4](NO3)2·11MeCN·2PhCN (2, triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) consists of a dicationic crown-like {Ni8} metallamacrocycle hosting two NO3− anions in the upper and lower cavities. The {Ni6} complex is characterised by antiferromagnetic exchange interactions, whereas {Ni8} reveals predominantly ferromagnetic exchange coupling between the spin-1 Ni(II) centres. The presence of terminal thioether anchoring groups at the periphery of complex 1, coupled with high thermal stability and solubility in several common organic solvents, renders this compound an interesting candidate for molecular surface-deposition from solution using electrospray ionisation mass spectrometry approach and, possibly, for supported-transition metal complex heterogeneous catalysis.
) consists of a dicationic crown-like {Ni8} metallamacrocycle hosting two NO3− anions in the upper and lower cavities. The {Ni6} complex is characterised by antiferromagnetic exchange interactions, whereas {Ni8} reveals predominantly ferromagnetic exchange coupling between the spin-1 Ni(II) centres. The presence of terminal thioether anchoring groups at the periphery of complex 1, coupled with high thermal stability and solubility in several common organic solvents, renders this compound an interesting candidate for molecular surface-deposition from solution using electrospray ionisation mass spectrometry approach and, possibly, for supported-transition metal complex heterogeneous catalysis.
 Paul Kögerler | Paul Kögerler graduated with a Dr rer. nat. degree with Prof. Achim Müller at the University of Bielefeld (Germany) in 2000, followed by a postdoctoral research stay at the Department of Physics and Astronomy at Iowa State University (USA). In 2003, he was appointed as a tenured Associate Scientist at the U.S. DOE Ames Laboratory, before returning to Germany in 2006 as Professor of Chemistry at the Institute of Inorganic Chemistry at RWTH Aachen University and Group Leader for Molecular Magnetism at the Peter Grünberg Institute, Research Centre Jülich. |
 Kirill Yu. Monakhov | Kirill Monakhov received his Dr rer. nat. degree with Prof. Gerald Linti at Heidelberg University (Germany) in 2010. After two years as a postdoctoral fellow of the German Research Foundation (DFG) and Cercle Gutenberg with Prof. Pierre Braunstein (University of Strasbourg, France), and after being awarded the Academia Europaea Burgen Scholarship in 2011, he returned to Germany in 2013 with a DFG postdoctoral reintegration fellowship to join the group of Prof. Paul Kögerler at RWTH Aachen University. In 2015 he received a DFG Emmy Noether fellowship and now leads a junior research group at the Institute of Inorganic Chemistry at RWTH Aachen. His research interests include coordination and supramolecular chemistry, redox-active magnetic molecules, molecular electronics and spintronics, computational chemistry, catalysis and surface science. |
Introduction
Fabrication of nanomaterials1 with well-defined morphology and properties derived from the contact of individual coordination complexes and clusters of magnetic, electrochemical, and catalytic interest with a metallic substrate (electrode) attracts great attention in modern surface chemistry2 and physics3 and is motivated by the continual search for multifunctional, compact, and biocompatible structures for renewable and sustainable energy. The peripheral functionalisation of stimuli-responsive metalorganic compounds with the electron-rich “anchoring” groups4 such as e.g. cyclic or acyclic thioether (–S–), 1,2-dithiolane (–S–S–) or thiol (–SH) constitutes one out of multiple chemical strategies5 to anchor molecules to macroscopic solid-state electrodes6 (e.g., gold). The ligand environment of the metal centres as well as the structural, thermal and redox stability, solubility (for solution-driven molecular deposition), and volatility/sublimability (for deposition from the solid phase) of molecules are the key features in multilateral surface-oriented investigation toward hybrid catalytic systems and electronic devices.The structural, spectroscopic, electrochemical, and catalytic properties of diamagnetic and paramagnetic S-terminated transition metal complexes and clusters were extensively studied during previous years: (i) electrochemical, electrocatalytic, and/or in situ infrared spectroscopic properties of some cobalt, iron, ruthenium, and osmium complexes adsorbed on gold electrodes via the sulfur-based binding groups (thiols, thiophenes, thiocyanates, etc.) were reported.7,8 (ii) Reactivity of a few iron and rhodium complexes toward thiolate-stabilised gold nanoparticles was discussed.9 (iii) The mono- and dinuclear Cr(III)10 and Ni(II)11 complexes bearing thioether groups at the periphery of the PNP and PNP = S chelates were exploited for the catalytic polymerisation and oligomerisation of ethylene. (iv) In view of the high interest for the selective formation of self-assembly of highly ordered and oriented metal–organic monolayers on surfaces or the integration of metal complexes into mesoporous materials,12 the deposition of S-terminated heterometallic complexes and clusters of Mo and Pd or Pt was studied on gold surfaces by scanning tunnelling microscopy (STM), ellipsometry, and X-ray photoelectron spectroscopy (XPS).13 (v) The tetrahedral mixed-metal {RuCo3} carbonyl clusters functionalised with the phosphane–thiol ligands were studied on a gold surface by STM, XPS and X-ray absorption spectroscopy.14 (vi) The chemical, structural, and thermodynamic characterisation of the mononuclear Pt(II) cis-complexes and the heterotrinuclear {Co2Pt} clusters containing N-substituted bis(diphenylphosphanyl)amine ligands with the pendant thiobenzyl ester groups15 as well as a trinuclear Ag(I) complex and a Ag(I) coordination polymer with a thioether function in their short-bite diphosphine ligands16 were described. (vii) Sulfur-assisted phenyl migration from phosphorus to platinum in molecular {PtW2} and {PtMo2} clusters with thioether-functionalised short-bite ligands of the bis(diphenylphosphanyl)amine-type was demonstrated.17 The synthesis and structural chemistry of several methylthio- or thiocyanate-furnished coordination complexes of Mn(II,III),18 Fe(II),19 and Fe(III)20 which exhibit ferromagnetic and antiferromagnetic exchange interactions were also reported. To the best of our knowledge, the potential of the above-mentioned metal complexes in catalytic processes on substrate surfaces has not been explored so far.
By contrast, surface studies towards nano and quantum computation which used the thioether-functionalised molecules exhibiting slow-relaxation of magnetisation (single-molecule magnets, SMMs) were performed and dealt with the {MnIII8MnIV4},21 {FeIII4}22 (see also gold nanoparticles functionalised with a 1,2-dithiolane-decorated FeIII4 SMM23), {FeIII3CrIII},24 and {NiII4}25 complexes. The chemical and physical behaviour of the {CrIII7NiII}-nuclearity rings26 (with antiferromagnetic coupling in bulk) and the {MnIII12MnII7}-nuclearity compound27 (with ferromagnetic coupling in bulk) upon deposition and anchoring to gold surfaces via sulfur bonds was assessed.
Our exploratory work is motivated by the potential use of isolated transition metal coordination complexes and clusters with the structurally exposed sulfur-based anchoring groups in industrially relevant catalytic processes on solid supports at low and high temperatures. Our strategy implies the synthesis and characterisation of polynuclear metal compounds with the functional nano-sized structures as well as their addressing from the perspectives of homogeneous catalysis, with the subsequent immobilisation of these molecules onto porous metallic substrates or noble metal-coated mesoporous silica nanoparticles28 (e.g., SBA-15) to eventually enable supported-transition metal complex heterogeneous (or heterogenised homogeneous29) catalysis. In particular, Ni(II) Schiff-base complexes,30 used as catalysts in the hydrogenation (see, e.g. reduction of benzene31) and oxidation reactions as well as for the ethylene polymerisation,32 constitute interesting target candidates for the above-mentioned functionalisation and application.
Herein, we report the preparation of two novel polynuclear nickel(II) complexes supported by a π-conjugated Schiff-base ligand with the chemically engineered –SMe functionalities (see H3L·SMe in Fig. 1), isolated as [Ni6(L·SMe)3(CO3)(MeOH)6(THF)2]Cl·2MeOH (1) and [Ni8(HL·SMe)2(L·SMe)2(OH)4(MeCN)4(H2O)4](NO3)2·11MeCN·2PhCN (2), which are of relevance to in situ ultra-high vacuum deposition on surfaces by electrospray ionisation33 (ESI). The obtained ionic compounds were characterised by infrared (IR) spectroscopy, ESI mass spectrometry (ESI-MS), and thermogravimetric analysis (TGA) and their solid-state molecular structures were determined by single-crystal X-ray diffraction. The magnetic properties were assessed via dc and ac magnetic susceptibility measurements and our study thus expands knowledge on existing topologies and magnetism in nickel coordination chemistry.34
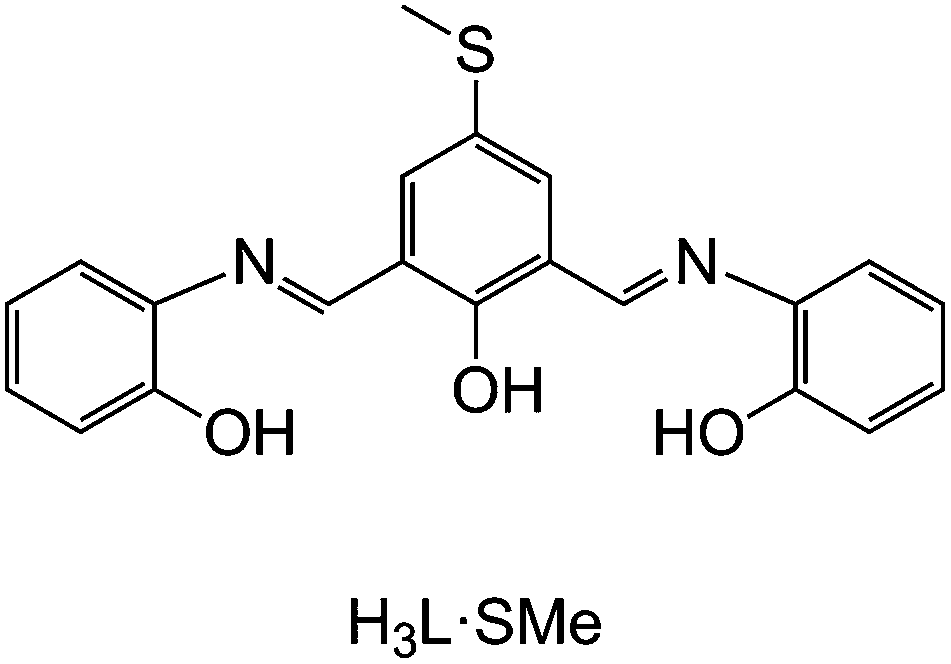 | ||
Fig. 1 The hexadentate Schiff base H3L·SMe (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C21H18N2O3S) used in this work. C21H18N2O3S) used in this work. | ||
Results and discussion
Synthesis and thermal stability
Compounds 1 and 2 were synthesised via one-pot four-component in situ reactions of n equivalents of NiX2·6H2O [n = 2 for X = Cl (1) and n = 1.4 for X = NO3 (2)] with two equivalents of 2-aminophenol, one equivalent of freshly prepared 2-hydroxy-5-(methylthio)isophthalaldehyde and two equivalents of triethylamine under reflux conditions in a mixture of organic solvents (Scheme 1). Note that upon increasing n to 2 in the synthesis reaction for compound 2, no solid product could be isolated. Likewise, changing or replacing the counteranions in the Ni(II) precursors also prevents precipitation of crystalline product. A methanol/tetrahydrofuran volume ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was used for the formation of the moisture stable, crystalline compound [Ni6(L·SMe)3(CO3)(MeOH)6(THF)2]Cl·2MeOH (1) and a acetonitrile/benzonitrile volume ratio of 10:1 for the moisture sensitive, crystalline compound [Ni8(HL·SMe)2(L·SMe)2(OH)4(MeCN)4(H2O)4](NO3)2·11MeCN·2PhCN (2). The aerobic reaction affording compound 1 is furthermore characterised by the uptake of atmospheric CO2. Dark-red compounds 1 and 2 with a potentially redox-active, fully-deprotonated L·SMe3− ligand (see Fig. S1 and synthesis of H3L·SMe in the ESI†) were obtained in ca. 39% and 55% yields, respectively. Compound 1 is well soluble in THF, DMF, and MeCN and modestly soluble in MeOH and CH2Cl2, while compound 2 is well soluble in THF, DMF, and MeOH and moderately soluble in MeCN and CH2Cl2.
:1 was used for the formation of the moisture stable, crystalline compound [Ni6(L·SMe)3(CO3)(MeOH)6(THF)2]Cl·2MeOH (1) and a acetonitrile/benzonitrile volume ratio of 10:1 for the moisture sensitive, crystalline compound [Ni8(HL·SMe)2(L·SMe)2(OH)4(MeCN)4(H2O)4](NO3)2·11MeCN·2PhCN (2). The aerobic reaction affording compound 1 is furthermore characterised by the uptake of atmospheric CO2. Dark-red compounds 1 and 2 with a potentially redox-active, fully-deprotonated L·SMe3− ligand (see Fig. S1 and synthesis of H3L·SMe in the ESI†) were obtained in ca. 39% and 55% yields, respectively. Compound 1 is well soluble in THF, DMF, and MeCN and modestly soluble in MeOH and CH2Cl2, while compound 2 is well soluble in THF, DMF, and MeOH and moderately soluble in MeCN and CH2Cl2.
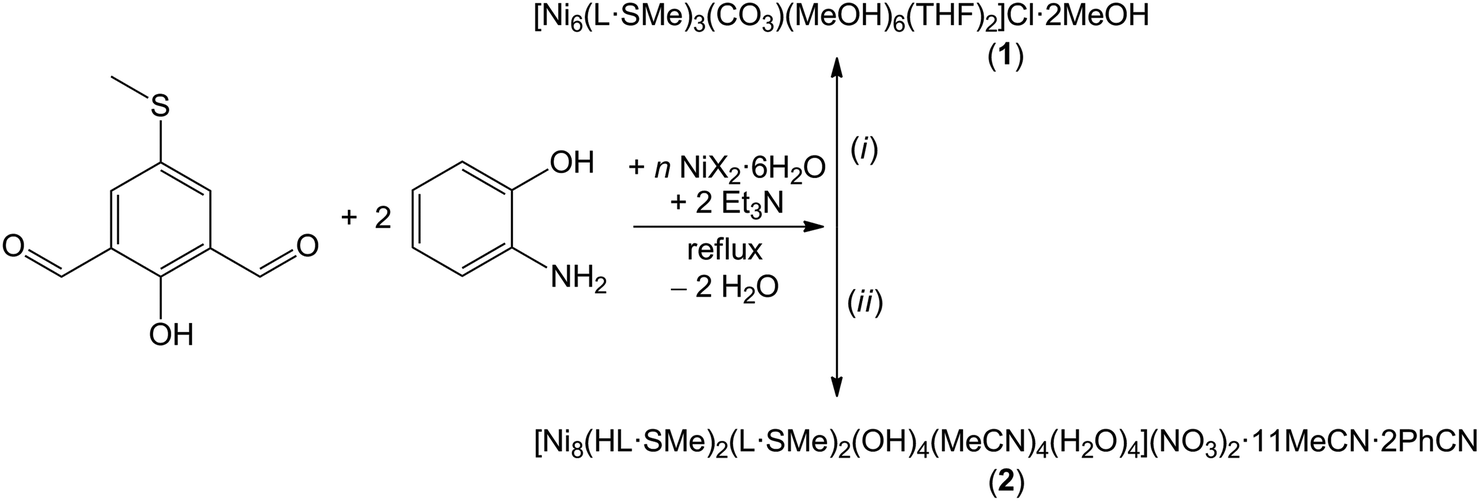 | ||
| Scheme 1 Synthesis of the hexanuclear and octanuclear nickel(II) Schiff-base complex compounds 1 and 2. Reaction conditions: (i) n = 2 for X = Cl and MeOH:THF = 4:1; (ii) n = 1.4 for X = NO3 and MeCN:PhCN = 10:1. For the IR spectra of these compounds, see Fig. S3 and S4.† | ||
In contrast to the gradual decomposition in TGA experiments of compound 2 occurring as an ill-defined mass loss step between 40 and 800 °C, the thermal stability of compound 1 in a N2 atmosphere is characterised by three distinctive steps: (i) the first step (40–210 °C) corresponds to a 13.8% mass loss of two uncoordinated MeOH solvent molecules (the compound was measured immediately following synthesis) as well as two THF and two MeOH ligands coordinated to the Ni1/Ni′1 and Ni2/Ni′2 atoms, respectively (Δmcalc = 13.8%). (ii) The second step shows a 6.6% mass loss of four remaining MeOH ligands coordinated to the Ni3/Ni′3 atoms (Δmcalc = 6.5%) between 210 °C and 390 °C. (iii) The third, small step (1.8% mass loss from 390 °C to 400 °C) indicates chloride counterion desorption (Δmcalc = 1.8%). Thus, the TGA suggests that the [Ni6(L·SMe)3(CO3)]+ core fragment is stable up to ca. 390 °C and this high thermal stability might enable the use of this magnetic complex in high-temperature catalytic reactions32 on solid supports. It is noteworthy that the molecular ion peak of [Ni6(L·SMe)3(CO3)H2]+ without coordinated solvent molecules could be detected by ESI mass spectrometry (Fig. S7†).
Interestingly, the use of 2-hydroxy-5-methylisophthalaldehyde (see Fig. S2† for the structure of the non-functionalised H3L ligand) instead of its methylthio derivate in the synthesis under less basic conditions (in the absence of Et3N; Scheme S1†) has resulted in the formation of mononuclear nickel(II) Schiff-base complexes with compositions [Ni(H3L)2]Cl2·4MeOH (A) and [Ni(H3L)2](NO3)2·4MeOH (B), see Fig. S8 and Tables S5–S8† for solid-state structure information. The sulfur ligand constituents and the presence of triethylamine thus appear to be crucial for the synthesis of compounds 1 and 2.
X-ray crystal structure analysis
The solid-state molecular structures of compounds 1 and 2 were established by single-crystal X-ray diffraction analyses and are illustrated in Fig. 2–4. Compound 1 crystallises in the monoclinic space group P2/c (Table S1, the ESI†) and consists of the cationic complex [Ni6(L·SMe)3(CO3)(MeOH)6(THF)2]+ (Fig. 2a), counterbalanced by a Cl− anion, and two crystal solvent (methanol) molecules. The nano-sized [Ni6(L·SMe)3(CO3)(MeOH)6(THF)2]+ complex contains a distorted horseshoe-shaped Ni6 core structure. The Ni atoms are connected to a μ6-CO32− group, a result of the fixation of atmospheric carbon dioxide (Fig. 2b). Molecular “horseshoe”-like structures have also been observed for {CrIII6}-nuclearity complexes.35 Sorption of atmospheric CO2 under aerobic reaction conditions is well-known in coordination chemistry;36 homonuclear and heteronuclear transition-metal and lanthanide complexes that incorporate the carbonate ion in a wide variety of bridging modes have been described.37 | ||
| Fig. 2 (a) Molecular structure of [Ni6(L·SMe)3(CO3)(MeOH)6(THF)2]+ in compound 1. (b) Different views of the Ni6 core templated by CO32− with a μ6–η1:η1:η1:η1:η1:η1 binding mode. (c) A segment of the extended solid-state structure of compound 1, highlighting the association of two {Ni6} coordination clusters via C–HL·SMe⋯Cl hydrogen bonding interactions (O–HMeOH⋯Cl interactions are also shown). Hydrogen atoms not involved in hydrogen bonds are omitted for clarity. Colour code: H, black; C, grey; N, blue; O, red; S, yellow; Cl, light green; Ni, dark green. The nickel and sulfur atoms, carbonate ion, and co-crystallised MeOH molecules are represented as ball-and-stick models. The chloride ion is depicted as space-filling model. See Table S2† for bond lengths and angles. | ||
 | ||
| Fig. 3 A fragment of compound 1 representing pseudo-octahedral coordination geometries of the Ni(II) centres. For clarity, only one chelating Schiff-base ligand L·SMe3− is explicitly depicted. The nickel, nitrogen, oxygen, and sulfur atoms are represented as ball-and-stick models. The terminal-coordinated solvent molecules are highlighted in different colours: MeOH, purple; THF, sky blue. Elemental colour code: C, grey; N, blue; O, red; S, yellow; Ni, dark green. | ||
 | ||
| Fig. 4 (a) Molecular structure of [Ni8(HL·SMe)2(L·SMe)2(OH)4(MeCN)4(H2O)4]2+ in compound 2. (b) Non-planar ring of eight nickel(II) atoms as metal core in compound 2. (c) Side view of the solid-state structure of compound 2 showing two NO3− anions in the top and down cavities. Hydrogen atoms and solvent molecules are omitted for clarity. The nickel and sulfur atoms as well as the C and N atoms of MeCN in (c) are represented as ball-and-stick models and the nitrate ions as space-filling models. Colour code: C, grey; N, blue; O, red; S, yellow; Ni, dark green. See Table S4† for bond lengths and angles. | ||
The {CO3 ⊂ Ni6} fragment in compound 1 is ligated by three in situ-formed Schiff bases (threefold-deprotonated L·SMe3−; see Fig. 1) acting as N2O3-chelates for the divalent nickel centres. Six MeOH and two THF solvent molecules complete the coordination spheres of metal ions (Fig. 3): Ni1 or Ni′1 site binds to one THF molecule, Ni2 or Ni′2 to one terminal MeOH molecule, and Ni3 or Ni′3 to two terminal MeOH molecules. Bond distances of the NO5-coordinated nickel ions (in Å): Ni1–OL·SMe: 2.0010(19)–2.158(2) Å; Ni1–NL·SMe: 1.976(2) Å; Ni1–OTHF: 2.098(2) Å; Ni1–OCO3: 2.0896(19); Ni2–OL·SMe: 1.9726(18)–2.143(2); Ni2–NL·SMe: 1.963(2); Ni2–OMeOH: 2.119(2); Ni2–OCO3: 2.0306(18); Ni3–OL·SMe: 2.000(2) and 2.0359(19); Ni3–NL·SMe: 1.995(2); Ni3–OMeOH: 2.095(2) and 2.112(2); Ni3–OCO3: 2.0856(19). Thus, each Ni(II) centre in compound 1 exhibits a pseudo-octahedral local geometry with axially distorted O positions. The [Ni6(L·SMe)3(CO3)(MeOH)6(THF)2]+ structure reveals the non-bonding Ni1⋯Ni2 (or Ni′1⋯Ni′2), Ni1⋯Ni3 (or Ni′1⋯Ni′3), and Ni2⋯Ni′2 separations of 2.9707(6), 3.1251(6), and 3.0323(7) Å, respectively (the sum of the covalent radii of two Ni atoms is 2.48 Å (ref. 38)). The magnetically relevant bond angles fall in the ranges 90.313(74)–90.828(78)° for Ni1–(μ3-O)–Ni2, 96.916(79)–101.451(84)° for Ni1–(μ3-O)–Ni3, and 96.608(17)–100.461(17)° for Ni2–(μ3-O)–Ni′2.
A further interesting feature of compound 1 is the infinite one-dimensional chain structure in the crystal lattice, made up from repeating [Ni6(L·SMe)3(CO3)(MeOH)6(THF)2]+ entites assembled via four C–HL·SMe⋯Cl (dC⋯Cl = 3.6385(4) Å and 3.7100(4) Å) and two identical O–HMeOH⋯Cl (dO⋯Cl = 3.0958(3) Å) hydrogen bonds, as shown in Fig. 2c.
Compound 2 crystallises in the triclinic space group P (Table S3†) and shows a dicationic {Ni8} metallamacrocycle [Ni8(HL·SMe)2(L·SMe)2(OH)4(MeCN)4(H2O)4]2+ (Fig. 4a), reminiscent of a rollercoaster, and counterbalanced by two NO3− anions. Eleven acetonitrile and two benzonitrile molecules co-crystallise in the unit cell. The central metal core in the nano-sized [Ni8(HL·SMe)2(L·SMe)2(OH)4(MeCN)4(H2O)4]2+ moiety displays a non-planar ring of eight nickel(II) atoms (Fig. 4b). This ring is maintained by two monoprotonated HL·SMe2− and two fully-deprotonated L·SMe3− Schiff-base ligands (see Fig. 1) as well as four bridging OH− groups. The coordination spheres of four nickel (Ni2, Ni3, Ni6 and Ni8) atoms are completed by terminal MeCN molecules, those of the other four (Ni1, Ni4, Ni5 and Ni7) only by terminal H2O molecules (one MeCN/H2O per Ni). As a result, each of the Ni(II) ions in the ring resides in a pseudo-octahedral N2O4 or NO5 coordination environment (ratio N2O4:NO5 = 4:4). The NiN2O4 bond distances (in Å) amount to Ni2–OL·SMe: 1.990(7)–2.151(7); Ni2–NL·SMe: 1.980(8); Ni2–OH: 2.045(7), Ni2–NMeCN: 2.091(8); Ni3–OL·SMe: 1.973(7)–2.171(7); Ni3–NL·SMe: 1.992(9), Ni3–OH: 2.061(7); Ni3–NMeCN: 2.075(10); Ni6–OL·SMe: 1.978(7)–2.162(7), Ni6–NL·SMe: 1.992(8); Ni6–OH: 2.029(7); Ni6–NMeCN: 2.112(9); Ni8–OL·SMe: 1.996(7)–2.141(7); Ni8–NL·SMe: 1.977(9), Ni8–OH: 2.056(7), Ni8–NMeCN: 2.080(10). For the NiNO5 environment: Ni1–OL·SMe: 2.027(7)–2.087(7); Ni1–NL·SMe: 2.013(8); Ni1–OH: 2.093(7), Ni1–OH2O: 2.071(7); Ni4–OL·SMe: 2.039(7)–2.077(7); Ni4–NL·SMe: 2.018(9); Ni4–OH: 2.138(6); Ni4–OH2O: 2.059(7); Ni5–OL·SMe: 2.038(7)–2.054(7); Ni5–NL·SMe: 2.010(8), Ni5–OH: 2.133(7); Ni5–OH2O: 2.071(7); Ni7–OL·SMe: 2.043(7)–2.063(7); Ni7–NL·SMe: 1.991(8); Ni7–OH: 2.116(7); Ni7–OH2O: 2.080(8). The non-equivalent, non-bonding Ni⋯Ni distances range from 3.0662(25) to 3.1001(25) Å. The magnetically relevant bond angles fall in the ranges 93.5(3)–100.7(3)° for Ni3–(μ3-O20L·SMe)–Ni8 and Ni3–(μ3-O12L·SMe)–Ni7, and 95.1(3)–96.6(3)° for Ni2–(μ2-O14OH)–Ni5 and Ni1–(μ2-O15OH)–Ni6.
In general, the [Ni8(HL·SMe)2(L·SMe)2(OH)4(MeCN)4(H2O)4]2+ architecture can be viewed as a metallocavitand39 hosting two NO3− anions in a nesting fashion (Fig. 4c). These nitrate groups are situated 2.3973(80) Å above and 2.6004(83) Å below the nickel ring plane and connected to the internal oxygen atoms of the dicationic complex via four H-bonds: (i) O21 or O26 of NO3− is linked to two bridging OH− groups (O21–OOH: 2.7958(123) Å–2.8692(117) Å; O26–OOH: 2.9280(101) Å–2.9428(123) Å) and one coordinated H2O molecule (O21–O5H2O: 3.0352(115) Å; O26–O9H2O: 2.9195(145) Å). (ii) The fourth H-bond appears between O22 or O25 of NO3− and the H2O ligands at the Ni1 and Ni4 centres (O22–O7H2O: 2.7542(125) Å; O25–O6H2O: 2.8609(123) Å).
We note that in the crystal structures of compounds 1 and 2, the S atoms at the periphery of the Schiff-base ligands L·SMe3− and HL·SMe2− are not involved in any intermolecular coordination bonds.
Magnetism and magnetochemical modelling
Magnetic properties of compounds 1 and 2 were studied based on the temperature- and field-dependent dc (static) and ac (dynamic) magnetic susceptibility. Since the ac susceptibilities do not reveal any out-of-phase components for compounds 1 nor 2, the experimental data of the magnetic dc measurements are presented as χmT vs. T and Mmvs. B curves in Fig. 5. For compound 1, a χmT value of 7.68 cm3 K mol−1 at 290 K is within the interval of 5.88–9.19 cm3 K mol−1 expected for six non-interacting high-spin Ni(II) centres.40 Lowering the temperature, χmT slowly decreases for T > 100 K and rapidly decreases below that point indicating predominantly antiferromagnetic exchange interactions between the Ni(II) centres. For compound 2, the corresponding value at 290 K is 9.29 cm3 K mol−1 being in the range expected for eight non-interacting high-spin Ni(II) centres (7.84–12.25 cm3 K mol−1).40 In contrast to compound 1, χmT slowly increases by decreasing the temperature, reaching a maximum at T ≈ 30 K. The χmT value rapidly decreases on further temperature decrease. Such a maximum results from predominantly ferromagnetic exchange interactions between the eight Ni(II) centres. Since both magnetisation curves of compounds 1 and 2 do not exhibit saturation, a reliable conclusion with respect to the upper limit of total effective spin ground state Stot is impossible. | ||
| Fig. 5 Temperature dependence of χmT at 0.1 Tesla for compounds 1 and 2; inset: field dependence of the molar magnetisation Mm at 2 K; circles: experimental data, solid graphs: least-squares fits. | ||
A model description of the magnetic susceptibilities was based on the following assumptions, in order to contain computational limitations and to restrict the number of independent fitting parameters: Since the ligand field of each Ni(II) centre is well approximated as octahedrally symmetric, the ground term is an orbital momentum singlet 3A2 originating from ground term 3F of the free high-spin Ni(II) ion – according to the crystal structure, the distortion of the octahedral symmetry is expected to be small. Therefore, the zero-field splitting is expected to be negligible and thus omitted. Hence, the magnetic features of compounds 1 and 2 are described in terms of an isotropic effective spin approach (S = 1) including an effective Heisenberg-Hamiltonian to characterise the exchange interactions. The number of exchange parameters is restricted in a further approximation: exchange pathways exhibiting similar geometries are assumed as identical, see Fig. 6 for the resulting coupling schemes.
 | ||
| Fig. 6 Coupling schemes used for the magnetochemical modelling for compounds 1 and 2. | ||
The exchange interaction Hamiltonian for compound 1
| Ĥex,1 = −2JA(Ŝ1Ŝ2 + Ŝ5Ŝ6) − 2JB(Ŝ2Ŝ3 + Ŝ4Ŝ5) − 2JCŜ3Ŝ4 |
in combination with the Zeeman Hamiltonian are employed to model the experimental data within the computational framework CONDON 2.0.41 The least-squares fit to the magnetic data of compound 1 (quality-of-fit parameter SQ = 0.9%) results in the exchange parameters JA = −7.7 ± 0.1 cm−1, JB = +7.2 ± 0.1 cm−1, JC = −2.6 ± 0.1 cm−1, and an isotropic g-value of giso = 2.25 ± 0.01. These parameters confirm the predominantly antiferromagnetic exchange interaction between the six Ni(II) centres. The magnitudes of the exchange interaction energies reveal weak interactions within the compound that result in a ground state of a total effective spin Stot = 0.
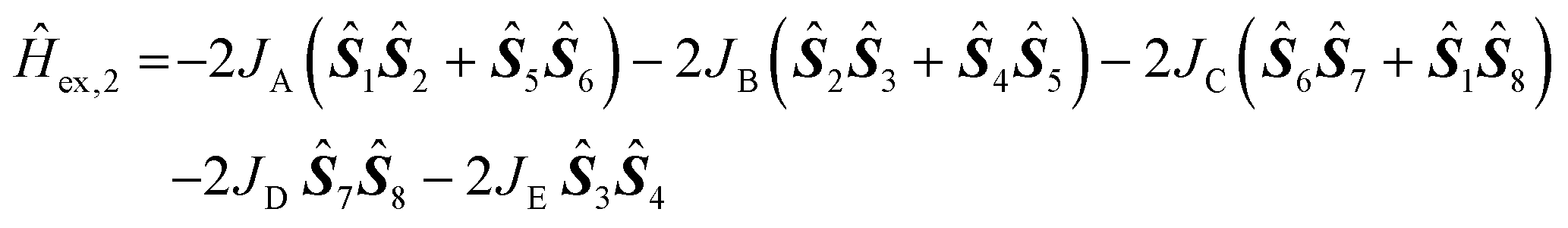
The least-squares fit to the magnetic susceptibility data of compound 2 is slightly worse (SQ = 1.5%) if a model including three independent exchange coupling parameters (JA = JD = −1.6 ± 0.1 cm−1, JB = −1.2 ± 0.1 cm−1, JC = JE = +5.2 ± 0.1 cm−1, giso = 2.13 ± 0.02) is employed. A slightly better goodness-of-fit is achieved (SQ = 1.2%) in the approximation shown in Fig. 6 based on the less-symmetric structure of the compound, although the margin of error of the fit parameter JE is larger. The corresponding least-squares parameters are JA = −1.5 ± 0.2 cm−1, JB = −1.1 ± 0.1 cm−1, JC = +4.6 ± 0.1 cm−1, JD = −1.6 ± 0.1 cm−1, JE = +4.9 ± 1.7 cm−1 and giso = 2.13 ± 0.02. Even though the calculated values exhibit weaker net exchange interactions in compound 2 than in compound 1, they confirm the predominantly ferromagnetic interaction between the eight Ni(II) centres. According to the calculated parameters, the ground state of compound 2 is characterised by a total effective spin Stot = 2.
Conclusion
We synthesised and characterised two polynuclear nickel(II) compounds 1 and 2 with structurally exposed thioether functional groups in the periphery of the cationic complexes [Ni6(L·SMe)3(CO3)(MeOH)6(THF)2]+ and [Ni8(HL·SMe)2(L·SMe)2(OH)4(MeCN)4(H2O)4]2+, respectively. These coordination clusters with distinct nickel topologies are characterised by different total ground-state spins and exhibit different types of exchange interactions: predominantly antiferromagnetic for the horseshoe-like {Ni6} with Stot = 0 and predominantly ferromagnetic for the rollercoaster-like {Ni8} with Stot = 2. Due to specific chemical properties (thermal stability, solubility and detectability following ESI-MS), the anion-templated self-assembly 1 appears to be a suitable candidate for molecular deposition and anchoring onto metallic surfaces via a thioether linkage and studies at solid–liquid interfaces. In forthcoming studies we will explore its potential as a heterogenised homogeneous catalyst, for example via the use of cation exchange42 to incorporate the solvent-free [Ni6(L·SMe)3(CO3)]+ fragment into large-pore metal–organic frameworks. We also plan to assess compound 1 and its carbonate-bonded metal core [Ni6(L·SMe)3(CO3)]+ as catalysts for ethylene oligomerisation where we expect the catalytic activity and selectivity to also depend on the pendant thioether groups.11b,c,43 Finally, we target the immobilisation of the horseshoe-like {Ni6} complex with its structurally exposed thioether anchoring groups onto noble metal-coated mesoporous silica nanoparticles (such as SBA-15),44 in order to probe industrially relevant catalytic oxidation reactions on solid supports at low and high temperatures.Experimental
Materials and methods
The synthesis of compounds 1, 2, A and B was carried out under aerobic conditions. All starting materials were from commercial sources and used as received. Solvents were used without further purification. The IR spectra of compounds 1, 2, A and B were recorded on a Nicolet Avatar 360 FTIR spectrometer (KBr pellets, v = 4000–400 cm−1). TG/DT analyses of compounds 1 and 2 were performed under N2 flow and a heating rate of 5 K min−1 in the temperature range 25–800 °C on a Mettler Toledo TGA/SDTA 851e instrument. The ESI-MS spectrum of compound 1 in the positive ion mode was recorded on a 4000 QTRAP mass spectrometer system, using the LC/LC-MS method with direct infusion.X-ray crystallography
Single-crystal X-ray diffraction data for all compounds were collected on a Bruker Apex II diffractometer with Mo Kα radiation (λ = 0.71073 Å) at 153 K. Crystals were mounted in a Hampton cryoloop with Paratone-N oil. Absorption corrections were applied numerically based on multifaceted crystal model using SADABS software.45 A summary of the crystal data and structure refinements for compounds 1, 2, A and B are given in Tables S1, S3, S5 and S7 in the ESI,† respectively. Details are described in the CIF files. CCDC 1440001–1440004 contain the supplementary crystallographic data for this paper.Magnetic susceptibility measurements
Magnetic susceptibility data of compounds 1 and 2 were recorded using a Quantum Design MPMS-5XL SQUID magnetometer for direct current (dc) and alternating current (ac) measurements. The polycrystalline samples were compacted and immobilised into cylindrical PTFE capsules. The dc susceptibility data were acquired as a function of the field (0.1–5.0 T) and temperature (2.0–290 K). The ac susceptibility data were measured in the absence of a static bias field in the 10–1000 Hz frequency range (T = 2.0–50 K, Bac = 3 G), but no out-of-phase signals were detected. Data were corrected for diamagnetic contributions from the sample holder and the compounds (χdia(1) = −0.96 × 10−3 cm3 mol−1, χdia(2): −1.20 × 10−3 cm3 mol−1).Acknowledgements
This work was supported by an ERC Starting Grant (MOLSPINTRON, 308051). K. Y. M. thanks the Excellence Initiative of the German federal and state governments for an RWTH Start-Up grant. We are grateful to Oliver Linnenberg for the synthesis and purification of the H3L·SMe ligand.References
- M. N. Rittner, J. of Metals, 2004, 56, 22 Search PubMed.
- Modern Surface Organometallic Chemistry, ed. J.-M. Basset, R. Psaro, D. Roberto and R. Ugo, Wiley-VCH, Weinheim, 2009 Search PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed.
- See, e.g.: (a) I. S. Kristensen, D. J. Mowbray, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2008, 20, 374101 CrossRef CAS PubMed; (b) Z. Li, M. Smeu, M. A. Ratner and E. Borguet, J. Phys. Chem. C, 2013, 117, 14890 CrossRef CAS; (c) V. Kaliginedi, A. V. Rudnev, P. Moreno-García, M. Baghernejad, C. Huang, W. Hong and T. Wandlowski, Phys. Chem. Chem. Phys., 2014, 16, 23529 RSC. See also: (d) S. Schmitz, J. van Leusen, A. Ellern, P. Kögerler and K. Yu. Monakhov, Inorg. Chem. Front., 2015, 2, 1095 RSC.
- A. Cornia, M. Mannini, P. Sainctavit and R. Sessoli, Chem. Soc. Rev., 2011, 40, 3076 RSC.
- J. J. Parks, A. R. Champagne, T. A. Costi, W. W. Shum, A. N. Pasupathy, E. Neuscamman, S. Flores-Torres, P. S. Cornaglia, A. A. Aligia, C. A. Balseiro, G. K.-L. Chan, H. D. Abruña and D. C. Ralph, Science, 2010, 328, 1370 CrossRef CAS PubMed.
- (a) T. T. T. Li, H. Y. Liu and M. J. Weaver, J. Am. Chem. Soc., 1984, 106, 1233 CrossRef CAS; (b) K. I. Ozoemena, T. Nyokong and P. Westbroek, Electroanalysis, 2003, 15, 1762 CrossRef CAS; (c) W. Zhou, S. Ye, M. Abe, T. Nishida, K. Uosaki, M. Osawa and Y. Sasaki, Chem. – Eur. J., 2005, 11, 5040 CrossRef CAS PubMed.
- (a) H. O. Finklea and D. D. Hanshew, J. Am. Chem. Soc., 1992, 114, 3173 CrossRef CAS; (b) M. S. Ravenscroft and H. O. Finklea, J. Phys. Chem., 1994, 98, 3843 CrossRef CAS; (c) H. O. Finklea and M. S. Ravenscroft, Isr. J. Chem., 1997, 37, 179 CrossRef CAS; (d) D. A. Brevnov and H. O. Finklea, J. Electrochem. Soc., 2000, 147, 3461 CrossRef CAS; (e) R. M. Haddox and H. O. Finklea, J. Phys. Chem. B, 2004, 108, 1694 CrossRef CAS; (f) N. Tuccitto, V. Torrisi, M. Cavazzini, T. Morotti, F. Puntoriero, S. Quici, S. Campagna and A. Licciardello, ChemPhysChem, 2007, 8, 227 CrossRef CAS PubMed.
- (a) M. Yamada and H. Nishihara, Langmuir, 2003, 19, 8050 CrossRef CAS; (b) M. Yamada, T. Tadera, K. Kubo and H. Nishihara, J. Phys. Chem. B, 2003, 107, 3703 CrossRef CAS; (c) T. Belser, M. Stoehr and A. Pfaltz, J. Am. Chem. Soc., 2005, 127, 8720 CrossRef CAS PubMed.
- Z. Weng, S. Teo and T. S. A. Hor, Dalton Trans., 2007, 3493 RSC.
- (a) K. Song, H. Gao, F. Liu, J. Pan, L. Guo, S. Zai and Q. Wu, Eur. J. Inorg. Chem., 2009, 3016 CrossRef CAS; (b) A. Ghisolfi, C. Fliedel, V. Rosa, R. Pattacini, A. Thibon, K. Yu. Monakhov and P. Braunstein, Chem. – Asian J., 2013, 8, 1795 CrossRef CAS PubMed; (c) A. Ghisolfi, C. Fliedel, V. Rosa, K. Yu. Monakhov and P. Braunstein, Organometallics, 2014, 33, 2523 CrossRef CAS.
- P. Van Der Voort, C. Vercaemst, D. Schaubroeck and F. Verpoort, Phys. Chem. Chem. Phys., 2008, 10, 347 RSC.
- P. Croizat, F. Müller, H. Mantz, A. Englisch, R. Welter, S. Hüfner and P. Braunstein, C. R. Chim., 2009, 12, 1228 CrossRef CAS.
- A. Naitabdi, O. Toulemonde, J. P. Bucher, J. Rosé, P. Braunstein, R. Welter and M. Drillon, Chem. – Eur. J., 2008, 14, 2355 CrossRef CAS PubMed.
- V. Gallo, P. Mastrorilli, C. F. Nobile, P. Braunstein and U. Englert, Dalton Trans., 2006, 2342 RSC.
- V. Rosa, C. Fliedel, A. Ghisolfi, R. Pattacini, T. Avilés and P. Braunstein, Dalton Trans., 2013, 42, 12109 RSC.
- S. Todisco, P. Mastrorilli, M. Latronico, V. Gallo, U. Englert, N. Re, F. Creati and P. Braunstein, Inorg. Chem., 2015, 54, 4777 CrossRef CAS PubMed.
- S. Mameri, A. M. Ako, F. Yesil, M. Hibert, Y. Lan, C. E. Anson and A. K. Powell, Eur. J. Inorg. Chem., 2014, 4326 CrossRef CAS.
- C. Fliedel, V. Rosa, A. Falceto, P. Rosa, S. Alvarez and P. Braunstein, Inorg. Chem., 2015, 54, 6547 CrossRef CAS PubMed.
- A. Abhervé, J. M. Clemente-Juan, M. Clemente-León, E. Coronado, J. Boonmak and S. Youngme, New J. Chem., 2014, 38, 2105 RSC.
- (a) A. Cornia, A. C. Fabretti, M. Pacchioni, L. Zobbi, D. Bonacchi, A. Caneschi, D. Gatteschi, R. Biagi, U. Del Pennino, V. De Renzi, L. Gurevich and H. S. J. Van der Zant, Angew. Chem., Int. Ed., 2003, 42, 1645 CrossRef CAS PubMed; (b) L. Zobbi, M. Mannini, M. Pacchioni, G. Chastanet, D. Bonacchi, C. Zanardi, R. Biagi, U. Del Pennino, D. Gatteschi, A. Cornia and R. Sessoli, Chem. Commun., 2005, 1640 RSC; (c) S. Voss, M. Burgert, M. Fonin, U. Groth and U. Rüdiger, Dalton Trans., 2008, 499 RSC.
- (a) M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194 CrossRef CAS PubMed; (b) F. Pineider, M. Mannini, C. Danieli, L. Armelao, F. M. Piras, A. Magnani, A. Cornia and R. Sessoli, J. Mater. Chem., 2010, 20, 187 RSC; (c) M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, Ph. Sainctavit, M.-A. Arrio, E. Otero, L. Joly, J. Criginski Cesar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417 CrossRef CAS PubMed; (d) M. Jesus Rodriguez-Douton, M. Mannini, L. Armelao, A.-L. Barra, E. Tancini, R. Sessoli and A. Cornia, Chem. Commun., 2011, 47, 1467 RSC.
- M. Perfetti, F. Pineider, L. Poggini, E. Otero, M. Mannini, L. Sorace, C. Sangregorio, A. Cornia and R. Sessoli, Small, 2014, 10, 323 CrossRef CAS PubMed.
- M. Mannini, E. Tancini, L. Sorace, P. Sainctavit, M.-A. Arrio, Y. Qian, E. Otero, D. Chiappe, L. Margheriti, J. C. Cezar, R. Sessoli and A. Cornia, Inorg. Chem., 2011, 50, 2911 CrossRef CAS PubMed.
- (a) A. Ghisolfi, K. Yu. Monakhov, R. Pattacini, P. Braunstein, X. López, C. de Graaf, M. Speldrich, J. van Leusen, H. Schilder and P. Kögerler, Dalton Trans., 2014, 43, 7847 RSC; (b) V. Heβ, F. Matthes, D. E. Bürgler, K. Yu. Monakhov, C. Besson, P. Kögerler, A. Ghisolfi, P. Braunstein and C. M. Schneider, Surf. Sci., 2015, 641, 210 CrossRef.
- (a) V. Corradini, R. Biagi, U. del Pennino, V. De Renzi, A. Gambardella, M. Affronte, C. A. Muryn, G. A. Timco and R. E. P. Winpenny, Inorg. Chem., 2007, 46, 4937 CrossRef CAS PubMed; (b) V. Corradini, F. Moro, R. Biagi, V. De Renzi, U. del Pennino, V. Bellini, S. Carretta, P. Santini, V. A. Milway, G. Timco, R. E. P. Winpenny and M. Affronte, Phys. Rev. B: Condens. Matter, 2009, 79, 144419 CrossRef; (c) V. Corradini, A. Ghirri, U. del Pennino, R. Biagi, V. A. Milway, G. Timco, F. Tuna, R. E. P. Winpenny and M. Affronte, Dalton Trans., 2010, 39, 4928 RSC; (d) H. Rath, G. A. Timco, V. Corradini, A. Ghirri, U. del Pennino, A. Fernandez, R. G. Pritchard, C. A. Muryn, M. Affronte and R. E. P. Winpenny, Chem. Commun., 2013, 49, 3404 RSC.
- J. Dreiser, A. M. Ako, C. Wäckerlin, J. Heidler, C. E. Anson, A. K. Powell, C. Piamonteze, F. Nolting, S. Rusponi and H. Brune, J. Phys. Chem. C, 2015, 119, 3550 Search PubMed.
- N. Song and Y.-W. Yang, Chem. Soc. Rev., 2015, 44, 3474 RSC.
- M. G. L. Petrucci and A. K. Kakkar, Adv. Mater., 1996, 8, 251 CrossRef CAS.
- A. R. Silva, C. Freire, B. de Castro, M. M. A. Freitas and J. L. Figueiredo, Microporous Mesoporous Mater., 2001, 46, 211 CrossRef CAS and references cited therein.
- V. Arun, N. Sridevi, P. P. Robinson, S. Manju and K. K. M. Yusuff, J. Mol. Catal. A: Chem., 2009, 304, 191 CrossRef CAS.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420 CrossRef CAS and references cited therein.
- C. Hamann, R. Woltmann, I.-Po Hong, N. Hauptmann, S. Karan and R. Berndt, Rev. Sci. Instrum., 2011, 82, 033903 CrossRef PubMed.
- For recent studies, see e.g.: (a) J. Esteban, L. Alcázar, M. Torres-Molina, M. Monfort, M. Font-Bardia and A. Escuer, Inorg. Chem., 2012, 51, 5503 CrossRef CAS PubMed; (b) J. Esteban, A. Escuer, M. Font-Bardia, O. Roubeau and S. J. Teat, Polyhedron, 2013, 52, 339 CrossRef CAS; (c) J. Esteban, M. Font-Bardia, J. S. Costa, S. J. Teat and A. Escuer, Inorg. Chem., 2014, 53, 3194 CrossRef CAS PubMed; (d) A. Massard, G. Rogez and P. Braunstein, Dalton Trans., 2014, 43, 42 RSC; (e) A. Escuer, J. Esteban, J. Mayans and M. Font-Bardia, Eur. J. Inorg. Chem., 2014, 5443 CrossRef CAS; (f) M. Pait, A. Bauzá, A. Frontera, E. Colacio and D. Ray, Inorg. Chem., 2015, 54, 4709 CrossRef CAS PubMed; (g) K. I. Alexopoulou, A. Terzis, C. P. Raptopoulou, V. Psycharis, A. Escuer and S. P. Perlepes, Inorg. Chem., 2015, 54, 5615 CrossRef CAS PubMed.
- (a) F. K. Larsen, J. Overgaard, S. Parsons, E. Rentschler, A. A. Smith, G. A. Timco and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 5978 CrossRef CAS PubMed; (b) M. Rancan, G. N. Newton, C. A. Muryn, R. G. Pritchard, G. A. Timco, L. Cronin and R. E. P. Winpenny, Chem. Commun., 2008, 1560 RSC; (c) A. McRobbie, A. R. Sarwar, S. Yeninas, H. Nowell, M. L. Baker, D. Allan, M. Luban, C. A. Muryn, R. G. Pritchard, R. Prozorov, G. A. Timco, F. Tuna, G. F. S. Whitehead and R. E. P. Winpenny, Chem. Commun., 2011, 47, 6251 RSC.
- (a) E. García-España, P. Gaviña, J. Latorre, C. Soriano and B. Verdejo, J. Am. Chem. Soc., 2004, 126, 5082 CrossRef PubMed; (b) L.-Y. Kong, Z.-H. Zhang, H.-F. Zhu, H. Kawaguchi, T.-A. Okamura, M. Doi, Q. Chu, W.-Y. Sun and N. Ueyama, Angew. Chem., Int. Ed., 2005, 44, 4352 CrossRef CAS PubMed; (c) L. Natrajan, J. Pécaut and M. Mazzanti, Dalton Trans., 2006, 1002 RSC; (d) P. C. Andrews, T. Beck, C. M. Forsyth, B. H. Fraser, P. C. Junk, M. Massi and P. W. Roesky, Dalton Trans., 2007, 5651 RSC; (e) S.-D. Bian, J.-H. Jia and Q.-M. Wang, J. Am. Chem. Soc., 2009, 131, 3422 CrossRef CAS PubMed; (f) X.-L. Tang, W.-H. Wang, W. Dou, J. Jiang, W.-S. Liu, W.-W. Qin, G.-L. Zhang, H.-R. Zhang, K.-B. Yu and L.-M. Zheng, Angew. Chem., Int. Ed., 2009, 48, 3499 CrossRef CAS PubMed; (g) H. Ke, L. Zhao, G. F. Xu, Y. N. Guo, J. Tang, X. Y. Zhang and H. J. Zhang, Dalton Trans., 2009, 10609 RSC; (h) A. M. García-Deibe, C. Portela-García, M. Fondo, A. J. Mota and J. Sanmartín-Matalobos, Chem. Commun., 2012, 48, 9915 RSC; (i) J.-B. Peng, Q.-C. Zhang, X.-J. Kong, Y.-Z. Zheng, Y.-P. Ren, L.-S. Long, R.-B. Huang, L.-S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2012, 134, 3314 CrossRef CAS PubMed; (j) T. D. Pasatoiu, A. Ghirri, A. M. Madalan, M. Affronte and M. Andruh, Dalton Trans., 2014, 43, 9136 RSC.
- (a) T. N. Hooper, R. Inglis, M. A. Palacios, G. S. Nichol, M. B. Pitak, S. J. Coles, G. Lorusso, M. Evangelisti and E. K. Brechin, Chem. Commun., 2014, 50, 3498 RSC and references cited therein; (b) A. K. Ghosh, M. Pait, M. Shatruk, V. Bertolasi and D. Ray, Dalton Trans., 2014, 43, 1970 RSC; (c) C. C. Wang, H. Y. Li, X. Q. Zhang, Y. M. Jiang and S. H. Zhang, J. Cluster Sci., 2015, 26, 1055 CrossRef CAS; (d) V. Velasco, D. Aguilà, L. A. Barrios, I. Borilovic, O. Roubeau, J. Ribas-Ariño, M. Fumanal, S. J. Teat and G. Aromí, Chem. Sci., 2015, 6, 123 RSC; (e) W. Sethi, S. Sanz, K. S. Pedersen, M. A. Sørensen, G. S. Nichol, G. Lorusso, M. Evangelisti, E. K. Brechin and S. Piligkos, Dalton Trans., 2015, 44, 10315 RSC; (f) X.-M. Zhang, P. Li, W. Gao, J.-P. Liu and E.-Q. Gao, Dalton Trans., 2015, 44, 13581 RSC; (g) M. Hołyńska, R. Clérac and M. Rouzières, Chem. – Eur. J., 2015, 21, 13321 CrossRef PubMed.
- (a) R. D. Shannon, Acta Crystallogr., 1976, 32, 751 CrossRef; (b) B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- See e.g.: (a) P. D. Frischmann and M. J. MacLachlan, Chem. Soc. Rev., 2013, 42, 871 RSC; (b) J.-J. Liu, Y.-F. Guan, Y. Chen, M.-J. Lin, C.-C. Huang and W.-X. Dai, Dalton Trans., 2015, 44, 9370 RSC.
- H. Lueken, Magnetochemie, B.G. Teubner, Stuttgart, 1999 Search PubMed.
- (a) M. Speldrich, H. Schilder, H. Lueken and P. Kögerler, Isr. J. Chem., 2011, 51, 215 CrossRef CAS; (b) J. van Leusen, M. Speldrich, H. Schilder and P. Kögerler, Coord. Chem. Rev., 2015, 289–290, 137–148 CrossRef CAS.
- D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586 CrossRef CAS PubMed and references cited therein.
- W. Keim, Angew. Chem., Int. Ed. Engl., 1990, 29, 235–244 CrossRef.
- (a) Y. T. Lai, T. C. Chen, Y. K. Lan, B. S. Chen, J. H. You, C. M. Yang, N. C. Lai, J. H. Wu and C. S. Chen, ACS Catal., 2014, 4, 3824–3836 CrossRef CAS; (b) H. Guerba, B. Djellouli, C. Petit and V. Pitchon, C. R. Chim., 2014, 17, 775–784 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of the H3L·SMe ligand and its proposed oxidation pathway, depiction of the non-functionalised H3L ligand, synthesis and solid-state molecular structures of compounds A and B, IR spectra and crystal data and structure refinement details of compounds 1, 2, A and B, and ESI-MS spectrum of compound 1. CCDC 1440001–1440004. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00278h |
| This journal is © the Partner Organisations 2016 |