
Shrimp-shell derived carbon nanodots as precursors to fabricate Fe,N-doped porous graphitic carbon electrocatalysts for efficient oxygen reduction in zinc–air batteries†
Xian
Zhang
ab,
Rongrong
Liu
ab,
Yipeng
Zang
ab,
Guoqiang
Liu
ab,
Shengwen
Liu
a,
Guozhong
Wang
a,
Yunxia
Zhang
a,
Haimin
Zhang
*a and
Huijun
Zhao
ac
aKey Laboratory of Materials Physics, Centre for Environmental and Energy Nanomaterials, Anhui Key Laboratory of Nanomaterials and Nanotechnology, Institute of Solid State Physics, Chinese Academy of Sciences, Hefei 230031, China. E-mail: zhanghm@issp.ac.cn; Fax: +86 (0)551 65591434; Tel: +86 (0)551 65591973
bUniversity of Science and Technology of China, Hefei 230026, China
cCentre for Clean Environment and Energy, Griffith University, Gold Coast Campus, QLD 4222, Australia
First published on 14th April 2016
Abstract
In this work, shrimp-shell derived N-doped carbon nanodots (N-CNs) as carbon and nitrogen sources are assembled into particle-like aggregates by a simple polymerization reaction of pyrrole in the presence of Fe3+ to form Fe containing N-CN/polypyrrole (PPY) composites (Fe–N-CN/PPy). The resulting composites are thermally treated by a facile pyrolysis approach under a N2 atmosphere to obtain an Fe,N-doped porous graphitic carbon (Fe-N-PGC) material. The results demonstrate that the pyrolytically converted carbon material at 800 °C (Fe-N-PGC-800) exhibits an approximately mesoporous structure with a pore size distribution centered at ∼1.97 nm and ∼2.8 nm and a surface area of 806.7 m2 g−1. As an electrocatalyst for oxygen reduction reaction (ORR) in alkaline media, Fe-N-PGC-800 shows superior ORR catalytic activity with an onset potential of −0.017 V and a limiting current density of 5.42 mA cm−2 (at −0.4 V, vs. Ag/AgCl), which is superior to that of commercial Pt/C catalysts (onset potential of −0.018 V and a limiting current density of 5.21 mA cm−2 at −0.4 V, vs. Ag/AgCl). Additionally, Fe-N-PGC-800 also exhibits good ORR activity in acidic media with an onset potential of 0.53 V and a limiting current density of 5.58 mA cm−2 (at 0.1 V, vs. Ag/AgCl), comparable to that of most reported Fe-based N-doped carbon electrocatalysts. An air cathode made from Fe-N-PGC-800 shows high performance and superior cycling durability in zinc–air batteries (gravimetric energy density of 752 Wh kg−1), comparable to that of commercial Pt/C-based batteries (gravimetric energy density of 774 Wh kg−1). This work demonstrates the feasibility of utilizing biomass as a starting material to fabricate Fe,N-doped carbon materials as high performance ORR electrocatalysts for practical application in ORR-relevant energy devices.
Introduction
It is critically important to develop non-precious metal electrocatalysts with high oxygen reduction reaction (ORR) performance for potential applications in ORR-relevant energy devices such as fuel cells and metal–air batteries.1–5 Although Pt-based materials as electrocatalysts have demonstrated the best ORR catalytic activity to date, the high cost and the source scarcity of Pt-based electrocatalysts with poor tolerance to small fuel molecules (e.g., methanol) in ORR have greatly limited their large-scale practical applications.1–7 Therefore, much effort has been devoted to developing cheap, abundant and high-efficiency ORR catalyst materials to replace Pt-based electrocatalysts.6–9 Among the variety of ORR catalysts reported to date, transition metal containing N-doped carbon materials (M@N–C, M = Fe, Co etc.) have demonstrated great potential as promising candidates for ORR under both alkaline and acidic conditions.10–17 Besides experimental evidence, theoretical calculation results also reveal that the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond on N-coordinated metal active sites of M@N–C can be effectively weakened, thus leading to highly efficient O2 reduction.1 Although lots of achievements have been made, it is still highly desirable to explore further transition metal containing N-doped carbon materials with respect to their structure, composition, catalytic activity and cost for practical applications in future ORR-relevant energy devices.
O bond on N-coordinated metal active sites of M@N–C can be effectively weakened, thus leading to highly efficient O2 reduction.1 Although lots of achievements have been made, it is still highly desirable to explore further transition metal containing N-doped carbon materials with respect to their structure, composition, catalytic activity and cost for practical applications in future ORR-relevant energy devices.
N-doped carbon materials containing different formed Fe active species have recently been extensively investigated, demonstrating their great potential as promising candidates for ORR due to their intrinsically high catalytic activity.5,10–12,16–24 In studies reported to date, a variety of Fe-based N-doped carbon materials with different structures and compositions have been developed, for example, Fe,N-doped carbon materials,17,25 Fe nanoparticle@N-doped carbon core–shell structures,5,20 iron oxide nanoparticle@N-doped carbon composites,11,24 and iron carbide (Fe3C) nanocrystals encapsulated in N-doped carbon nanostructures.12,16,18,22,26 No matter what the structure and composition are, the aforementioned Fe-based N-doped carbon materials as electrocatalysts all exhibit superior ORR catalytic activities under alkaline or both alkaline and acidic conditions.5,10–12,16–26 However, the origin of the ORR catalytic activity of these Fe-based N-doped carbon electrocatalysts is still under debate. It has been generally accepted that Fe-Nx moieties in Fe,N-doped carbon structures are major catalytically active sites promoting ORR performance,1,17,23,25 whereas a synergetic coupling effect between N-doped carbon and iron oxide nanoparticles may contribute the high ORR catalytic activities of iron oxide@N-doped carbon composites.11,24 A recent experimental study reveals the presence of three Fe–N4 like ORR catalytic centers with distinctly different Fe–N switching behaviors in the Fe,N-doped carbon structure, contributing the ORR catalytic activities governed by the dynamic structure associated with the Fe2+/3+ redox transition.27 For Fe/Fe3C nanocrystal@N-doped carbon core–shell materials, some related studies have demonstrated that the encapsulated Fe/Fe3C nanocrystals in carbon structures cannot directly come into contact with the electrolyte, but possibly activate the surrounding graphitic layers to enable the outer surface of the carbon to become active toward ORR.5,12,16,20,22 Although the ORR active mechanisms have not been very clear, it is believed that the superior ORR catalytic activity of these Fe-based N-doped carbon materials is ascribed to not only their intrinsically catalytically active sites but also their structural properties such as high surface area and porous structure. It has been demonstrated that the high surface area and porous structure of electrocatalysts are favorable for the exposure of catalytically active sites and the ORR-related mass transport, thus improving ORR performance.16,28 Up to now, almost all reported Fe-based N-doped carbon materials have been synthesized by using fossil oil derived chemicals as carbon and nitrogen sources,5,10–12,16–24 which enhances undoubtedly the cost of practical applications. Therefore, it is highly desirable to utilize inexpensive and abundant precursors as carbon and nitrogen sources to fabricate high performance Fe-based N-doped carbon electrocatalysts for ORR and ORR-related energy devices.
Biomass (e.g., grass, soy milk)-derived N-doped carbon nanodots (N-CNs) not only possess small nanodot sizes and surface rich O-containing functional groups, but also contain naturally doped nitrogen atoms in carbon structures, which have shown good ORR performance as electrocatalysts.29–31 Furthermore, it should be feasible to use biomass-derived N-CNs as carbon and nitrogen sources to fabricate Fe-based N-doped carbon materials for ORR and the corresponding energy devices (e.g., zinc–air batteries). As far as we know, related studies have not been reported in the literature. Herein, Fe containing N-CN/polypyrrole (PPY) composites (Fe–N-CN/PPy) were first fabricated by a simple polymerization reaction of pyrrole in the presence of Fe3+ and shrimp-shell derived N-CNs. Subsequently, the Fe–N-CN/PPy composites were pyrolytically treated at different temperatures in N2 to obtain Fe,N-doped porous graphitic carbon (Fe-N-PGC) materials, as shown in Fig. 1. As an ORR electrocatalyst, Fe-N-PGC obtained at 800 °C (Fe-N-PGC-800) exhibits superior catalytic activity with a close four-electron-transfer pathway under both alkaline and acidic conditions. Additionally, Fe-N-PGC-800 also demonstrates great potential for use as an air cathode material in zinc–air batteries with high performance and cycling durability.
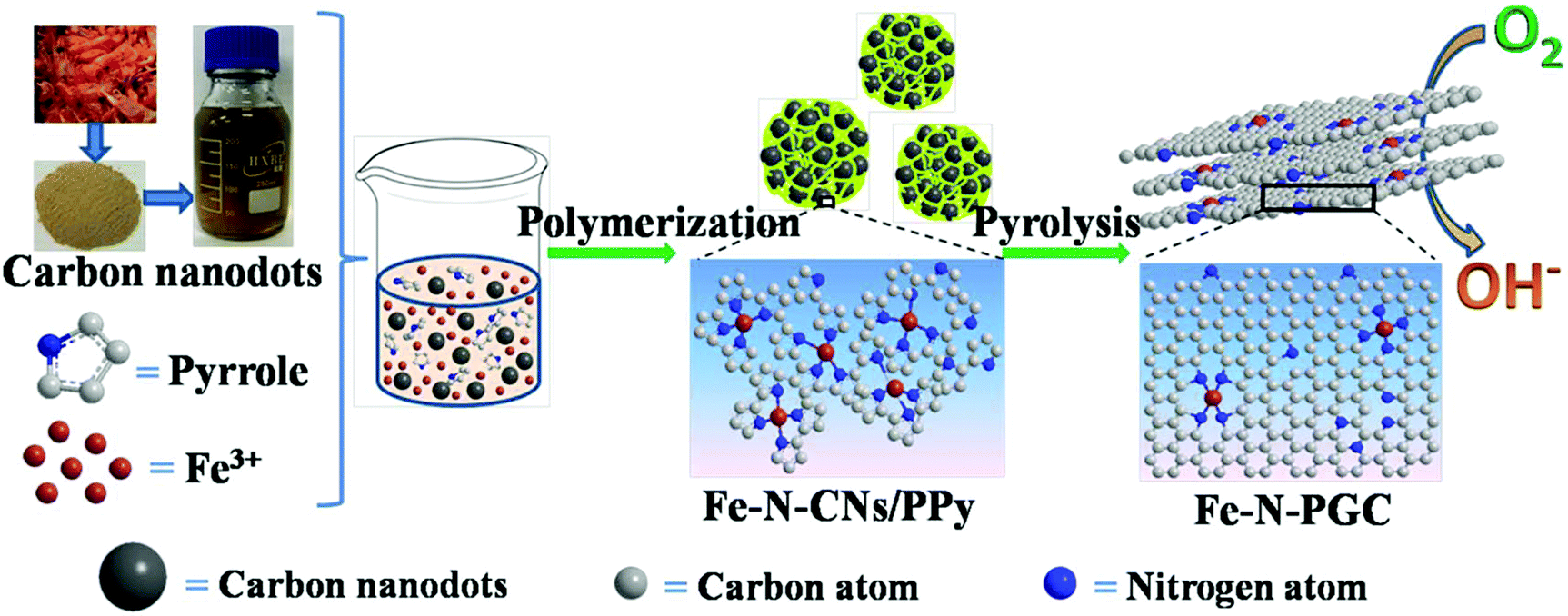 | ||
| Fig. 1 Schematic illustration of the preparation process of Fe-N-PGC using shrimp-shell derived N-doped carbon nanodots as precursors. | ||
Experimental section
Material synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min, and the supernatant was then collected for further characterization and use. The product yield of N-CNs is ∼15% and the concentration of the N-CN solution is ∼15 mg mL−1.
000 rpm for 15 min, and the supernatant was then collected for further characterization and use. The product yield of N-CNs is ∼15% and the concentration of the N-CN solution is ∼15 mg mL−1.
Fe,N-doped porous graphitic carbon (Fe-N-PGC) samples were fabricated by pyrolysis of the polymerized product (Fe–N-CNs/PPy) at different temperatures (600, 700, 800, 900 °C) for 1 h with a heating rate of 5 °C min−1 under a N2 atmosphere. To remove ORR-nonreactive substances, an acid preleaching procedure was performed for the pyrolytic product in a 0.5 M H2SO4 solution at 80 °C for 5 h, followed by centrifugation and thorough washing with deionized water. Then, the acid preleached sample was again pyrolyzed under a N2 atmosphere by the same procedure as that for the first pyrolysis process for 2 h to obtain Fe-N-PGC products with different temperatures. The pyrolytic product was denoted as Fe-N-PGC-X (X represents pyrolysis temperatures of 600, 700, 800, and 900 °C, respectively).
Characterization
The crystalline structures of samples were identified by X-ray diffraction analysis (XRD, Philips X'pert PRO) using Ni filtered monochromatic CuKα radiation (λKα1 = 1.5418 Å) at 40 kV and 40 mA. The morphology and structure of samples were characterized by field emission scanning electron microscopy (FESEM, Quanta 200FEG) and transmission electron microscopy (TEM, JEOL 2010) with an energy dispersive X-ray spectrometer (EDS Oxford, Link ISIS). An X-ray photoelectron spectroscopy (XPS) analysis was performed on an ESCALAB 250 X-ray photoelectron spectrometer (Thermo, America) equipped with AlKα1, 2 monochromatized radiation at a 1486.6 eV X-ray source. All binding energies were carefully aligned referenced to the C 1s peak (284.6 eV) arising from the surface hydrocarbon of the sample. The surface area and porosity of samples were measured using a Surface Area and Porosity Analyzer (Tristar3020M). Fourier transform infrared (FT-IR) spectroscopy of the sample was performed on a Perkin-Elmer TGA7 infrared spectrometer to identify the functional groups of the sample.Electrochemical measurements
Electrochemical measurements were performed on an electrochemical workstation (CHI 760D, CH Instruments, Inc., Shanghai, China) coupled with a PINE rotating disk electrode (RDE) system (Pine Instruments Co. Ltd USA). A standard three-electrode electrochemical cell equipped with a gas flow system was employed during measurements. Prior to measurements, a rotating disk electrode (RDE, 5.0 mm in diameter) was first polished with 5.0, 3.0 and 0.05 μm alumina slurry sequentially and then washed ultrasonically with water and ethanol for 1 min, respectively. The cleaned electrode was dried with a high-purity nitrogen steam. The Fe-N-PGC catalyst ink was prepared by dispersing catalyst powder (5.0 mg) into a mixture containing 100 μL of Nafion solution (0.5 wt%) and 900 μL of ethanol, followed by ultrasonic treatment for 2 min. After that, 12 μL of catalyst ink was then cast onto a glassy carbon (GC) electrode surface, leading to a catalyst loading amount of 305 μg cm−2. For comparison, a commercial Pt/C catalyst ink was also prepared by the same procedure as that for Fe-N-PGC catalyst ink.The ORR performance of catalysts was investigated by cyclic voltammogram (CV) and linear sweep voltammogram (LSV) measurements in O2 (or N2)-saturated 0.1 M KOH solution. CV curves were measured at a scan rate of 50 mV s−1. LSV curves were measured at a scan rate of 10 mV s−1 under different disk rotation rates of 400, 625, 900, 1225, 1600 and 2025 rpm. All the potentials in this work were recorded with respect to the Ag/AgCl reference electrode. The electron transfer number (n) per oxygen molecule in an ORR process was calculated using the Koutecky–Levich (K–L) equation:9
| J−1 = Jk−1 + (Bω½)−1 | (1) |
| B = 0.62nF(D0)2/3ν−1/6C0 | (2) |
| Jk = nFkCo | (3) |
485 C mol−1), D0 is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1), v is the kinetic viscosity (0.01 cm2 s−1), and C0 is the bulk concentration of O2 (1.2 × 10−3 mol L−1). The ORR performance of catalysts was also evaluated in O2 (or N2)-saturated 0.1 M HClO4 solution with other test conditions kept the same. In 0.1 M HClO4 solution, the diffusion coefficient D0 is 2.0 × 10−5 cm2 s−1, and the bulk concentration of O2 and C0 is 1.5 × 10−3 mol L−1. For comparison, a N-CN derived carbon material was fabricated by direct pyrolysis of freeze dried N-CNs in the absence of pyrrole and Fe3+ in N2 at 800 °C for 2 h (denoted as N-CNs-800); N-CN/pyrrole composites without Fe3+ were pyrolyzed in N2 at 800 °C for 2 h to obtain a N-doped carbon material (denoted as N-CNs/P-800); Fe3+ triggered polypyrrole without N-CNs was pyrolyzed in N2 at 800 °C for 2 h to obtain an Fe-containing N-doped carbon material (denoted as Fe-P-800).
The measurements of zinc–air batteries were performed on home-built electrochemical cells.16 All data were collected from the as-fabricated cells with a CHI 760D (CH Instruments, Inc., Shanghai, China) electrochemical workstation at room temperature. Briefly, zinc foil was used as the anode and catalysts loaded on the gas diffusion layer (Teflon-coated carbon fiber paper with a geometric area of 1.0 cm2 and a catalyst loading amount of 2.0 mg cm−2) were used as the air cathode. The electrolyte was 6.0 M KOH.
Results and discussion
Recently, Fe-based N-doped carbon materials fabricated by a simple pyrolysis approach have been widely investigated as ORR electrocatalysts.5,10–12,16–26 However, the use of different carbon sources as precursors results in the formation of various Fe active species (e.g., zero-valent Fe, iron oxides, and iron carbide) in graphitic carbon structures, thus leading to dramatically different ORR performance.5,10–12,16–26 In this work, as shown in Fig. 1, we utilize shrimp-shell derived N-doped carbon nanodots as carbon and nitrogen sources to fabricate Fe,N-doped porous graphitic carbon (Fe-N-PGC) materials as ORR electrocatalysts by a simple combined approach of polymerization and pyrolysis. Fig. 2a shows the TEM image of shrimp-shell derived N-CNs. As shown, the hydrothermally fabricated carbon nanodots possess a size distribution of 1.5–5.5 nm (inset in Fig. 2a), exhibiting a lattice spacing of 0.26 nm which is smaller than the interlayer distance between graphene layers in graphite (inset in Fig. 2a).33 The FT-IR analysis indicates that shrimp-shell derived carbon nanodots contain rich surface O and N functional groups such as O–H, N–H, CO/CN, C–O/C–N, similar to previous reports (Fig. 2b).30,33 Further, XPS survey spectra of the N-CNs confirm the presence of C, N and O elements with a nitrogen content of 14.4% (Fig. S1, ESI†). The high resolution N 1s spectrum of the N-CNs reveals the existence of three types of doped N atoms, namely, pyridinic-N (398.7 eV), pyrrolic-N (399.7 eV) and graphitic-N (400.8 eV) (Fig. 2c).30,34 The N-CNs with small nanodot sizes and surface rich O- and N-containing functional groups can be fully utilized as carbon and nitrogen sources to fabricate high performance ORR electrocatalysts.29,30,35 After the assembly of N-CNs by a polymerization reaction of pyrrole in the presence of Fe3+, particle-like aggregates can be obtained (Fe–N-CNs/PPy), as shown in Fig. 2d. Further, Fe–N-CNs/PPy can be pyrolytically transformed into Fe-based graphitic carbon material for ORR.
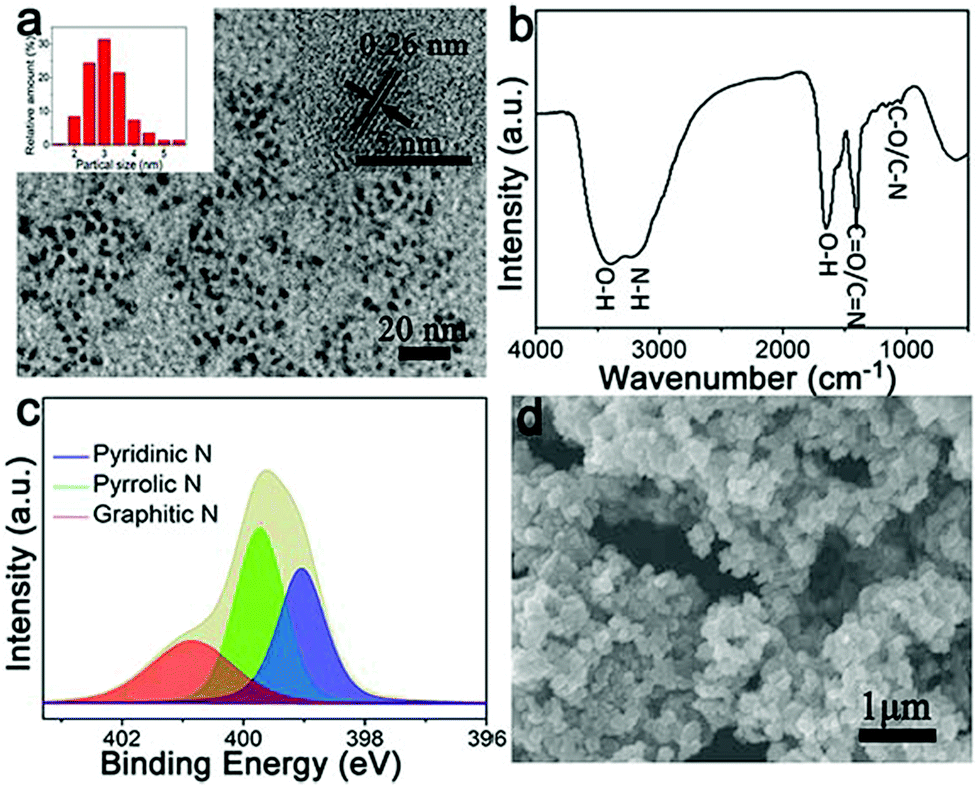 | ||
| Fig. 2 (a) TEM image of N-CNs (insets of nanodot size distribution and HRTEM image of an individual nanodot). (b) FT-IR spectrum of N-CNs. (c) High resolution N 1s XPS spectrum of N-CNs. (d) SEM image of Fe–N-CNs/PPy. | ||
The Fe–N-CNs/PPy was treated by a pyrolysis approach at different temperatures to obtain Fe,N-doped porous graphitic carbon materials (Fe-N-PGC-X, X represents pyrolysis temperatures of 600, 700, 800 and 900 °C, respectively). Fig. 3a shows the XRD patterns of pyrolytic samples at different temperatures. As shown, all pyrolytic samples exhibit strong inter-plane (002) diffraction at around 24.7° and relatively weak inner-plane (101) diffraction at around 43.7°, ascribed to graphitic carbon.28 No diffraction peaks of Fe-related species such as zero-valent iron, iron carbide and iron oxides can be observed in the XRD patterns, indicating that the acid leaching process removes almost most of the unstable Fe-related species (e.g., iron oxides) in this work (Fig. S2, ESI†). After pyrolysis treatment, the SEM images of samples at different temperatures show similar carbon aggregation morphologies (Fig. S3, ESI†). Detailed structure information on the pyrolytic sample was obtained by TEM characterization. As shown in Fig. 3b and inset, a stacking layered structure of graphitic carbon can be clearly observed with a highly distorted lattice (take Fe-N-PGC-800 as an example). Moreover, no Fe-related particles were found, consistent with XRD results. The energy-filtered TEM (EFTEM) imaging confirms the presence of C, N, O and Fe elements in Fe-N-PGC-800 with homogeneously distributed characteristics (Fig. 3c). Importantly, the elemental mapping signals of homogeneously distributed Fe and N in graphitic carbon structures are highly overlaid with each other, implying the presence of Fe,N doping in graphitic carbon structures.16,35,36 These Fe,N-doped graphitic carbon structures have demonstrated superior ORR catalytic activity.16,35,37 Fig. S4 (ESI†) shows a surface survey XPS spectra of Fe-N-PGC-800, further indicating the presence of C (87.73%), O (6.96%), N (4.95%) and Fe (0.36%) elements. The high resolution N 1s spectrum (Fig. 3d) can be divided into four peaks at 398.5, 399.6, 400.8, and 404.6 eV, corresponding to pyridinic-N (25.0%), pyrrolic-N (22.9%), graphitic-N (40.4%), and quaternary N+–O− (11.7%).17,37 It has been generally accepted that pyridinic-N and pyrrolic-N can serve as metal-coordination sites owing to their lone-pair electrons, whereas pyridinic-N and graphitic-N are responsible for oxygen reduction.38 By comparing the high resolution N 1s spectrum of N-CNs and Fe-N-PGC-800 (Fig. 2c and 3d), it can be clearly seen that pyrrolic-N is predominant in shrimp-shell derived N-doped carbon nanodots, while pyridinic-N and graphitic-N are dominant in Fe-N-PGC-800, indicating high temperature pyrolysis treatment facilitating the transformation of N doping type. The pyridinic-N and graphitic-N are of high content in Fe-N-PGC-800, possibly leading to high ORR catalytic activity. The composition analysis of all pyrolytic samples by the XPS technique indicates that the N doping concentration in graphitic carbon decreases with pyrolysis temperature, as shown in Table S1 (ESI†). At higher pyrolysis temperature (e.g., 900 °C), the significantly decreased N doping level in graphitic carbon may be unfavourable for high ORR catalytic activity owing to the decreased catalytically active sites resulting from N doping. Besides, the high resolution C 1s XPS spectrum of Fe-N-PGC-800 exhibits the presence of CC–C and C–N/O (Fig. S5, ESI†).32 The high resolution Fe 2p spectrum (Fig. 3e) shows five peaks at 710.9, 713.5, 718.5, 724.7, and 730.5 eV. Among them, the peak at 718.5 eV is a satellite peak; the peaks at 710.9 and 713.5 eV can be ascribed to the binding energies of the 2p3/2 orbits of Fe2+ and Fe3+ species, respectively, whereas the peaks at 724.7 and 730.5 eV correspond to the binding energies of the 2p1/2 bands of Fe2+ and Fe3+, respectively.16,17 Additionally, the peak at 710.9 eV in the Fe 2p3/2 XPS spectrum may also be due to Fe–N bonding resulting from Fe ions coordinated to N.16,17Fig. 3f shows the N2 adsorption–desorption isotherm of Fe-N-PGC-800. The Brunauer–Emmett–Teller surface area and total pore volume of Fe-N-PGC-800 are 806.7 m2 g−1 and 0.205 cm3 g−1, respectively. The pore size distribution (inset of Fig. 3f) of Fe-N-PGC-800 is mainly centered at ∼1.97 nm and ∼2.8 nm, exhibiting an approximately mesoporous structure. Also, the surface area and total pore volume (Fig. S6 and Table S2, ESI†) of Fe-N-PGC-600, Fe-N-PGC-700 and Fe-N-PGC-900 are 677.3 m2 g−1 and 0.198 cm3 g−1, 786.8 m2 g−1 and 0.203 cm3 g−1, and 837.7 m2 g−1 and 0.242 cm3 g−1, respectively. Apparently, an increase in pyrolysis temperature leads to the increase of surface area and pore volume of the pyrolytic sample, which is very beneficial for the exposure of catalytically active sites.16 The pore size distribution curves (Fig. S6b, ESI†) indicate that Fe-N-PGC-600 and Fe-N-PGC-700 mainly display microporous structures with pore size distribution concentrated at ∼1.4 nm and ∼1.6 nm, respectively, while Fe-N-PGC-800 and Fe-N-PGC-900 exhibit mesoporous structures (inset of Fig. 3f and S6b, ESI†). Obviously, the pyrolytic samples obtained at 800 °C and 900 °C possess mesoporous structures with high surface area and large pore volume, which are favorable for ORR-related mass transport and catalytically active site exposure, and thus high ORR catalytic performance.16
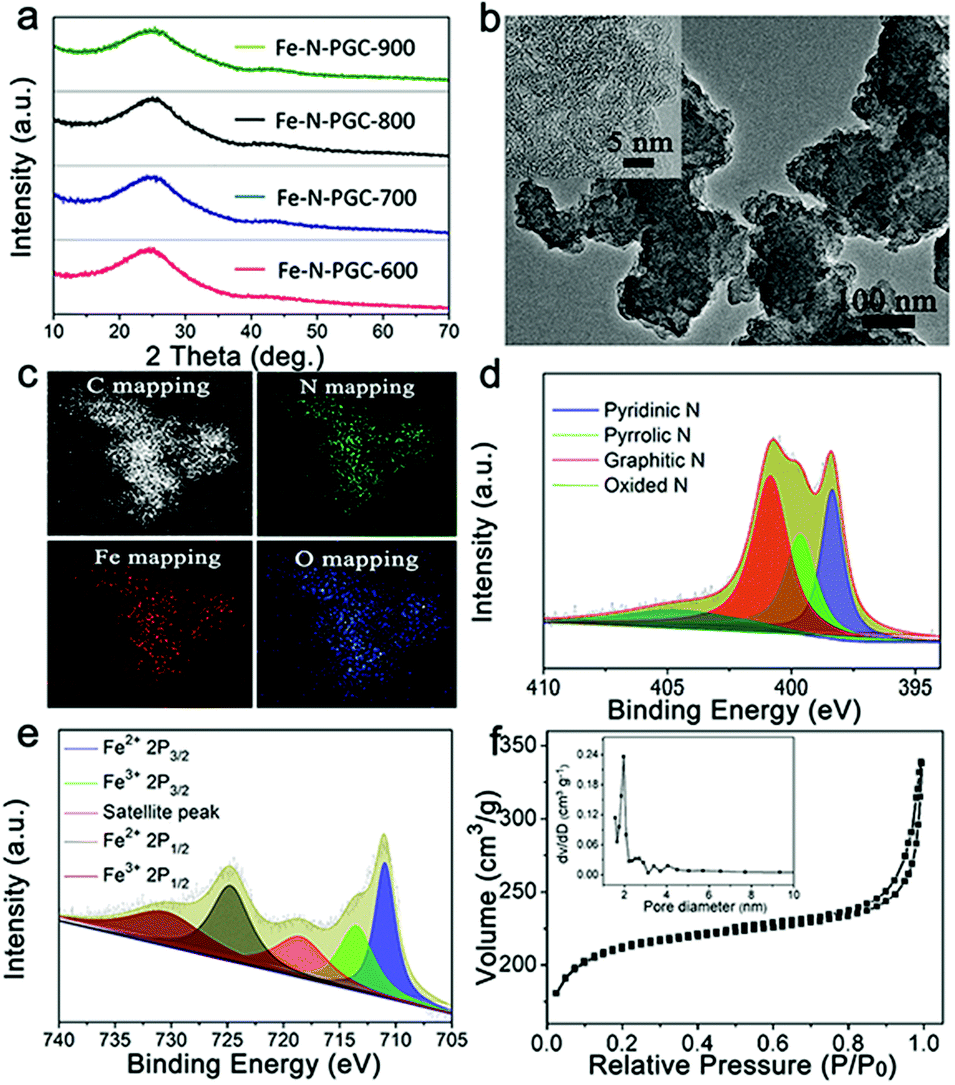 | ||
| Fig. 3 (a) XRD patterns of Fe-N-PGC-600, Fe-N-PGC-700, Fe-N-PGC-800 and Fe-N-PGC-900. (b) TEM image of Fe-N-PGC-800. (c) EDX elemental mapping images of Fe-N-PGC-800. (d) High resolution N 1s XPS spectrum of Fe-N-PGC-800. (e) High resolution Fe 2p XPS spectrum of Fe-N-PGC-800. (f) N2 adsorption–desorption isotherm of Fe-N-PGC-800 and the corresponding pore size distribution. | ||
To evaluate the ORR electrocatalytic activity, cyclic voltammetry (CV) measurements were first performed for Fe-N-PGC-800 and commercial Pt/C catalysts in O2- and N2-saturated 0.1 M KOH solution under identical experimental conditions. As shown in Fig. 4a, no O2 reduction peaks can be observed in N2-saturated 0.1 M KOH solution for both catalysts, while O2 reduction peaks at −0.18 V and −0.17 V appear for Fe-N-PGC-800 and commercial Pt/C in O2-saturated 0.1 M KOH solution, respectively. Very similar O2 reduction potentials of Fe-N-PGC-800 and Pt/C indicate that Fe-N-PGC-800 possesses superior ORR catalytic activity. To obtain a meaningful comparison, N-CNs without Fe3+ and pyrrole, N-CN/pyrrole composites without Fe3+ and Fe3+ triggered polypyrrole without N-CNs were pyrolyzed at 800 °C to obtain the corresponding carbon products, denoted as N-CNs-800, N-CNs/P-800 and Fe-P-800, respectively. Fig. 4b shows the linear sweep voltammetric (LSV) responses of N-CNs-800, N-CNs/P-800, Fe-P-800, Fe-N-PGC-800 and commercial Pt/C catalysts in an O2-saturated 0.1 M KOH solution under a rotation rate of 1600 rpm. As shown, the onset potentials are −0.17 V, −0.09 V, −0.09 V, −0.017 V and −0.018 V for N-CNs-800, N-CNs/P-800, Fe-P-800, Fe-N-PGC-800 and Pt/C, respectively, and the half-wave potential is −0.27 V, −0.19 V, −0.185 V, −0.15 V and −0.16 V for N-CNs-800, N-CNs/P-800, Fe-P-800, Fe-N-PGC-800 and Pt/C, respectively. In general, the N-CN-800 sample shows good ORR catalytic activity.32 On introducing the pyrrole or Fe, the ORR catalytic activity of the corresponding catalyst was obviously improved. The introduction of pyrrole can contribute N doping sites to the resulting carbon structures, while the introduction of the Fe element can help to form Fe,N doped active sites.16 These are favourable for improving the electrocatalytic activity of the electrocatalyst. Comparatively, the Fe-N-PGC-800 sample possessing Fe,N doped active sites and porous structure exhibits a smaller overpotential value close to that of commercial Pt/C catalysts, indicating its highly intrinsic ORR catalytic activity. More importantly, Fe-N-PGC-800 also shows higher limiting current density (5.42 mA cm−2 at −0.4 V), superior to the commercial Pt/C catalyst (5.21 mA cm−2 at −0.4 V), further manifesting the superior ORR activity of Fe-N-PGC-800. In this work, Fe-N-PGC samples at different pyrolysis temperatures were evaluated for comparative purpose. The LSV responses (Fig. S7, ESI†) demonstrate that Fe-N-PGC samples at lower and higher pyrolysis temperatures (e.g., Fe-N-PGC-600 and Fe-N-PGC-900) show decreased ORR catalytic performance. The pyrolytic sample obtained at 600 °C possesses a relatively low surface area and microporous structure (Fig. S6 and Table S2, ESI†) that may be unfavourable for the exposure of more catalytically active sites and mass transport, thus decreasing ORR activity. However, although the sample obtained at 900 °C exhibits larger surface area and mesoporous structure, a significantly decreased N doping level in graphitic carbon (Table S1, ESI†) may result in the decrease of catalytically active sites caused by Fe–N bonding (the absolute Fe content in graphitic carbon should be constant under the given experimental conditions). In this work, the Fe-N-PGC samples obtained at 700 °C and 800 °C exhibit very similar ORR catalytic performances possibly attributed to their suitable Fe,N doping level and advantageous graphitic carbon structure (such as large surface area and porous structure), favourable for creating more catalytically active sites, catalytically active site exposure and ORR-relevant mass transfer.16Fig. 4c shows the LSV curves of Fe-N-PGC-800 obtained from an O2-saturated 0.1 M KOH solution under different rotation rates. With increasing rotation rate, an increase in the cathodic current demonstrates a mass transfer controlled process.39 The linearity of the Koutecky–Levich (K–L) plots for Fe-N-PGC-800 derived from Fig. 4c indicates first-order reaction kinetics with regard to the concentration of dissolved oxygen and similar transferred electron numbers at different potentials (Fig. 4d). The average electron transfer number (n) at −0.35 V–0.60 V was calculated to be 3.98 (Fig. 4e), approximate to the theoretical value of Pt/C catalysts (n = 4.0, Fig. S6a, ESI†), indicating a near four electron ORR process. Under identical experimental conditions, the average values of transferred electron number (n) were calculated to be 3.72, 3.96 and 3.15 for Fe-N-PGC-600, Fe-N-PGC-700 and Fe-N-PGC-900, respectively (Fig. S8b–d, ESI†), further confirming the important role of a suitable pyrolysis temperature to fabricate highly catalytically active ORR catalysts. The Tafel plots shown in Fig. 4f exhibit Tafel slopes of 66.8 mV dec−1 and 69.7 mV dec−1 for Fe-N-PGC-800 and Pt/C catalysts, respectively, demonstrating a superior kinetic process of Fe-N-PGC-800 for ORR.
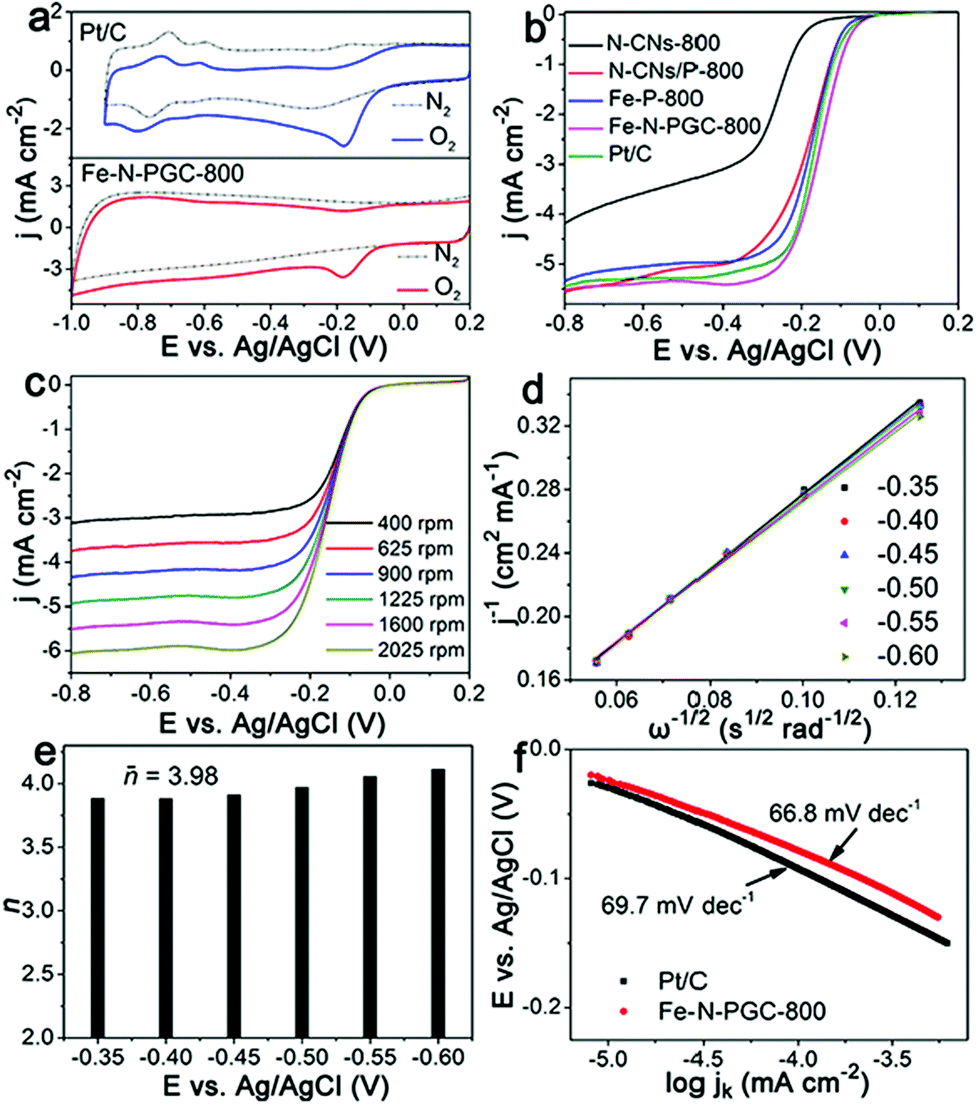 | ||
| Fig. 4 (a) Cyclic voltammetry (CV) curves of Fe-N-PGC-800 and commercial Pt/C catalysts in N2- or O2-saturated 0.1 M KOH solution; the scan rate was 50 mV s−1. (b) Linear sweep voltammogram (LSV) curves of N-CNs-800, N-CNs/P-800, Fe-P-800, Fe-N-PGC-800 and commercial Pt/C catalysts in O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1 and a rotation speed of 1600 rpm. (c) Linear sweep voltammogram (LSV) curves of Fe-N-PGC-800 obtained at different rotating rates in O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1. (d) Koutecky–Levich (K–L) plots of Fe-N-PGC-800 derived from c at different potentials. (e) Electron transfer number (n) of Fe-N-PGC-800 derived from d at different potentials. (f) Tafel plots of Fe-N-PGC-800 and commercial Pt/C catalysts. | ||
The resistance to fuel molecule interference and durability are important parameters used to evaluate an ORR catalyst for practical application. Fig. 5a shows the influence of methanol addition into O2-saturated 0.1 M KOH solution on the instant current curves of Fe-N-PGC-800 and Pt/C catalysts under identical experimental conditions. Apparently, no important influence can be found for Fe-N-PGC-800 when 3.0 M methanol was introduced, while methanol addition results in significantly decreased instant current for Pt/C catalysts, indicating the high tolerance of Fe-N-PGC-800 to the methanol crossover effect. Fig. 5b shows the durability test of Fe-N-PGC-800 and Pt/C catalysts. As shown, only 9.8% decrease in the initial activity was observed from Fe-N-PGC-800 after a 11 h run, but almost 30% decrease was achieved from Pt/C catalysts, confirming a high applicable stability of Fe-N-PGC-800. Recently, Fe-based N-doped carbon materials have demonstrated great potential for use as ORR electrocatalysts under both alkaline and acidic conditions.16,17,22 To explore the applicability, the ORR performance of Fe-N-PGC-800 was also evaluated in a 0.1 M HClO4 electrolyte under a rotation rate of 1600 rpm in this work. As shown in Fig. 5c, the LSV curves demonstrate that Fe-N-PGC-800 possesses a good ORR catalytic activity with an onset potential of 0.53 V, a half wave potential 0.29 V and a large limiting current density of 5.91 mA cm−2 at 0 V (limiting current density of 5.84 mA cm−2 for Pt/C at 0 V). Fig. 5d shows the LSV curves of Fe-N-PGC-800 with different rotation rates in an O2-saturated 0.1 M HClO4 electrolyte. Also, the cathodic currents obviously increase with rotation rates under given experimental conditions, indicating a mass transfer controlled process.39 Based on the K–L plots (Fig. 5e) derived from Fig. 5d, the average value of transferred electron number (n) was calculated to be 3.66 at 0.15 V–0.35 V, suggesting that the Fe-N-PGC-800 possesses a 4e ORR process in acidic media (Fig. 5f). The electrocatalytic activity measurements mentioned above demonstrate that Fe-N-PGC-800 possesses high ORR activity under both alkaline and acidic conditions, which can be ascribed to the formed Fe-Nx moieties in the graphitic carbon structure contributing catalytically active sites and the structural characteristics of high surface area and porous structure favourable for catalytically active site exposure and ORR-related mass transfer.16,17,27
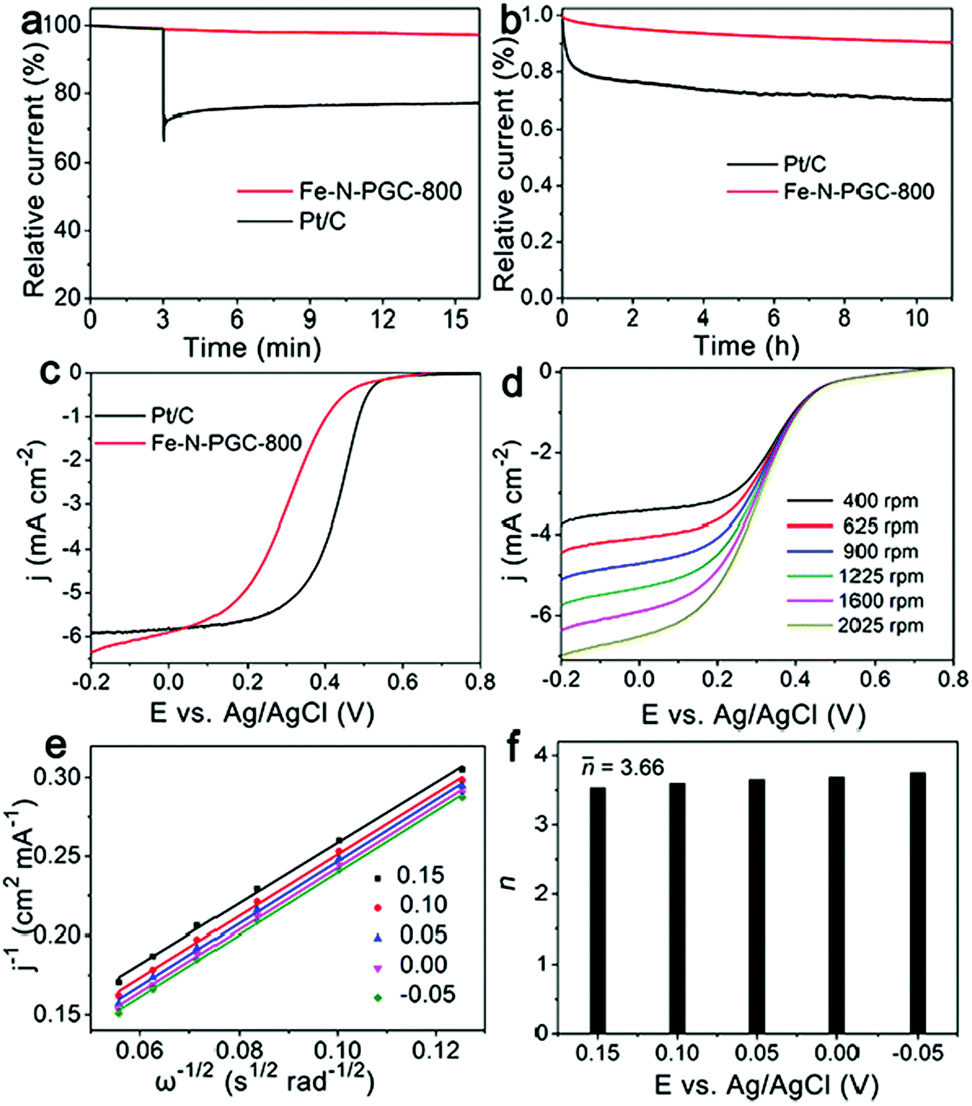 | ||
| Fig. 5 (a) Crossover effect measurements of Fe-N-PGC-800 and commercial Pt/C catalysts. (b) Durability tests of Fe-N-PGC-800 and commercial Pt/C catalysts at an applied potential of −0.35 V and a rotation rate of 1600 rpm. (c) Linear sweep voltammogram (LSV) curves of Fe-N-PGC-800 and commercial Pt/C catalysts in O2-saturated 0.1 M HClO4 solution at a scan rate of 10 mV s−1 and a rotation rate of 1600 rpm. (d) Linear sweep voltammogram (LSV) curves of Fe-N-PGC-800 obtained at different rotating rates in O2-saturated 0.1 M HClO4 solution at a scan rate of 10 mV s−1. (e) K–L plots of Fe-N-PGC-800 derived from d at different potentials, (f) electron transfer number (n) of Fe-N-PGC-800 derived from e at different potentials. | ||
It is critically important for an ORR electrocatalyst to evaluate its practical applicability in a real energy device. In this work, a zinc–air battery device assembled with a zinc foil anode and an Fe-N-PGC-800 loaded cathode was tested in a 6.0 M KOH electrolyte. As shown in Fig. 6a, the open-circuit voltage was determined to be 1.45 V for Fe-N-PGC-800, comparable to that (1.49 V) of commercial Pt/C catalysts. Also, the Fe-N-PGC-800 catalyst exhibits a high voltage of 1.32 V at the discharge current density of 10 mA cm−2, almost identical with commercial Pt/C catalysts (1.34 V), further indicating the high ORR catalytic performance of Fe-N-PGC-800 in a zinc–air battery device (Fig. 6b). In the discharge process at different current densities, Fe-N-PGC-800 shows a high discharge current density of 112 mA cm−2 at 1.0 V, slightly lower than that (120 mA cm−2) of commercial Pt/C catalysts (Fig. 6c). At a current density of 200 mA cm−2, the power densities are 140 mW cm−2 and 150 mW cm−2 for Fe-N-PGC-800 and commercial Pt/C catalysts, corresponding to the voltage values at 0.70 V and 0.75 V, respectively (Fig. 6c). Further, the specific capacities of Fe-N-PGC-800 and commercial Pt/C catalysts normalized to the mass of consumed zinc were calculated to be 578 mA h g−1 and 596 mA h g−1, respectively, at the discharge current density of 10 mA cm−2, corresponding to the gravimetric energy densities of 752 Wh kg−1 for Fe-N-PGC-800 and 784 Wh kg−1 for commercial Pt/C. The zinc–air battery performance of Fe-N-PGC-800 is comparable to most results obtained from recently reported Fe-based N-doped carbon catalysts.5,16,40,41 Therefore, the Fe-N-PGC-800 presented in this work shows great potential for use in future zinc–air battery devices.
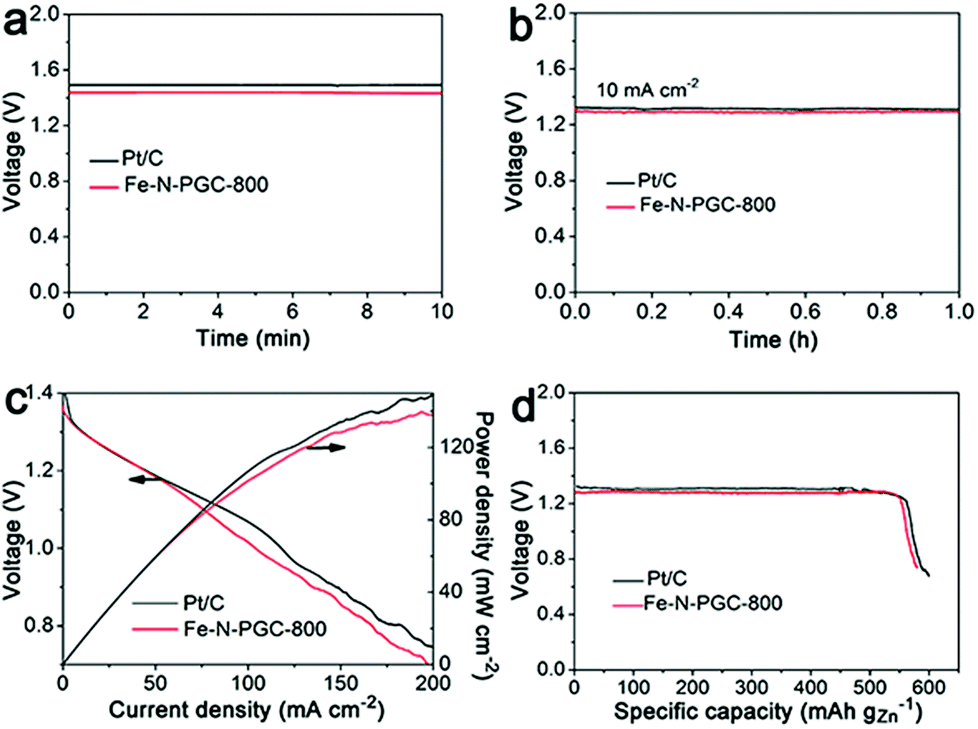 | ||
| Fig. 6 (a) Open circuit voltage measurements of zinc–air batteries with Fe-N-PGC-800 and Pt/C as the cathode catalysts. (b) Typical galvanostatic discharge curves of zinc–air batteries with Fe-N-PGC-800 and Pt/C as cathode catalysts at current densities of 10 mA cm−2. (c) A polarization curve (V–i) and the corresponding power density plot of the battery using Fe-N-PGC-800 as the cathode catalyst compared with the battery using commercial Pt/C catalysts. (d) Long-term galvanostatic discharge curves of zinc–air batteries until complete consumption of the Zn anode. The specific capacity was normalized to the mass of consumed zinc. | ||
Conclusions
In summary, Fe,N-doped porous graphitic carbon materials were successfully fabricated using shrimp-shell derived N-doped carbon nanodots as carbon and nitrogen sources by a combined approach of polymerization and pyrolysis. As an electrocatalyst, the pyrolytic product obtained at 800 °C (Fe-N-PGC-800) exhibited superior ORR catalytic activity closely comparable to the state-of-the-art Pt/C catalysts in alkaline media, and good ORR catalytic activity in acidic media. As a highly efficient oxygen reduction electrocatalyst, Fe-N-PGC-800 also demonstrated a great potential for use as an air cathode material in zinc–air battery devices with high performance and excellent cycling durability. The findings of this work demonstrated the feasibility of using cheap and abundant biomass as the starting material to fabricate high performance oxygen reduction electrocatalysts for zinc–air battery applications.Acknowledgements
This work was financially supported by the CAS Pioneer Hundred Talents Program, the Users with Potential Program (Hefei Science Center, CAS), the Natural Science Foundation of China (Grant No. 51372248 and 51432009), and the CAS/SAFEA International Partnership Program for Creative Research Teams of Chinese Academy of Sciences, China.Notes and references
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- D. S. Su and G. Sun, Angew. Chem., Int. Ed., 2011, 50, 11570–11572 CrossRef CAS PubMed.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 CAS.
- J. Wang, H. Wu, D. Gao, S. Miao, G. Wang and X. Bao, Nano Energy, 2015, 13, 387–396 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- J. T. Zhang, Z. H. Zhao, Z. H. Xia and L. M. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- D. Geng, Y. Chen, Y. Chen, Y. Li, R. Li, X. Sun, S. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760–764 CAS.
- L. M. Dai, Y. H. Xue, L. T. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- C. W. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- W. Yang, X. Liu, X. Yue, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436–1439 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- Z.-Y. Wu, P. Chen, Q.-S. Wu, L.-F. Yang, Z. Pan and Q. Wang, Nano Energy, 2014, 8, 118–125 CrossRef CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- Z. Y. Wu, X. X. Xu, B. C. Hu, H. W. Liang, Y. Lin, L. F. Chen and S. H. Yu, Angew. Chem., Int. Ed., 2015, 127, 8297–8301 CrossRef.
- L. Lin, Q. Zhu and A.-W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- H. Jiang, Y. Yao, Y. Zhu, Y. Liu, Y. Su, X. Yang and C. Li, ACS Appl. Mater. Interfaces, 2015, 7, 21511–21520 CAS.
- X. Fan, Z. Peng, R. Ye, H. Zhou and X. Guo, ACS Nano, 2015, 9, 7407–7418 CrossRef CAS PubMed.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- G. Zhong, H. Wang, H. Yu and F. Peng, J. Power Sources, 2015, 286, 495–503 CrossRef CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- S. Q. Chen, P. Bao and G. X. Wan, Nano Energy, 2013, 2, 425–434 CrossRef CAS.
- H. Peng, Z. Mo, S. Liao, H. Liang, L. Yang, F. Luo, H. Song, Y. Zhong and B. Zhang, Sci. Rep., 2013, 3, 1765–1772 Search PubMed.
- T. X. Wu, H. M. Zhang, X. Zhang, Y. X. Zhang, H. J. Zhao and G. Z. Wang, Phys. Chem. Chem. Phys., 2015, 17, 27527–27533 RSC.
- Q. Wang, L. Jiao, H. Du, Y. Si, Y. Wang and H. Yuan, J. Mater. Chem., 2012, 22, 21387–21391 RSC.
- Y. B. Li, H. M. Zhang, Y. Wang, P. R. Liu, H. G. Yang, X. D. Yao, D. Wang, Z. Y. Tang and H. J. Zhao, Energy Environ. Sci., 2014, 7, 3720–3726 CAS.
- H. M. Zhang, J. Y. Chen, Y. B. Li, P. R. Liu, Y. Wang, T. C. An and H. J. Zhao, Electrochim. Acta, 2015, 165, 7–13 CrossRef CAS.
- H. Zhang, Y. Wang, D. Wang, Y. Li, X. Liu, P. Liu, H. Yang, T. An, Z. Tang and H. Zhao, Small, 2014, 10, 3371–3378 CrossRef CAS PubMed.
- C. Zhu, J. Zhai and S. Dong, Chem. Commun., 2012, 48, 9367–9369 RSC.
- R. Liu, H. Zhang, S. Liu, X. Zhang, X. Ge, Y. Zang, H. Zhao and G. Wang, Phys. Chem. Chem. Phys., 2016, 18, 4095–4101 RSC.
- J. Zhou, C. Booker, R. Li, X. Zhou, T.-K. Sham, X. Sun and Z. Ding, J. Am. Chem. Soc., 2007, 129, 744–745 CrossRef CAS PubMed.
- S. Liu, J. Tian, L. Wang, Y. Zhang, X. Qin, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Adv. Mater., 2012, 24, 2037–2041 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2012, 134, 15–18 CrossRef CAS PubMed.
- Y. G. Li, W. Zhou, H. L. Wang, L. M. Xie, Y. Y. Liang, F. Wei, J. C. Idrobo, S. J. Pennycook and H. J. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- H.-W. Liang, W. Wei, Z.-S. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- J. S. Lee, G. S. Park, S. T. Kim, M. Liu and J. Cho, Angew. Chem., 2013, 125, 1060–1064 CrossRef.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. G. Kim, Q. Li, N. Park, M. Liu and J. Cho, Nat. Commun., 2013, 4, 2076–2083 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed and additional figures. See DOI: 10.1039/c6qi00059b |
| This journal is © the Partner Organisations 2016 |