
On the fly multi-modal observation of ligand synthesis and complexation of Cu complexes in flow with ‘benchtop’ NMR and mass spectrometry†
Luzian
Porwol
,
Alon
Henson
,
Philip J.
Kitson
,
De-Liang
Long
and
Leroy
Cronin
*
WestCHEM, School of Chemistry, University of Glasgow, University Avenue, Glasgow, G12 8QQ, UK. E-mail: Lee.Cronin@glasgow.ac.uk; Web: http://www.croninlab.com
First published on 11th May 2016
Abstract
Exploring complex chemical systems requires reproducible and controllable ways to access non-equilibrium conditions. Herein we present a programmable flow system that can do both ligand synthesis and complexation on the fly, and the conditions of the reaction can be monitored using two simultaneous techniques, namely NMR and mass spectrometry. By using this approach we monitored the formation of unknown complexes, followed by crystallization that resulted in the characterisation of their structures giving 5 new compounds (4 isolated and fully characterised) which can be formulated as: Cu2(L1)4(μ-CO3)](BF4)2 (2); [Cu3(L1)6(μ-CO3)](PF6)2(OH)2 (3) [Cu2(L2)2](BF4)2 (4) and [Cu(L2)2](BF4)2·CH3CN (5).
Introduction
Complex molecular architectures can often form through multi-component coordination chemistry,1 but often this chemistry is restricted to one-pot reactions.2 Under most conditions the mechanism of the underlying process of self-assembly is well known being based on the rules3 of acceptor–donor interactions and are therefore understood and often predictable.4,5 However the principle of pre-organisation and self-sorting, although well established to control the outcome of complex molecular architectures,6 do not apply under non-equilibrium conditions.7 This means that there is a pressing need to combine the design of coordinating ligands, via coordination programming,8 with non-equilibrium dynamics9 to explore the reproducible formation of more complex and structurally diverse architectures. This has not been possible since the approach requires real-time observation of the reaction process. Success could allow the ‘digitalisation’ of chemical discovery and synthesis. Indeed, this digitalisation is long overdue with respect to the development of methods that allow synthesis to be followed more closely, gaining a deeper insight in experiments to be performed more reliably without extensive investment of time and money for so called ‘routine’ literature procedures.Herein we realise this idea by automating and monitoring the self-assembly of coordination complexes under non-equilibrium conditions.10 A purpose built flow system allows for automation and live control of multi-step reactions (see Fig. 1) under semi-inert atmospheric reaction conditions. This platform not only permits us to follow the reaction progress in real-time, but also to optimise, as well as reliably reproduce, the self-assembly process via in-line analysis using a benchtop NMR spectrometer and a portable MS. To explore this concept, we initially chose to study a simple ligand-complex model system as a proof of concept that we can detect, follow, understand and processes for new and complex coordination architectures. This study lead to the identification of an intermediate species [Cu(I)(L1)2] (1) – which is not possible to isolate or observe using classical synthetic procedures – important for the self-assembly of two new complexes isolated after crystallization: [Cu2(L1)4(μ-CO3)](BF4)2 (2); [Cu3(L1)6(μ-CO3)](PF6)2(OH)2 (3). It was also possible to identify a binuclear Cu-complex [Cu2(L2)2](BF4)2 (4) and [Cu(L2)2](BF4)2·CH3CN (5) in situ.
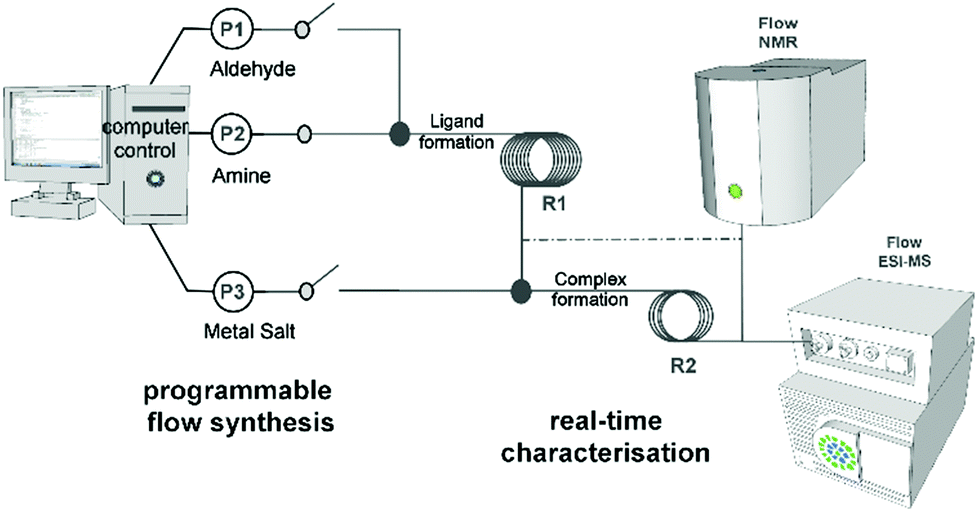 | ||
| Fig. 1 Schematic representation of the digital-programmable-flow-platform to perform ligand-complex synthesis in an automated way. The syringe pumps are computer controlled to deliver the chosen reagents in a well synchronized way into the flow reactors. New compounds are formed in situ and detected by real-time analyses. | ||
The structures of four compounds (2–5) discovered in this study were confirmed by single crystal X-ray diffraction, however we also studied the synthesis of two different ligands and their coordination behaviour with Cu(I)-precursors under flow.
Results and discussion
To develop our system, we opted to investigate the synthesis of a pyridine-moiety with bidentate and tridentate ligands, followed by a complexation reaction employing a metal precursor in the second step. This model system consists of two different ligands – a bidentate (L1 = N-(2-pyridinylmethylene)-benzenemethanamine) and a tridentate (L2 = 2,6-bis(N-benzyliminomethyl)pyridine) ligand – and of two different Cu salts – Cu(CH3CN)4BF4 and Cu(CH3CN)4PF6 (Scheme 1). The two ligands are prepared in situ by reacting benzylamine with 2-pyridinecarboxaldehyde and 2,6-pyridinedicarboxaldehyde to obtain L1 and L2 respectively. Both ligands are characterised by strong N-donor properties, which allow us to predict the formation of complex structures according to the metal-cation precursor chosen.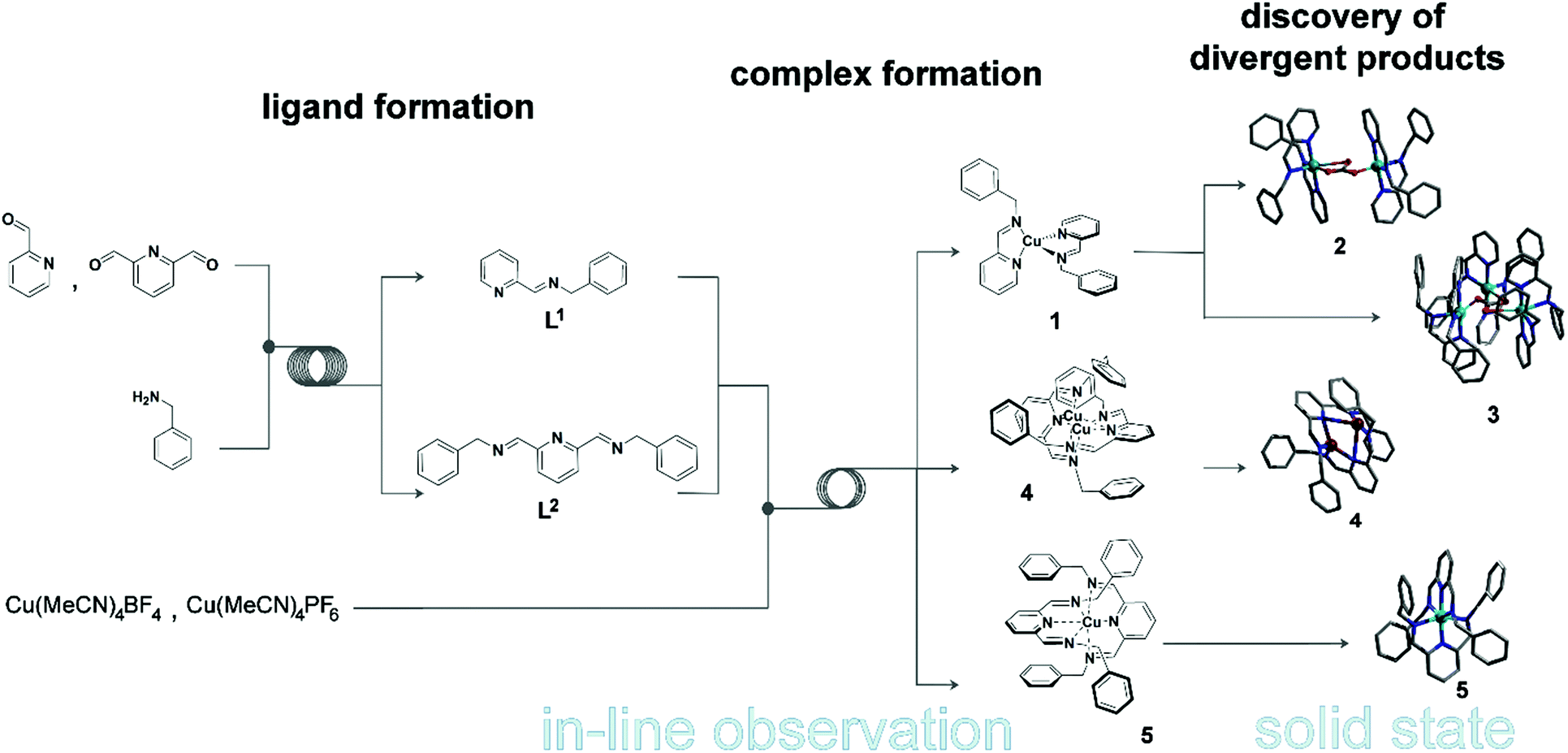 | ||
| Scheme 1 Flow synthesis of N-(2-pyridinylmethylene)-benzenemethanamine (L1), 2,6-bis(N-benzyliminomethyl)pyridine (L2) and their complexation with metal precursors Cu(CH3CN)4BF4 and Cu(CH3CN)4PF6. See ESI† for full details. | ||
The platform was designed to follow the formation of both ligands and complexes by in-line NMR and MS spectrometers. Therefore, the platform consists of six 5 mL syringe pumps, two reactors (VR1 with volume of 2.1 mL and VR2 with volume of 0.7 mL) and the two in-line analytics. A custom designed LabVIEW™ interface allows for the automation and synchronization of multi-step reactions, data acquisition and simultaneous screening of reaction conditions.11,12 This platform allows us to operate in continuous and under semi-inert atmosphere conditions, which is of great advantage working with Cu salts/complexes. Copper(I) chemistry13 in particular, is well known for producing interesting molecular structures and the flow platform reported here permits easy manipulation of these reagents in a way that is more difficult under batch conditions.14
The first screening consisted in the flow synthesis of L1 and its complexation with Cu(CH3CN)4BF4 or Cu(CH3CN)4PF6 is illustrated in Scheme 1. The synthesis of L1 was obtained in 10 minutes, by mixing into VR1 2-pyridinecarboxaldehyde with benzylamine (both as 0.4 M solutions in acetonitrile (MeCN)), having a molar aldehyde![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amine ratio of 1:1. The formation of L1 was screened in real-time by integrating the flow set-up described with a benchtop NMR spectrometer, allowing us to analyse reaction mixtures in non-deuterated solvents. This synthesis was investigated using different flow rates and residence times (see Fig. 2), showing a lower conversion to the imine with increasing flow-rate and decreasing reaction time. The disappearance of the signal corresponding to the aldehyde moiety at 10.00 ppm, and the appearance of the imine proton (as a broad band signal overlapping an aromatic proton signal) at 8.76–8.39 ppm, in addition to the low field shift from 3.86 ppm to 4.82 ppm of the signal relating to the benzylic protons are evidences for the formation of the expected imine (L1). The synthesis of L1 was further confirmed by substituting the flow NMR with a flow ESI-MS which identified the presence of the imine as a signal having a m/z = 197.2 (Fig. S3 in ESI†) corresponding to the protonated ligand (L1 + H)+.
:amine ratio of 1:1. The formation of L1 was screened in real-time by integrating the flow set-up described with a benchtop NMR spectrometer, allowing us to analyse reaction mixtures in non-deuterated solvents. This synthesis was investigated using different flow rates and residence times (see Fig. 2), showing a lower conversion to the imine with increasing flow-rate and decreasing reaction time. The disappearance of the signal corresponding to the aldehyde moiety at 10.00 ppm, and the appearance of the imine proton (as a broad band signal overlapping an aromatic proton signal) at 8.76–8.39 ppm, in addition to the low field shift from 3.86 ppm to 4.82 ppm of the signal relating to the benzylic protons are evidences for the formation of the expected imine (L1). The synthesis of L1 was further confirmed by substituting the flow NMR with a flow ESI-MS which identified the presence of the imine as a signal having a m/z = 197.2 (Fig. S3 in ESI†) corresponding to the protonated ligand (L1 + H)+.
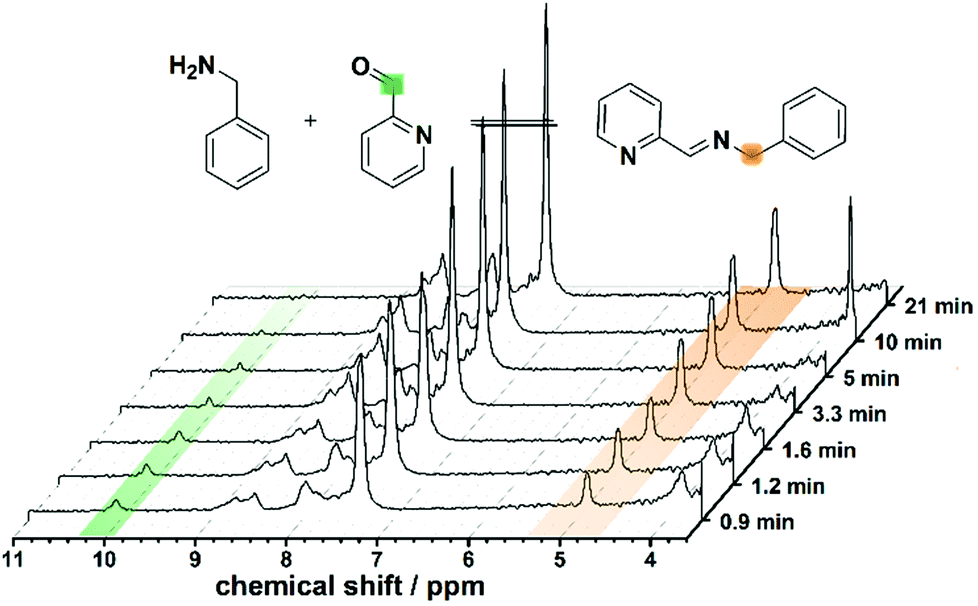 | ||
| Fig. 2 Schematic of the real-time observation in the flow synthesis of L1 at different residence times showing a comparison of the normalised 1H-NMR spectra collected during the in-line benchtop NMR study. | ||
Complex 1 was synthesised by mixing Cu(CH3CN)4BF4 or Cu(CH3CN)4PF6 (as 0.1 M solutions in MeCN) with L1, having a molar ligand:metal ratio equal to 2:1. Also in this case, the formation of 1 was confirmed by in-line 1H-NMR and MS, giving the same results for both metal precursors. Notably, complex 1 was only ever identified using in-line methods whilst under flow conditions and was not crystallised as an isolated product. This synthesis was thus repeated at different flow rates to see how this parameter affects the assembly of compound 1. Complexation occurred at all flow rates and we were able to observe the template effect of the metal-precursor for the ligand-synthesis and the complex itself. From the in-line 1H-NMR measurements, the formation of the complex 1 could be observed in the shift of the proton signals corresponding to the imine. The pyridine protons signal shifted from 8.76–8.39 and 8.18–7.19 ppm to 8.21–6.93 ppm; for the imine proton, new signals appeared at 8.78–8.52 ppm and shifted from 8.76–8.39 ppm and the benzylic protons shifted from 4.82 to 4.81–4.56 ppm, which is higher field compared to the imine (Fig. S2 in ESI†). Similarly, complex 1 could also be detected by in-line MS (m/z = 455.4) (Fig. S3 in ESI†).
As such, this work has allowed us to probe the formation of a transient monomer complex (1), which serves as a common ‘building block’ precursor in the formation of two new multinuclear complexes. Crystallisation of the reaction mixtures containing 1 led to two different compounds according to the Cu precursor used, which result from the assembly of this monomeric sub-unit into different multinuclear complexes. Thus, the brownish-red solution corresponding to 1 obtained using Cu(CH3CN)4BF4 crystallised to yield the dimeric structure 2 after 14 days at 7 °C whilst employing Cu(CH3CN)4PF4 gives the trimeric product 3 after 4 days at 18 °C (see Fig. 3). Here, both complexes are found to contain Cu(II) as indicated by their green-blue colour (opposed to the red-brown colour of the non-oxidised reaction mixture) and their characterisation by X-ray diffraction, which clearly indicates that the tetrahedral Cu(I) centre in the precursor (1) has oxidised in air to Cu(II). Notably, the in-line analyses, in particular NMR, only showed the formation of the sub-unit complex (1) under flow conditions. The sub-unit 1 thus assembles into dimeric (2) and a trimeric (3) structures through inclusion of a bridging carbonate moiety.15 Not only the spectroscopic (IR for complex 2 and Raman for complex 3), magnetic and analytical data is consistent with the assignment of carbonate (see ESI†), but also control experiments show that these structures can be easily reproduced by adding dry ice to the crystallisation solution.
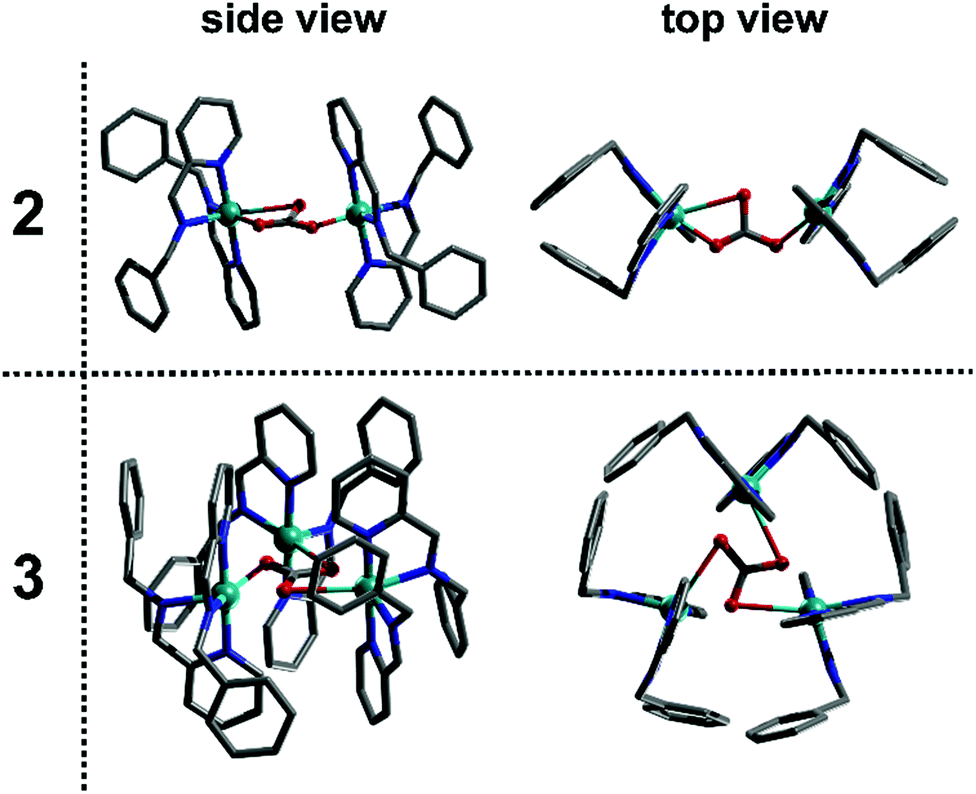 | ||
| Fig. 3 Molecular structures of dimer complex 2 and trimer complex 3. A distorted carbonate ion templates the assembly of two or three monomer complexes into dimeric and trimeric structures, respectively. | ||
It should be noted that adoption of the flow-platform reported here confers a significant advantage in both the ease with which 2 and 3 can be synthesised from the transient monomer 1 (i.e. no requirement for inert atmosphere) and the reproducibility of this approach, in which the major synthetic variables are closely controlled by automation. The ‘batch’ crystallisation process could be easily reproduced in terms of both yield and purity, as indicated by PXRD (Fig. 4).
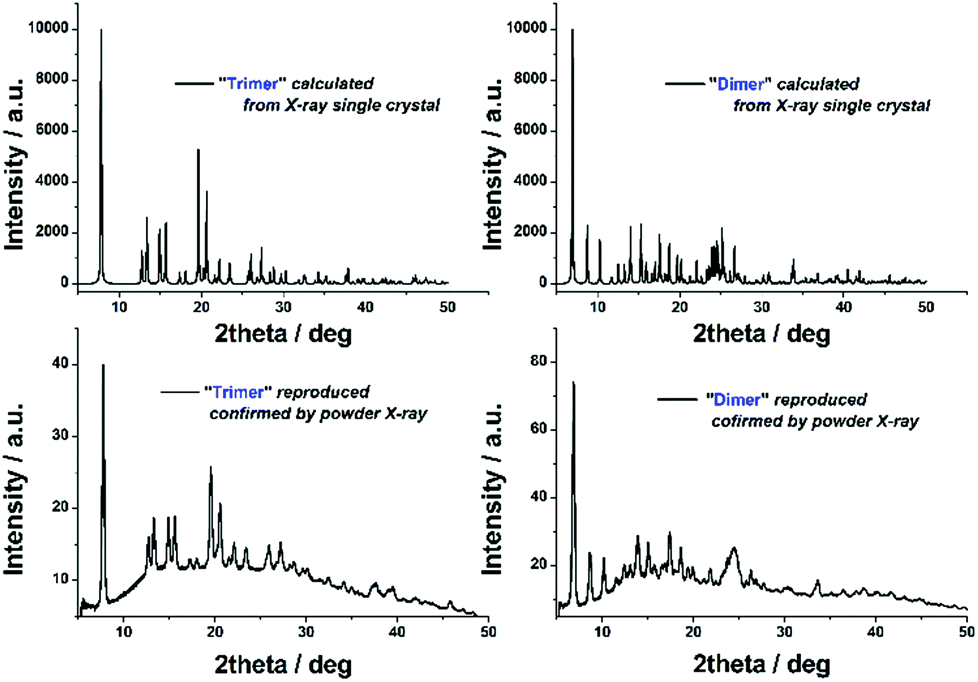 | ||
| Fig. 4 PXRD: top left: simulated pattern from single crystal [Cu2(L1)4(μ-CO3)](BF4)2 (2), top right, simulated pattern from single crystal [Cu3(L1)6(μ-CO3)](PF6)2(OH)2 (3), bottom left PXRD measurement of reproduced material 2, bottom right, PXRD measurement of reproduced material 3. | ||
The second screening consisted on the flow synthesis of L2 and its complexation with Cu(CH3CN)4BF4 (Scheme 1). The formation of L2 was obtained in 4.5 minutes using both reactors (total reactor volume of 2.8 ml as the results of VR1 + VR2). For the formation of this tridentate ligand, 2-pyridinecarboxaldehyde was substituted with 2,6-pyridinedicarboxaldehyde (as 0.5 M solutions in MeCN) and subsequently reacted with benzylamine, having a molar aldehyde:amine ratio of 2:1. This reaction step was monitored both by in-line NMR spectroscopy and ESI-MS that confirmed the formation of the imine. In the 1H-NMR two new signals appeared at 8.50 and 4.86 ppm and the signal corresponding to the aldehyde moiety disappeared at 10.08 ppm, which is indicative of imine formation (Fig. S11a in ESI†). Similarly, in-line MS showed the formation of L2 corresponding to the appearance of a signal with m/z = 314.4 (L2 + H)+ (Fig. S12a in ESI†).
The complexation of L2 with Cu(CH3CN)4BF4 (as 0.1 M solutions in MeCN) was obtained using a molar ligand:salt ratio equal to 2:1. As in the case of compound 1, this complexation yields a deep red-brown coloured solution, indicative of the formation of Cu(I) complexes. The complete synthesis could be monitored by in-line NMR and ESI-MS. The disappearance of the peak at 10.08 ppm corresponds to the aldehyde protons of 2,6-pyridinedicarboxaldehyde (see Fig. 5). Fig. 5A–D shows full conversion of the aldehyde into the di-imino ligand with the appearance of the imine signal at 8.50 ppm. In addition, a shift of the bands corresponding to the aromatic ring of the pyridine (from 8.17 to 7.98 ppm, in spectra A and C respectively) and the CH2 band from the benzylamine (from 3.86 to 4.86 ppm, in spectra B and C respectively) confirm the formation of L2.
 | ||
| Fig. 5 Real-time monitoring of the multi-step synthesis of a coordination compound. NMR spectra corresponding to: 2,6-pyridinedicarboxaldehyde (A), benzylamine (B), diimine ligand (C) and Cu(I) complex (D). All spectra are the averaged results of 40 scans. Solvent indicated by dotted line. | ||
The addition of Cu(CH3CN)4BF4 had a significant effect in shifting the bands corresponding to the imine signal, the aromatic rings (from 8.50, 7.98 and 7.34 ppm to 8.30–7.86 and 7.41–6.88 ppm) and the CH2 bands (from 4.86 ppm to 4.57 ppm), which is clearly indicative of a change in the environment of the ligand due to complexation (confirmed by controls, see ESI†). Also, the in-line MS of the reaction solution showed three different signals; two with m/z = 689.4 and 344.8, and a main peak having m/z = 376.2 corresponding to the Cu(I) and Cu(II) analogue of compound 5 and directly to compound 4, respectively. Following this, from the reddish brown solution two species could be crystallised, which were subsequently analysed crystallographically (Fig. 6). The two copper complexes identified as [Cu(L2)2]+ (5) and also [Cu2(L2)2]2+ (4) via the flow-MS measurements crystallise after several days upon which 5 oxidises to the analogous copper(II) complex (Fig. 6, left) whereas 4, remarkably, remains in the same oxidation state as detected in solution (Fig. 6, middle).
 | ||
| Fig. 6 Molecular structure of the Cu(II)-complex 5 (left) and the corresponding MS envelope for its Cu(I) species in solution (right). Molecular structure and MS envelope of the dimetallic Cu(I) complex 4 (middle). | ||
It is therefore important to note that under flow conditions, the main observed complexes are always Cu(I), due to the red-brownish colour observed and the representative signals in the in-line NMR. The subsequent oxidation occurs during the crystallisation process to form, in the case of 5, a chiral complex (space group P212121). Interestingly, the binuclear Cu(I) complex (4) shows a remarkably short copper–copper distance (2.5864(5) Å),16 which is unusual, even in the well-studied area of Cu(II) Schiff-base complexes.17
Conclusions
In summary a new strategy for setting up non-equilibrium reaction conditions for both the synthesis of ligands, and their complexation, was showed in a computer controlled flow system. The use of in-line 1H-NMR and ESI-MS analysis enables the direct observation of reactions in real-time and allowed us to discover five new coordination complexes. It is important to note that no additional steps are taken to perform these reactions under anaerobic conditions and so the flow reactor can be considered to provide an easily controllable pseudo-inert atmosphere for the manipulation of air sensitive reagents. We believe that platforms such as this will be increasingly important in the emerging field of systems chemistry,15 not to mention the digitalisation of chemistry for chemical discovery, synthesis, and reliable repetition.19 The platform allows the reaction steps to be handled in an automated way and it is possible to follow the reaction as it proceeds, permitting the discovery of compounds prior to their isolation. The significant advantage of this modular flow set-up is therefore the ability to control self-assembly processes in situ by altering the reaction parameters in real-time (e.g. flow rate, temperatures, and reagents)18 and observe new compounds in-line which are critical to isolate in solid state.Acknowledgements
This work was supported by the EPSRC, University of Glasgow, and WestCHEM. This work was supported by the EPSRC grants (No. EP/J015156/1; EP/L023652/1; EP/I033459/1; EP/K023004/1), EC grant 318671 MICREAGENTS, LC thanks the Royal Society/Wolfson Foundation for a Merit Award and the ERC for an Advanced Grant (ERC-ADG, 670467 SMART-POM).Notes and references
- A. J. Metherell and M. D. Ward, Chem. Commun., 2014, 50, 6330 RSC
.
- G. N. Newton, K. Mitsumoto, R. J. Wei, F. Iijima, T. Shiga, H. Nishikawa and H. Oshio, Angew. Chem., Int. Ed., 2014, 53, 2941 CrossRef CAS PubMed
- H. Werner, Angew. Chem., Int. Ed., 2013, 52, 6146 CrossRef CAS PubMed
- R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794 CrossRef CAS PubMed
- A. R. de la Oliva, V. Sans, H. N. Miras, J. Yan, H. Zang, C. J. Richmond, D.-L. Long and L. Cronin, Angew. Chem., Int. Ed., 2012, 51, 12759 CrossRef CAS PubMed
- M. Lal Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651 CAS
- J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222 CrossRef CAS PubMed
- H. Nishihara and H. Oshio, Dalton Trans., 2013, 42, 15825 RSC
- H. N. Miras, G. J. T. Cooper, D.-L. Long, H. Bögge, A. Müller, C. Streb and L. Cronin, Science, 2010, 327, 72 CrossRef CAS PubMed
- R. Makki and O. Steinbock, J. Am. Chem. Soc., 2012, 134, 15519 CrossRef CAS PubMed
- J. S. Mathieson, M. H. Rosnes, V. Sans, P. J. Kitson and L. Cronin, Beilstein J. Nanotechnol., 2013, 4, 285 CrossRef PubMed
- J. S. Mathieson, J. T. Cooper, A. L. Pickering, M. Keller, D.-L. Long, G. N. Newton and L. Cronin, Chem. – Asian J., 2009, 4, 681 CrossRef CAS PubMed
- S. Roy, B. Sarkar, D. Bubrin, M. Niemeyer, S. Záliš, G. K. Lahiri and W. Kaim, J. Am. Chem. Soc., 2008, 130, 15230 CrossRef CAS PubMed
- A. A. Salaudeen, C. A. Kilner and M. A. Halcrow, Chem. Commun., 2008, 41, 5200 RSC
- R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101 RSC
- C. Harding, J. Nelson, M. C. R. Symons and J. Wyatt, J. Chem. Soc., Chem. Commun., 1994, 21, 2499 RSC
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003 RSC
- S. Newton, C. F. Carter, C. M. Pearson, L. D. Alves, H. Lange, P. Thansandote and S. V. Ley, Angew. Chem., Int. Ed., 2014, 53, 4915 CrossRef CAS PubMed
- V. Sans, L. Porwol, V. Dragone and L. Cronin, Chem. Sci., 2014, 2, 1258 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Full details of the platform set up, software, flow synthesis, and full analytical data. CCDC 1011860, 1011861, 1011858 and 1011863. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00079g |
| This journal is © the Partner Organisations 2016 |