
Synthesis of multi-shelled MnO2 hollow microspheres via an anion-adsorption process of hydrothermal intensification†
Mengjie
Chen‡
ac,
Jiangyan
Wang‡
ac,
Hongjie
Tang
a,
Yu
Yang
ac,
Bao
Wang
a,
Huijun
Zhao
b and
Dan
Wang
*ab
aNational Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, No. 1 Beiertiao, Zhongguancun, Beijing 100190, P. R. China. E-mail: danwang@ipe.ac.cn
bCentre for Clean Environment and Energy, Gold Coast Campus Griffith University, Queensland 4222, Australia
cUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, P. R. China
First published on 31st May 2016
Abstract
In this study, multi-shelled manganese oxide hollow microspheres with controlled valence were successfully synthesized by varying the Mn-precursor and using an anion-adsorption process for hydrothermal intensification. Used as the supercapacitor electrode material, the multi-shelled MnO2 hollow microspheres achieved superior specific capacitance (1457 F g−1 at the discharge current density of 0.5 A g−1) and excellent cycling stability (91.2% retention of the initial capacitance after 4000 cycles), benefiting from the superiorities of these unique hierarchical structures such as increased active sites, shortened ion and electron transport lengths, better contact between the electrolyte and active materials, as well as better protection of interior shells by the exterior shell.
Introduction
Nowadays, with many desired characteristics, such as high power density, long cycle life and high charging/discharging rates, supercapacitors, have been widely used in electrical equipment and hybrid electric vehicles.1 Pseudocapacitors store the energy through a fast or a reversible faradaic reaction (redox reaction), resulting in higher specific capacitance and larger energy density compared with double-layer supercapacitors.2–5 Various materials can undertake redox reactions such as RuO2,6 MnO2,7 NiO8 and Co3O4.9 Among them, MnO2 turns out to be one of the most promising pseudocapacitor electrode materials with advantages of high theoretical capacity (1370 F g−1), wide potential window, low cost, low toxicity and environmental friendliness.10–13 Unfortunately, the practical application of MnO2 pseudocapacitor electrode materials still suffer from some drawbacks14 as follows: (1) low specific surface area, thus few faradic active sites, resulting in low practical accessible capacitance; (2) poor electron and ion conductivities, thus limited rate capability, leading to a low power density; and (3) partial dissolution in electrolytes, thus undesirable cycling stability.An effective solution to conquer the abovementioned disadvantages is replacing bulk solid structures with hollow micro-/nano-structures given their structural and performance superiorities:15–20 (1) the hollow structure possesses a higher specific surface area and higher energy density; (2) unique hollow structure with porous shells enables better accessibility of electrolyte to electrode surface and provides shortened ions and electrons transport length, thus it improves rate capability and power density; (3) an accurately designed multi-shelled hierarchical structure can significantly support and protect the structure during the charge/discharge processes, resulting in better structural and cycling stabilities.
According to our previous study, a universal method, the sequential template method based on the electrostatic attraction between negatively-charged carbonaceous microsphere (CMS) templates and metal cations, was developed to synthesize hollow micro/nanostructured metal oxides.21 As a result, many metal oxides, such as Co3O4,22 α-Fe2O3,23 SnO2,24 ZnO,25 TiO2,26 LiMn2O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 multi-shelled hollow microspheres, were successfully achieved through this method. Unfortunately, the approach failed when extended to synthesize multi-shelled MnO2 hollow microspheres (MS-MnO2-HMSs) because cationic Mn ions (mainly Mn2+) are low-valent, whereas high-valent Mn usually exists as oxygen-containing anions (such as MnO42− and MnO4−). Although Mn cations can be largely adsorbed by negatively-charged CMS templates, only MS-Mn2O3-HMS can be produced due to insufficient driving force for low-valent Mn(II) cations to step over the energy barrier to a much higher valence state.
27 multi-shelled hollow microspheres, were successfully achieved through this method. Unfortunately, the approach failed when extended to synthesize multi-shelled MnO2 hollow microspheres (MS-MnO2-HMSs) because cationic Mn ions (mainly Mn2+) are low-valent, whereas high-valent Mn usually exists as oxygen-containing anions (such as MnO42− and MnO4−). Although Mn cations can be largely adsorbed by negatively-charged CMS templates, only MS-Mn2O3-HMS can be produced due to insufficient driving force for low-valent Mn(II) cations to step over the energy barrier to a much higher valence state.
Herein, an anion-adsorption (KMnO4 was used as the Mn-precursor) process by negatively-charged CMS templates, as opposed to the conventional cation-adsorption, is proposed and validated. In addition, considering the large radii of MnO4− ions, hydrothermal adsorption instead of simple adsorption at ambient condition was adopted to enhance the Mn adsorption amount and depth into the CMS templates.28 Followed with an annealing process to remove the CMS templates, multi-shelled MnO2 hollow microspheres with controlled valence, shell number and shell roughness were achieved. Benefitting from the superiorities of multi-shelled hollow structures, the as-prepared quadruple-shelled MnO2 hollow microspheres exhibited an impressive specific capacitance as high as 1457 F g−1 at a 0.5 A g−1 current density, and remarkable cycling stability with 91.2% capacitance retention even after 4000 consecutive cycles. Moreover, the rate capability was also impressive, showing a specific capacitance as high as 1037 F g−1 at a high 10 A g−1 current density. To the best of our knowledge, the performance of MS-MnO2-HMSs reported here are superior to all previously reported MnO2 materials with various nanostructures as supercapacitor electrodes.
Results and discussion
Previously, given the negatively-charged nature of CMS templates, only cation-adsorption was adopted. However, only MS-Mn2O3-HMS can be produced through cation-adsorption process due to the insufficient driving force for low-valent Mn(II) cations to step over the energy barrier to a much higher valence state, as shown in Route I in Fig. 1.29 Through Fourier transform infrared (FTIR) (Fig. S1†) characterization of CMSs, we found that CMSs are rich with –OH, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –C–O–C groups, which means that to some extent CMS templates could be considered as large colloidal particles that tend to preferentially adsorb ions with similar elemental compositions. Therefore, OH− or other oxygen-containing groups (such as MnO42− and MnO4−) were preferentially adsorbed by CMS templates, endowing CMS templates with negative charge and then followed by cation adsorption through electrostatic interactions. In other words, anionic MnO42− and MnO4− can be adsorbed by CMS templates, making it possible to achieve high-valent MnO2 quadruple-shelled hollow microspheres.
O and –C–O–C groups, which means that to some extent CMS templates could be considered as large colloidal particles that tend to preferentially adsorb ions with similar elemental compositions. Therefore, OH− or other oxygen-containing groups (such as MnO42− and MnO4−) were preferentially adsorbed by CMS templates, endowing CMS templates with negative charge and then followed by cation adsorption through electrostatic interactions. In other words, anionic MnO42− and MnO4− can be adsorbed by CMS templates, making it possible to achieve high-valent MnO2 quadruple-shelled hollow microspheres.
 | ||
| Fig. 1 Comparing synthesis mechanisms for the conventional solution adsorption method under ambient conditions (Routes I, II and III) and the hydrothermal intensification method (Route IV) with various Mn-precursors. | ||
When chosen as a Mn-precursor in the solution adsorption method under ambient conditions, as shown in Route II in Fig. 1, K2MnO4 had a higher valence state of Mn than cationic Mn2+. The TEM image in Fig. S2a† shows that only single-shelled hollow microspheres can be synthesized with the big radii of MnO42− ions. Furthermore, the XRD pattern in Fig. S2b† shows that the sample was mixed-phase with both α-MnO2 and Mn2O3 phases. Herein, KMnO4 was considered as the Mn-precursor with even higher valence state of Mn via the same anion-adsorption method.
Due to the large radii of MnO4− ions, it is hard for MnO4− to penetrate into the deep inside of CMS templates through conventional ion-adsorption under ambient conditions, as shown in Route III, thus only thick single-shelled hollow microspheres (Fig. S3†) could be obtained. Herein, a hydrothermal intensification method with higher temperature and pressure was adopted to enhance the Mn-adsorption amount and depth into CMS templates, resulting in multi-shelled MnO2 hollow microspheres (Fig. 1 (Route IV)).
XPS characterization was used to analyze the sample acquired after hydrothermal intensification adsorption (Fig. S4†). Each of the two main characteristic broad peaks could be deconvoluted into three components that represent different Mn valence states, i.e., Mn(IV) (642.2 eV and 653.8 eV), Mn(III) (641.8 eV and 653.3 eV) and Mn(VII) (645.2 eV and 657.7 eV).30 The distance between Mn 2p1/2 peak and its satellite (Δ2p1/2) of Mn(IV) was 11.8 eV and Mn(III) was 10.0 eV, which all agreed with a previous report.31 It was reported that KMnO4 has a strong oxidability to oxidize groups such as –OH and –CO of the CMS templates.32 Therefore, in the process of hydrothermal adsorption, CMS templates with oxygen groups, which work as a hard template and sacrificial reductant reduce MnO4− to Mn(IV) and other manganese compounds, all of which will finally deposit on the CMSs. Those MnOx compounds will be immobilized inside CMS templates effectively, which can be further oxidized to MnO2 by MnO4− during the annealing process.
After the annealing process, XPS characterization was also used to confirm the exact valence states of Mn in Fig. S6† using a quadruple-shelled MnO2 hollow microspheres as an example. The peaks of Mn 2p, Mn 3p, Mn 3s, O 1s and C 1s could be observed and two characteristic peaks representing the Mn(IV) 2p1/2 at 653.8 eV and Mn 2p3/2 at 642.2 eV in the Mn 2p magnification region. The distance between Mn 2p1/2 peak and its satellite (Δ2p1/2) of Mn(IV) was 11.8 eV, which are all in good agreement with the previous reported MnO2 results.31,33
Furthermore, according to Fick's first law,34 a higher precursor concentration can provide higher chemical potential for diffusion, thus facilitating the ions adsorption amount and penetration depth within the CMS templates. As a result, thin single-, double-, and triple-shelled hollow microspheres (Fig. 2b–d and S8†) were attained with the KMnO4 adding amount being 0.05 g, 0.08 g and 0.111 g, respectively. Nevertheless, the shell number could not be further increased by increasing the KMnO4 added amount because Mn-adsorption tended to saturate and MnO4− ions tended to accumulate on the CMS templates surface. Thus triple-shelled hollow microspheres with some large particles formed when further increasing the KMnO4 added amount to 0.2 g (Fig. S11†).
 | ||
| Fig. 2 (a) SEM image of quadruple-shelled MnO2 hollow microspheres and (b, c, d, e) TEM images of multi-shelled MnO2 hollow microspheres. (f) HRTEM image of the single MnO2 nanoparticle taken from a quadruple-shelled MnO2 hollow microsphere and (g) the corresponding SAED patterns. (h) XRD patterns of quadruple-shelled MnO2 hollow microspheres (i, ii, iii and iv represent single-, double-, triple- and quadruple-shelled MnO2 hollow microspheres, respectively). | ||
According to our previous study, the annealing process is also a key parameter that controls the morphology and structure of the final microsphere products. TGA–DTA (Fig. S12†) curves showed that the weight loss stopped at around 300 °C. Considering the real heat treatment conditions (450 °C, 1 °C min−1, 2 h), one can assert that the obtained multi-shelled hollow microspheres comprises high crystallinity Mn oxides without any remaining carbon (Fig. S7†). In addition, the heating rate is also one of the most important factors, which can cause great discrepancy between the rate of metal oxide shell formation (v1) and CMS contraction (v2).25 Normally, a slow heating rate would facilitate a higher shell number because under a slow heating rate (1 °C min−1) both the speed of oxide-crystallization and the difference between v1 and v2 decrease.24 Thus the outer oxide will shrink along with the shrinking of the inner CMS core during the early period of annealing with insufficient oxide to sustain the formation of oxide shell, thus accumulating to form a thick shell. Then, the thick shell further shrinks and separates to double shells and finally results in quadruple-shelled MnO2 hollow microspheres (Fig. 2e).
The morphology of the single-, double-, triple- and quadruple-shelled MnO2 hollow microspheres were observed by SEM and TEM (Fig. 2a–e and S8).†Fig. 2a, S8 and S9† show that there are numerous nanoparticles on the surface of the hollow microspheres with uniform ∼2 μm diameters. This hierarchical structure with loading nanoparticles can help improve the specific surface area and specific capacitance greatly and protect the interior shells from electrochemical dissolution to achieve better cycling performance.
XRD analysis (Fig. 2h) demonstrates that the multi-shelled MnO2 hollow microspheres were all pure α-MnO2 phase (JCPDS card no. 44-141) without the existence of other impurity Mn species.35 The reason could be ascribed to the unique high temperature and pressure under hydrothermal conditions, as well as the strong oxidizing capability of KMnO4 in the calcination process. The resulting pure α-MnO2 possesses a tunnel structure that comprises a double-chained edge-sharing MnO6 octahedral, as well as a ∼0.46 nm cavity36 In this way, water molecules and large cations such as K+ can easily enter the tunnel cavity without apparent volume inflation to ensure the electrode structure stability.30
More characterizations about the crystal structure and composition of resulted product are shown by HRTEM images (Fig. 2f) and SAED patterns (Fig. 2g). HRTEM shows clear lattice fringes with 0.31 nm inter-planar distances in correspondence with the (310) crystal plane of α-MnO2 and the SAED patterns revealed the existence of (310) and (220) α-MnO2 crystal planes, which were consistent with the previous XRD analysis.
The superiority of as-prepared multi-shelled MnO2 hollow microspheres for charge storage compared with MnO2 nanoparticles are shown below. Fig. 3, S14 and S15† show the representative CV curves at various scan rates (5–200 mV s−1) and the galvanostatic charge/discharge curves at various current densities (0.5–10 A g−1) of the multi-shelled MnO2 hollow microspheres, i.e., two pairs of broad redox peaks (too close to be identified), which represent two reversible redox reactions:37
| Mn(IV) ↔ Mn(III) − e− and Mn(III) ↔ Mn(II) − e− |
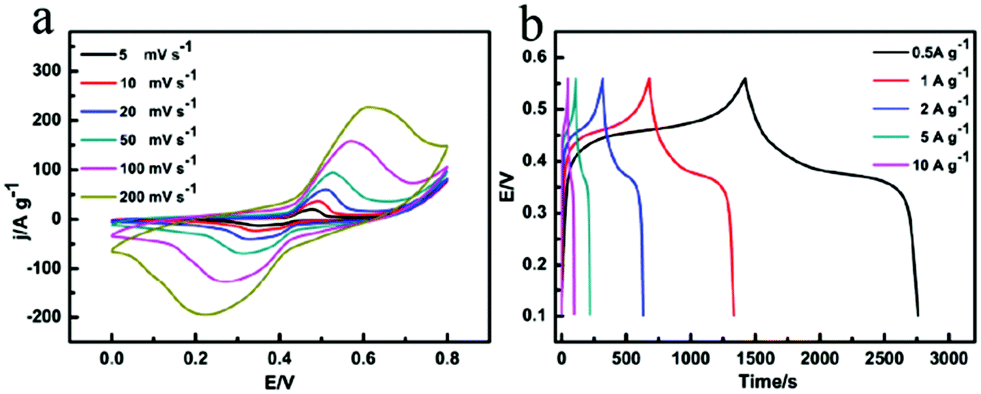 | ||
| Fig. 3 (a) CV curves of quadruple-shelled MnO2 hollow microspheres at different scan rates of 5–200 mV s−1. (b) Galvanostatic charge/discharge curves of quadruple-shelled MnO2 hollow microspheres at different current densities. | ||
Among all the five samples, quadruple-shelled hollow microspheres showed the highest specific capacitance (1457 F g−1 at a 0.5 A g−1 current density). Therefore, it could be confirmed that the Faradic capacitance played the main role in charge storage of the as-prepared MnO2 hollow microspheres.38–40 Without obvious IR drop even at high current densities such as 10 A g−1, the multi-shelled MnO2 hollow microspheres electrode showed superior capacitive characteristics of rapid I–V response, a rapid and reversible faradic reaction and low internal resistance.41,42
To evaluate the influence of shell number on capacitive performance, CV curves of MnO2 single-, double-, triple-, quadruple-shelled hollow microspheres and MnO2 nanoparticles at the same 50 mV s−1 scan rate were compared and are shown in Fig. 4a. It was obviously determined that the quadruple-shelled MnO2 hollow microspheres electrode achieved the highest specific capacitance among all the as-prepared MnO2 samples, and the same tendency was observed in the galvanostatic charge/discharge curves (Fig. 4c).
 | ||
| Fig. 4 (a) Comparing the CV curves of MnO2 multi-shelled hollow microspheres and MnO2 NPs, at a 50 mV s−1 scan rate. (b) Comparison of the galvanostatic charge/discharge curves of MnO2 multi-shelled hollow microspheres and MnO2 NPs at 1 A g−1 current density. (c) Specific capacitances of MnO2 multi-shelled hollow microspheres and MnO2 NPs at different current densities. (d) Cyclic performance of MnO2 multi-shelled hollow microspheres and MnO2 NPs for 4000 cycles at 5 A g−1 current density. | ||
It is obvious that this type of unique hierarchical structure with porous thin shells greatly contributes to the supercapacitor electrodes specific capacitance improvement because the pseudocapacitors charge-storage process is the surface reaction between the electrode materials and the electrolyte ions.5,7 The BET characterization (Fig. S17 and Table S2†) showed that the specific surface area and the pore volume both increased along with the increased shell number, wherein the quadruple-shelled MnO2 hollow microspheres with the highest specific capacitance exhibited the highest specific surface area (≈99.36 m2 g−1) and largest pore volume (≈0.3397 cm3 g−1). This is very reasonable because a higher specific surface area provides more actual accessible active sites and larger pore volume facilitating the electrolyte to penetrate into the materials, thus achieving higher capacitance and better power density.
Good cycling stability is also an important index for supercapacitors. Fig. 4d clearly shows that the specific capacitance of the quadruple-shelled MnO2 hollow microspheres was still maintained at 1082 F g−1 with only 8.8% loss after 4000 consecutive cycles, which is excellent compared with all the previously reported results (Table 1). In contrast, the MnO2 nanoparticles lost 15.8% of the original capacitance after only 2000 cycles. There are two reasons of the capacitance loss for multi-shelled MnO2 hollow microspheres and MnO2 nanoparticles. One reason is the partial dissolution of MnO2 in the electrolyte during cycling. Multi-shelled MnO2 hollow microspheres exhibited a much better cycling stability than MnO2 nanoparticles with the advantages of the multi-shelled hollow structures and this results in better cycling stability than MnO2 nanoparticles, as shown in Fig. S16.† Another reason is the irreversible redox reaction between Mn(IV) and Mn(III), which means that not all the Mn(III) could be oxidized to Mn(IV) during cycling and the specific capacitance was sure to decrease.5 Obviously, the quadruple-shelled MnO2 hollow microspheres exhibited the best cycling stability among the five samples due to the best protection from the four-layer porous thin shells, which plays the crucial role of providing support and protection, even in alkaline electrolyte, during the charge/discharge processes.
| Electrode material | Specific capacitance | Current load or scan rate | Electrolyte | Voltage window | Capacitance retention | Ref. |
|---|---|---|---|---|---|---|
| Co3O4 nanowire@MnO2 ultrathin nanosheet core/shell arrays | 480 F g−1 | 2.67 A g−1 | 1 M LiOH | −0.2 to 0.6 V (vs. Ag/AgCl) | 97.3% after 5000 cycles at 11.25 mA cm−2 | 43 |
| 267 F g−1 | 29.8 A g−1 | |||||
| Flexible Zn2SnO4/MnO2 core/shell nanocable-carbon microfiber | 642.4 F g−1 | 1 A g−1 | 1 M Na2SO4 | 0–0.8 V (vs. Ag/AgCl) | 98.8% after 1000 cycles at 10 A g−1 | 44 |
| 413.9 F g−1 | 40 A g−1 | |||||
| Ni(OH)2 nanowire-MnO2 nanoflakes core–shell nanostructure | 487.4 F g−1 | 1 A g−1 | 1 M KOH | −0.1 to 0.9 V (vs. Ag/AgCl) | 97.1% after 3000 cycles at 10 A g−1 | 45 |
| 269 F g−1 | 10 A g−1 | |||||
| Graphene nanoplate-MnO2 composites | 308.5 F g−1 | 5 mV s−1 | 1 M Na2SO4 | 0–1.0 V (SCE) | 87.4% after 100 cycles at 5 mV s−1 | 46 |
| — | — | |||||
| Au–MnO2–graphene nanocomposites | 575 F g−1 | 2.5 | 0.5 M NaOH | −0.4 to 0.5 V (vs. Ag/AgCl) | 74% after 1500 cycles at 2.5 A g−1 | 47 |
| 400 F g−1 | 10 | |||||
| MnO2 nanonest modified ionic liquid (IL) functionalized graphene paper | 415 F g−1 | 1 A g−1 | 1 M Na2SO4 | −0.2 to 0.8 V (SCE) | 85% after 10000 cycles at 10 A g−1 |
48 |
| 315 F g−1 | 20 A g−1 | |||||
| Co3O4@MnO2 hierarchical nanoneedle arrays | 603.2 F g−1 | 1 A g−1 | 1 M LiOH | −0.2 to 0.6 V (vs. Ag/AgCl) | 89.8% after 5000 cycles at 2 A g−1 | 49 |
| — | — | |||||
| MnO2 nanopillars | 603 F g−1 | 10 A g−1 | 1 M Na2SO4 | 0–0.8 V (SCE) | 93% after 5000 cycles at 1000 mV s−1 | 50 |
| 293 F g−1 | 100 A g−1 | |||||
| MnO2/Mn/MnO2 sandwich-structured nanotube arrays | 955 F g−1 | 1.5 A g−1 | 1 M Na2SO4 | 0–0.8 V (SCE) | 98.5% after 3000 cycles at 1.5 A g−1 | 51 |
| 660 F g−1 | 24 A g−1 | |||||
| Quadruple-shelled MnO2 hollow microspheres | 1457 F g−1 | 0.5 A g−1 | 1 M NaOH | 0.1–0.56 V (vs. Hg/HgO) | 91.2% after 4000 cycles at 5 A g−1 | This work |
| 1037 F g−1 | 10 A g−1 |
Conclusions
In summary, multi-shelled manganese oxide hollow microspheres with controlled valence, shell number and shell roughness were successfully achieved via an anion-adsorption process during hydrothermal intensification treatment. The hydrothermal intensification treatment not only could enhance the MnO4− adsorption amount and depth into the CMS templates, but also could immobilize the Mn element inside CMSs effectively. As supercapacitor electrode materials, the quadruple-shelled MnO2 hollow microspheres electrode achieved superior specific capacitance (1457 F g−1) at a 0.5 A g−1 discharge current density and excellent cyclic stability (91.2% retention of the initial capacitance after 4000 cycles), benefiting from the superiorities of these unique hierarchical structures such as increased number of active sites, shortened ions and electrons transport length, better contact between the electrolyte and active materials, as well as better protection of interior shells by the exterior shell. Given the enhanced electrochemical performance with high specific capacitance and excellent cyclic stability, multi-shelled MnO2 hollow microspheres are sure to be a promising material for supercapacitor electrodes.Acknowledgements
This study was supported financially by the National Natural Science Foundation of China (No. 21590795, 51572261, 51302266, 21401199, 21201167, 51372245, 51202248, 51472244, 51372245, 51272165, 51541206), the National Science Fund for Distinguished Young Scholars (No. 21325105) and Australian Research Council (ARC) Discovery Project (No. 160104817).References
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- S. L. Candelaria and G. Z. Cao, Sci. Bull., 2015, 60, 1587–1597 CrossRef CAS.
- S. L. Candelaria, E. Uchaker and G. Cao, Sci. China Mater., 2015, 58, 521–533 CrossRef CAS.
- H. B. Liu, Y. H. Tian, R. Amal and D. W. Wang, Sci. Bull., 2016, 61, 368–377 CrossRef CAS.
- G. P. Wang, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- N. Soin, S. S. Roy, S. K. Mitra, T. Thundat and J. A. McLaughlin, J. Mater. Chem., 2012, 22, 14944–14950 RSC.
- Y. Hou, Y. W. Cheng, T. Hobson and J. Liu, Nano Lett., 2010, 10, 2727–2733 CrossRef CAS PubMed.
- C. Y. Cao, W. Guo, Z. M. Cui, W. G. Song and W. Cai, J. Mater. Chem., 2011, 21, 3204–3209 RSC.
- D. W. Wang, Q. H. Wang and T. M. Wang, Inorg. Chem., 2011, 50, 6482–6492 CrossRef CAS PubMed.
- L. H. Bao, J. F. Zang and X. D. Li, Nano Lett., 2011, 11, 1215–1220 CrossRef CAS PubMed.
- K. Jin, A. Chu, J. Park, D. Jeong, S. E. Jerng, U. Sim, H. Y. Jeong, C. W. Lee, Y. S. Park, K. D. Yang, G. K. Pradhan, D. Kim, N. E. Sung, S. H. Kim and K. T. Nam, Sci. Rep., 2015, 5, 1–11 Search PubMed.
- J. Masa, W. Xia, I. Sinev, A. Q. Zhao, Z. Y. Sun, S. Grutzke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- D. Jeong, K. Jin, S. E. Jerng, H. Seo, D. Kim, S. H. Nahm, S. H. Kim and K. T. Nam, ACS Catal., 2015, 5, 4624–4628 CrossRef CAS.
- K. Zhang, X. P. Han, Z. Hu, X. L. Zhang, Z. L. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- N. Djilali, Sci. Bull., 2016, 1–2, DOI:10.1007/s11434-016-1047-5.
- W. Chen, Q. Kuang and Z. Xie, Sci. China Mater., 2015, 58, 281–288 CrossRef CAS.
- H. M. Du, L. F. Jiao, Q. H. Wang, J. Q. Yang, L. J. Guo, Y. C. Si, Y. J. Wang and H. T. Yuan, Nano Res., 2013, 6, 87–98 CrossRef CAS.
- C. Guan, X. H. Xia, N. Meng, Z. Y. Zeng, X. H. Cao, C. Soci, H. Zhang and H. J. Fan, Energy Environ. Sci., 2012, 5, 9085–9090 Search PubMed.
- W. X. Guo, W. W. Sun and Y. Wang, ACS Nano, 2015, 9, 11462–11471 CrossRef CAS PubMed.
- L. F. Shen, L. Yu, X. Y. Yu, X. G. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 1868–1872 CrossRef CAS PubMed.
- J. Qi, X. Y. Lai, J. Y. Wang, H. J. Tang, H. Ren, Y. Yang, Q. Jin, L. J. Zhang, R. B. Yu, G. H. Ma, Z. G. Su, H. J. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., Int. Ed., 2013, 125, 6545–6548 CrossRef.
- S. M. Xu, C. M. Hessel, H. Ren, R. B. Yu, Q. Jin, M. Yang, H. J. Zhao and D. Wang, Energy Environ. Sci., 2014, 7, 632–637 Search PubMed.
- Z. H. Dong, H. Ren, C. M. Hessel, J. Y. Wang, R. B. Yu, Q. Jin, M. Yang, Z. D. Hu, Y. F. Chen, Z. Y. Tang, H. J. Zhao and D. Wang, Adv. Mater., 2014, 26, 905–909 CrossRef CAS PubMed.
- Z. H. Dong, X. Y. Lai, J. E. Halpert, N. L. Yang, L. X. Yi, J. Zhai, D. Wang, Z. Y. Tang and L. Jiang, Adv. Mater., 2012, 24, 1046–1049 CrossRef CAS PubMed.
- H. Ren, R. B. Yu, J. Y. Wang, Q. Jin, M. Yang, D. Mao, D. Kisailus, H. J. Zhao and D. Wang, Nano Lett., 2014, 14, 6679–6684 CrossRef CAS PubMed.
- F. Wang, J. Y. Wang, H. Ren, H. J. Tang, R. B. Yu and D. Wang, Inorg. Chem. Front., 2016, 3, 365–369 RSC.
- P. F. Xu, R. B. Yu, H. Ren, L. B. Zong, J. Chen and X. R. Xing, Chem. Sci., 2014, 5, 4221–4226 RSC.
- J. Y. Wang, H. J. Tang, H. Ren, R. B. Yu, J. Qi, D. Mao, H. J. Zhao and D. Wang, Adv. Sci., 2014, 1, 6 Search PubMed.
- S. W. Zhang and G. Z. Chen, Energy Mater., 2008, 3, 186–200 CrossRef CAS.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- S. W. Donne, A. F. Hollenkamp and B. C. Jones, J. Power Sources, 2010, 195, 367–373 CrossRef CAS.
- W. Y. Li, G. Li, J. Q. Sun, R. J. Zou, K. B. Xu, Y. G. Sun, Z. G. Chen, J. M. Yang and J. Q. Hu, Nanoscale, 2013, 5, 2901–2908 RSC.
- D. Straub, R. Graue, F. Heitmeir, P. Nebendahl and T. K. Wurst, Propellants, Explos., Pyrotech., 1987, 12, 105–112 CrossRef CAS.
- X. Su, L. Yu, G. Cheng, H. Zhang, M. Sun and X. Zhang, Appl. Energy, 2015, 153, 94–100 CrossRef CAS.
- W. Chen, R. B. Rakhi, Q. X. Wang, M. N. Hedhili and H. N. Alshareef, Adv. Funct. Mater., 2014, 24, 3130–3143 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- S. A. Klankowski, G. P. Pandey, G. Malek, C. R. Thomas, S. L. Bernasek, J. Wu and J. Li, Nanoscale, 2015, 7, 8485–8494 RSC.
- J. Yan, Q. Wang, T. Wei and Z. J. Fan, Adv. Energy Mater., 2014, 4, 43 Search PubMed.
- D. D. Zhu, Y. D. Wang, G. L. Yuan and H. Xia, Chem. Commun., 2014, 50, 2876–2878 RSC.
- J. A. Yan, E. Khoo, A. Sumboja and P. S. Lee, ACS Nano, 2010, 4, 4247–4255 CrossRef CAS PubMed.
- Y. F. Zhao, W. Ran, J. He, Y. Z. Huang, Z. F. Liu, W. Liu, Y. F. Tang, L. Zhang, D. W. Gao and F. M. Gao, Small, 2015, 11, 1310–1319 CrossRef CAS PubMed.
- J. P. Liu, J. Jiang, C. W. Cheng, H. X. Li, J. X. Zhang, H. Gong and H. J. Fan, Adv. Mater., 2011, 23, 2076–2081 CrossRef CAS PubMed.
- L. H. Bao, J. F. Zang and X. D. Li, Nano Lett., 2011, 11, 1215–1220 CrossRef CAS PubMed.
- H. Jiang, C. Z. Li, T. Sun and J. Ma, Chem. Commun., 2012, 48, 2606–2608 RSC.
- H. J. Huang and X. Wang, Nanoscale, 2011, 3, 3185–3191 RSC.
- V. Veeramani, B. Dinesh, S. M. Chen and R. Saraswathi, J. Mater. Chem. A, 2016, 4, 3304–3315 RSC.
- Y. M. Sun, Z. Fang, C. X. Wang, K. Ariyawansha, A. J. Zhou and H. W. Duan, Nanoscale, 2015, 7, 7790–7801 RSC.
- D. Z. Kong, J. S. Luo, Y. L. Wang, W. N. Ren, T. Yu, Y. S. Luo, Y. P. Yang and C. W. Cheng, Adv. Funct. Mater., 2014, 24, 3815–3826 CrossRef CAS.
- Z. N. Yu, B. Duong, D. Abbitt and J. Thomas, Adv. Mater., 2013, 25, 3302–3306 CrossRef CAS PubMed.
- Q. Li, Z. L. Wang, G. R. Li, R. Guo, L. X. Ding and Y. X. Tong, Nano Lett., 2012, 12, 3803–3807 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qi00083e |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2016 |