
LiAlH4 supported on TiO2/hierarchically porous carbon nanocomposites with enhanced hydrogen storage properties†
Yaran
Zhao
,
Mo
Han
,
Haixia
Wang
,
Chengcheng
Chen
and
Jun
Chen
*
Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: chenabc@nankai.edu.cn; Fax: +86-22-23506808; Tel: +86-22-23506808
First published on 30th September 2016
Abstract
We report the synthesis of LiAlH4 supported on TiO2/hierarchically porous carbon (LAH–TiO2/HPC) nanocomposites using a one-step solvent method and their enhanced catalytic dehydrogenation performance. The as-prepared TiO2/HPC nanocomposites show that TiO2 nanoparticles (∼10 nm) are homogeneously distributed on the surface of hierarchically porous carbon (HPC). The results show that TiO2/HPC nanocomposites exhibit better catalytic performance for the dehydrogenation of LiAlH4 and rehydrogenation of the dehydrided sample than that of TiO2 nanoparticles and HPC. The dehydrogenation temperature of 37LAH–25TiO2/38HPC with 37 wt% LiAlH4, 25 wt% TiO2 and 38 wt% HPC is the lowest. Hydrogen started to be released at 64 °C, which is about 100 °C lower than that of pure LiAlH4. In addition, 4.3 wt% of hydrogen could be released from 37LAH–25TiO2/38HPC within 40 min at 130 °C, indicating fast kinetics with an activation energy of 47.1 ± 3.5 kJ mol−1. Furthermore, it can re-adsorb H2 at 300 °C under a hydrogen pressure of 4 MPa. The nanoconfinement of LiAlH4 into hierarchically porous carbon with high surface areas and the high distribution of TiO2 nanoparticles with a Ti(4+)/Ti(3+)/Ti(2+) defect site play a synergistic role in improving the hydrogen storage properties of LiAlH4.
Introduction
Hydrogen is regarded as a promising energy carrier due to its high energy density and environmental friendliness.1–6 However, the storage of hydrogen remains a bottleneck in the widespread utilization of hydrogen energy. It is still urgent to develop a safe and efficient technique to store hydrogen.7–10 Among all hydrogen storage materials, solid-state complex hydrides have attracted more attention because of their high gravimetric/volumetric hydrogen content, adequate kinetics, as well as reversible dehydrogenation/rehydrogenation properties.11–13 Particularly, light metal complex hydride LiAlH4 is considered to be a promising hydrogen storage material owing to the high theoretical hydrogen content (10.5 wt%).14–16 The process of hydrogen generation from LiAlH4 is presented below:17| LiAlH4 → 1/3Li3AlH6 + 2/3Al + H2↑ 187–218 °C | (1) |
| 1/3Li3AlH6 → LiH + 1/3Al + 1/2H2↑ 228–282 °C | (2) |
| LiH + Al → LiAl + 1/2H2↑ 380–430 °C | (3) |
Studies mainly focus on the former two steps because the decomposition temperature of the third step is too high.18 However, the thermodynamics and kinetics of reactions (1) and (2) are not appropriate for practical applications. Recently, metal catalysts have been widely applied to the former two steps.5,19 Among various metal catalysts, Ti-based catalysts are safe, of low cost and highly efficient.20–23 For example, Amama and coworkers24 investigated the effects of Ti, TiCl3 and TiO2 additives on the dehydrogenation properties of LiAlH4. The onset temperature (Tonset) decreased from 185 °C for pure LiAlH4 to 108 °C for 0.5 mol% TiO2 doped LiAlH4. Additionally, Chen et al. have reported the reversibility of LiAlH4 with the catalyst of TiCl3–1/3AlCl3 under a low hydrogen pressure of 4 MPa.7 In order to improve the hydrogen storage properties, the interaction between catalysts and LiAlH4 is a key factor.25,26 However, in previous studies, bulk LiAlH4 and Ti-based additives were mainly mixed by high-energy ball milling, which always caused the aggregation of catalysts and thus resulted in the loss of active sites.27,28 It is of great significance to disperse the catalysts on supporting materials with high surface areas; carbon matrices would be good candidates.29–31 Tan et al. reported the decoration of multiwall carbon nanotubes with TiCl3 particles (TiCl3-MWCNTs) as catalysts for the decomposition of LiAlH4. They found that the composite of LiAlH4-20 wt% TiCl3-MWCNTs began to desorb hydrogen at 75 °C, which is 96 °C lower than that of pure LiAlH4.32 Moreover, nanoconfining the hydrides to a carbon scaffold could reduce the particle size of hydrides to the nanoscale33–35 and prevent the de/rehydrogenated products from agglomerating in de/rehydrogenation cycles.36–38 In our previous work, we prepared hierarchically porous carbon (HPC) with large surface areas and multiscale pores, which is in favour of dispersing metal nanoparticles.39 Thus, TiO2/HPC nanocomposites as synergistic catalysts for the dehydrogenation of LiAlH4 are interesting.
Herein, we synthesized LiAlH4-supported on TiO2/HPC (LAH–TiO2/HPC) composites using a one-step solvent method at room temperature and further investigated their enhanced hydrogen storage properties. For the LAH–TiO2/HPC composites, LiAlH4 was homogeneously supported on the surface of TiO2/HPC. TiO2/HPC nanocomposites show better catalytic performance on de/rehydrogenation of LiAlH4 than individual TiO2 nanoparticles and HPC. The composite with 37 wt% LiAlH4, 25 wt% TiO2 and 38 wt% HPC (denoted as 37LAH–25TiO2/38HPC) exhibited superior performance. It started to release H2 at 64 °C and generate 4.3 wt% H2 within 40 min at 130 °C. Moreover, it can partially re-adsorb hydrogen at 300 °C under a hydrogen pressure of 4 MPa. HPC with high surface areas could downsize the hydride and restrain the dehydrogenated products from aggregation, while the highly distributed TiO2 nanoparticles with close contact of LiAlH4 provide a lot of active sites. The synergistic effect of the TiO2/HPC nanocomposites guarantees the enhanced hydrogen storage properties of LiAlH4.
Experiment
Synthesis of TiO2/HPC nanocomposites
HPC was prepared via a silica source/carbon source self-assembly approach and the associated details are shown in the ESI.† To prepare the TiO2/HPC nanocomposites, 50 mg HPC was dispersed in 15 mL absolute ethanol under ultrasonic treatment for 6 min. Then, 142 μL tetra-n-butyl titanate (TBT, 98%, Tianjin Guangfu Fine Chemical Research Institute) and 15 μL HNO3 (65%, Tianjin 5th Chemical Plant) were added under ultrasonic treatment for another 4 min in an ice–water bath. The mixture was then stirred for 0.5 h, followed by addition of 45 μL deionized water (the molar ratio of TBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3:H2O = 1:0.8:6)40 and kept under stirring for 6.5 h at room temperature. The solvent was evaporated under powerful stirring at 50 °C. The obtained powder was calcined at 500 °C for 3 h in an argon atmosphere. The content of TiO2 in TiO2/HPC nanocomposites is 40 wt%. For comparison, TiO2 nanoparticles and TiO2/HPC nanocomposites with TiO2 loading of 20, 50 and 60 wt% were synthesized by the same method.
:HNO3:H2O = 1:0.8:6)40 and kept under stirring for 6.5 h at room temperature. The solvent was evaporated under powerful stirring at 50 °C. The obtained powder was calcined at 500 °C for 3 h in an argon atmosphere. The content of TiO2 in TiO2/HPC nanocomposites is 40 wt%. For comparison, TiO2 nanoparticles and TiO2/HPC nanocomposites with TiO2 loading of 20, 50 and 60 wt% were synthesized by the same method.
Preparation of the LAH–TiO2/HPC composite
The composite of LiAlH4 loaded on TiO2/HPC was prepared using a one-step solvent method. Firstly, 100 mg TiO2/HPC composites was dispersed in 10 mL THF. A certain amount of LiAlH4 (97%, Alfa) was dissolved in 30 mL distilled THF under vigorous stirring for 1 h. After that, the LiAlH4–THF solution was added dropwise into a TiO2/HPC–THF mixture. The mixture was then stirred for about 7 h at room temperature. Finally, LAH–TiO2/HPC with various LiAlH4 contents (29, 37, 45, and 55 wt%) was obtained after evaporation under vacuum overnight. All the above operations were carried out in an argon filled glove box (Mikrouna Universal 2440/750) with H2O and O2 content below 1 ppm.Material characterization
The phase of all the samples was analyzed by powder X-ray diffraction (XRD) using a Mini Flex 600 X-ray generator with Cu-Kα radiation (λ = 1.5406 Å) between 10° and 80° at a scan rate of 2° min−1. The sample of LAH–TiO2/HPC was covered with a parafilm to prevent it from reacting with oxygen and water in the air during measurement. The morphology and structure were observed by field-emission scanning electron microscopy (SEM, JEOL JSM7500F) and transmission electron microscopy (TEM, Philips Tecnai-F20). Fourier transform infrared (FTIR) spectroscopy was performed to analyze the chemical bonding using a Tensor II (Bruker) at a resolution of 4 cm−1. N2 adsorption–desorption isotherms were measured with a BELSORP-Mini instrument at 77 K. The surface areas were calculated by the Brunauer–Emmett–Teller (BET) method. The pore size distributions were derived from the desorption branches of the isotherms using the Barrett–Joyner–Halenda (BJH) model. The oxidation state of the titanium component was detected with X-ray photoelectron spectroscopic (XPS) measurement in a Versa Probe PHI 5000 system (Al Kα radiation of 1486.6 eV). The binding energy of the C 1s core level is at 484.8 eV.Hydrogen storage property testing
The decomposition performance was tested on a temperature-programed desorption-mass spectroscopy (TPD-MS, Quantachrome) instrument. Typically, about 20 mg powder was loaded into the sample chamber, which is heated from room temperature to 300 °C with a heating rate of 5 °C min−1 under an Ar flow. The isothermal dehydrogenation kinetics and rehydrogenation properties were analysed on automated Sieverts’ apparatus (PCTPro-2000). For the dehydrogenation process, about 100 mg powder was firstly loaded on the reactor in the glove box and then transferred to the apparatus. After evacuating the system to vacuum, the sample was rapidly heated and maintained at a given temperature. For the rehydrogenation process, the fully dehydrogenated sample was heated at 300 °C under the hydrogen pressure of 4 MPa.Results and discussion
The morphology of HPC is shown in Fig. 1a, which is composed of honeycomb-like hollow hemispheres with an average diameter of 600 nm. Fig. 1b displays the SEM image of TiO2/HPC nanocomposites, in which TiO2 nanoparticles are homogeneously dispersed on the surface of HPC. The loading content of TiO2 is 40 wt%. The TEM image (Fig. 1c) demonstrates TiO2 nanoparticles with a narrow size distribution of ∼10 nm (Fig. S1†). The HRTEM image (inset of Fig. 1c) shows that the d-spacing of 0.3520 nm is attributed to the (111) lattice plane of TiO2. Fig. 1d shows XRD patterns of HPC, TiO2 nanoparticles and TiO2/HPC nanocomposites with 40 wt% TiO2. The TiO2/HPC nanocomposites exhibit distinct diffraction peaks of TiO2 indexing to an anatase phase (JCPDS card no. 21-1272). Furthermore, the Raman spectra of the TiO2/HPC nanocomposite in Fig. 1e displays four characteristic Raman scattering peaks at 147 cm−1 (Eg), 399 cm−1 (B1g), 516 cm−1 (B1g) and 639 cm−1 (Eg) assigned to anatase.41 XPS analysis (Fig. 1f) shows that the peaks at 463.9 eV and 458.2 eV correspond to Ti4+.42 When the loading content of TiO2 in the TiO2/HPC nanocomposites was increased to 50–60 wt%, the TiO2 nanoparticles suffered from serious aggregation (Fig. S2†). Since the highly dispersed catalysts are favourable for the production of hydrogen, the TiO2/HPC nanocomposites with 40 wt% TiO2 (the weight ratio of TiO2:HPC is 2:3) are selected to be further studied as catalysts for improving the hydrogen storage properties of LiAlH4.
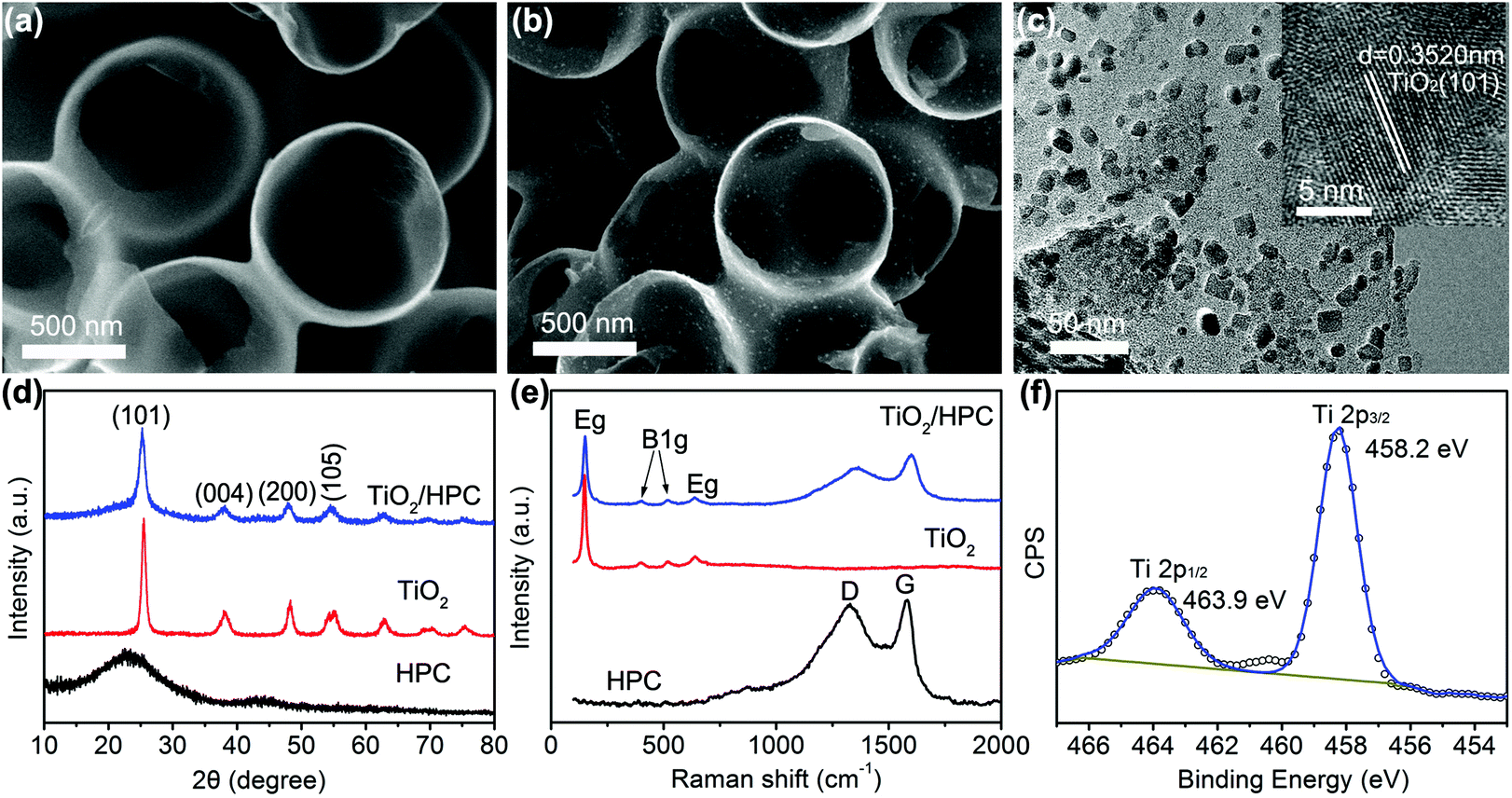 | ||
| Fig. 1 (a) SEM image of HPC, (b) SEM and (c) TEM images of the TiO2/HPC nanocomposite with 40 wt% TiO2 (inset of c is the HRTEM image), (d) XRD patterns and (e) Raman spectra of HPC and TiO2 nanoparticles and the TiO2/HPC nanocomposite with 40 wt% TiO2, (f) XPS spectra of the TiO2/HPC nanocomposite with 40 wt% TiO2. | ||
Fig. 2a shows the SEM image of the 37LAH–25TiO2/38HPC composites with 37 wt% LiAlH4, indicating that LiAlH4 is uniformly coated on the surface of TiO2/HPC. Fig. 2b displays that the wall of HPC becomes thicker, suggesting that LiAlH4 is successfully supported on the surface of TiO2/HPC. This is proved by Fig. 2c, in which the elements Al, Ti and O are evenly distributed on the carbon matrix. Furthermore, N2 adsorption/desorption analysis (Fig. S3†) shows that the BET specific surface area and the total pore volume of 37LAH–25TiO2/38HPC are much smaller than that of HPC, which demonstrates that LiAlH4 was successfully impregnated into the hierarchical pores of HPC.43,44 The morphologies of pure LiAlH4 and the LAH–TiO2/HPC composites with other loading weights (29, 45 and 55 wt%) of LiAlH4 are displayed in Fig. S4.† It is obvious that the particle size of LiAlH4 in LAH–TiO2/HPC is decreased in comparison with that of pure LiAlH4. A high degree of contact between nanosized LiAlH4 and TiO2 nanoparticles would be beneficial for dehydrogenation.
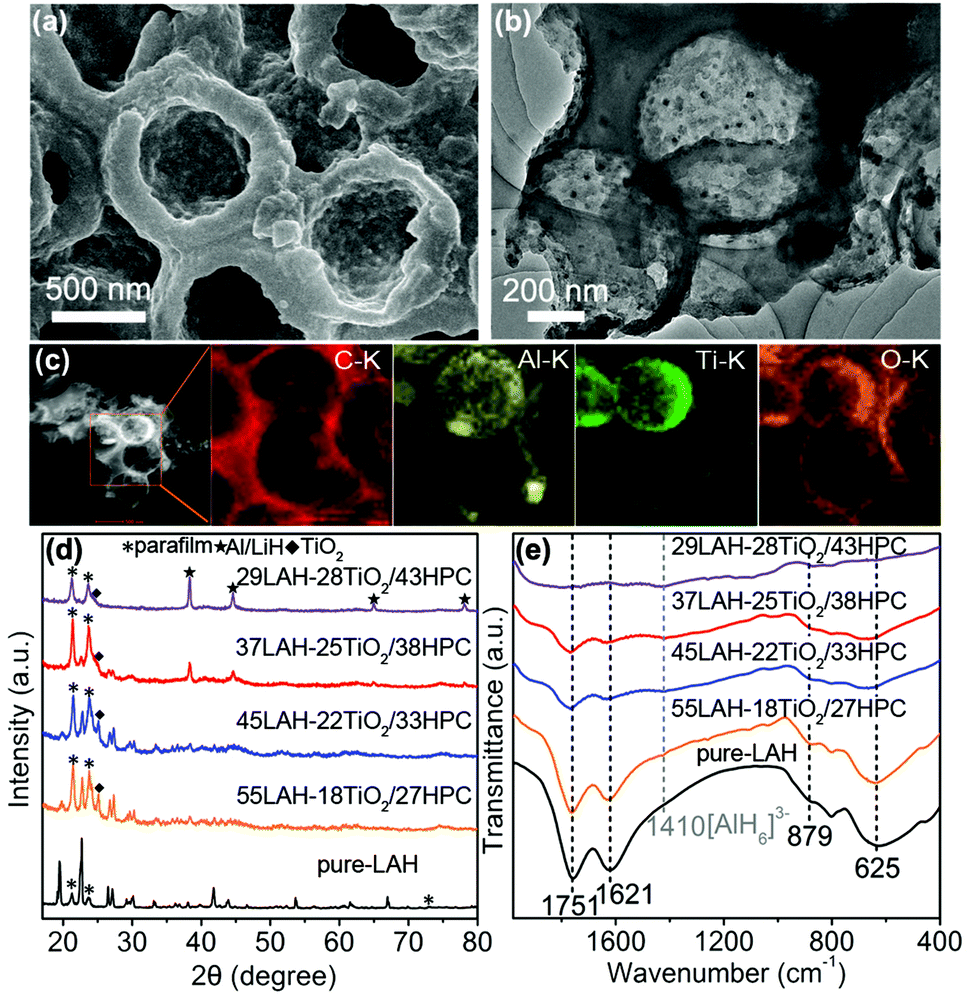 | ||
| Fig. 2 (a) SEM image, (b) TEM image and (c) TEM elemental mapping images of 37LAH–25TiO2/38HPC. (d) XRD patterns and (e) FTIR spectra of pure-LAH and LAH–TiO2/HPC nanocomposites with different loading weights of LiAlH4. | ||
XRD was employed to investigate the phase structure of pure LiAlH4 and LAH–TiO2/HPC nanocomposites with LiAlH4 loading of 29, 37, 45 and 55 wt%. In Fig. 2d, for pure-LAH, all diffraction peaks correspond to LiAlH4 (JCPDS card no. 12-473) except for the diffraction peaks of parafilm at 21.4°, 23.8° and 73°. With the decrease of the LiAlH4 loading weight, the peaks of LiAlH4 become weaker. When the loading weight of LiAlH4 decreases to 29 wt% in 29LAH–28TiO2/43HPC, the peaks of Al/LiH start to appear. This indicates that the decomposition of LiAlH4 occurred during the process of impregnation with abundant TiO2/HPC. Meanwhile, diffraction peaks of TiO2 are observed at 25.1°. To further confirm the decomposition process of LiAlH4, LAH–TiO2/HPC composites are further investigated by FTIR spectroscopy. In Fig. 2e, the infrared vibrations of Al–H stretching modes (1757 cm−1 and 1621 cm−1) and Li–Al–H bending modes (879 cm−1 and 625 cm−1) attribute to LiAlH4.15,18 In addition, a weak stretching mode at about 1410 cm−1 appears and becomes stronger with the increasing amount of TiO2/HPC, which can be assigned to Li3AlH6. This result demonstrates that a certain amount of LiAlH4 was decomposed into Li3AlH6 during the impregnation process, indicating the high reactivity of the TiO2/HPC nanocomposites. Furthermore, the stability of the LAH–TiO2/HPC nanocomposites is studied by FTIR spectroscopy after aging for 5 months in a glovebox, as shown in Fig. S5.† The representative peaks belonging to LiAlH4 indicate that the LAH–TiO2/HPC nanocomposites are relatively stable at room temperature.
The effect of the TiO2/HPC nanocomposites on the dehydrogenation behaviour of LiAlH4 was investigated by TPD at a constant heating rate. As shown in Fig. 3, two main peaks before 300 °C represent the first two steps of hydrogen desorption of LiAlH4. For pure-LAH, it begins to release hydrogen at 163 °C and reaches the largest hydrogen-desorption rate at 210 °C for step one. With the increasing content of the TiO2/HPC catalyst, a remarkable drop in the dehydrogenation temperature can be found. In particular, the 37LAH–25TiO2/38HPC nanocomposite with 37 wt% LiAlH4, 25 wt% TiO2 and 38 wt% HPC displays the optimal performance. It starts releasing hydrogen at 64 °C, which is about 100 °C lower than that of pure LiAlH4. The dehydrogenation properties of 37LAH–25TiO2/38HPC are superior to many LiAlH4-based systems (Table 1). It is noted that the desorption peak of 29LAH–28TiO2/43HPC is weak and broad, indicating slow kinetics of hydrogen release.44 To illustrate the good thermodynamic performance of 37LAH–25TiO2/38HPC, LiAlH4 mixed with TiO2 (LAH–TiO2, the weight ratio of LiAlH4:TiO2 = 37:25) or HPC (LAH–HPC, the weight ratio of LiAlH4:HPC = 37:28) were also prepared (Fig. S6†). The desorption temperatures are summarized in the line chart in Fig. S7.† The LAH–TiO2/HPC nanocomposites display much better thermodynamic properties than those of LAH–HPC and LAH–TiO2. Therefore, TiO2/HPC has a more efficient catalytic effect than that of HPC and TiO2.
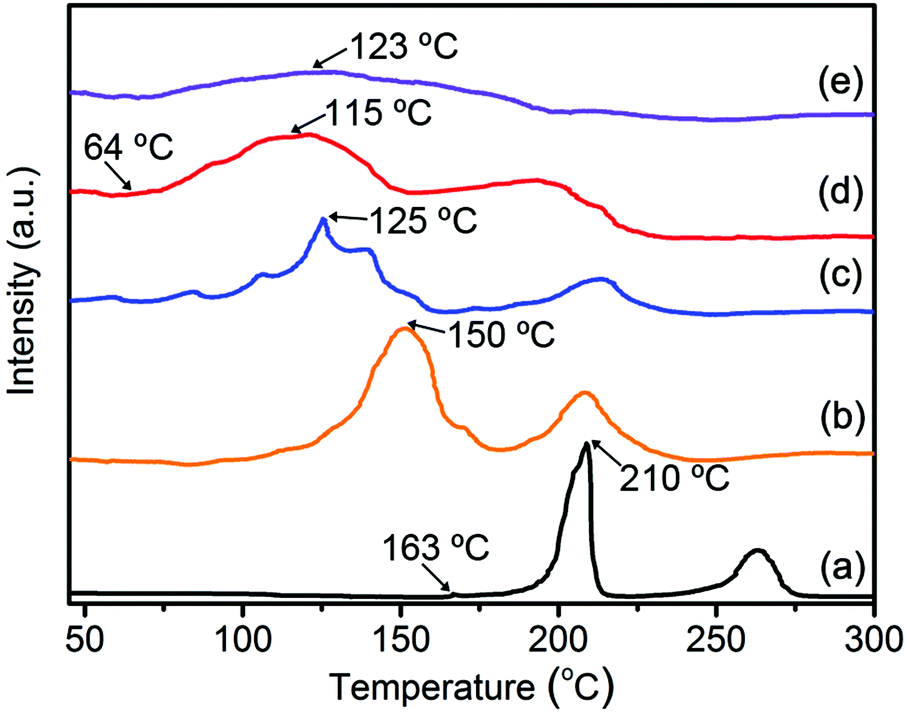 | ||
| Fig. 3 TPD hydrogen desorption curves of (a) pure-LAH, (b) 55LAH–18TiO2/27HPC, (c) 45LAH–22TiO2/33HPC, (d) 37LAH–25TiO2/38HPC, (e) 29LAH–28TiO2/43HPC. | ||
| Sample | Thermodynamics (°C) | Method | Kinetics | Ref. | |
|---|---|---|---|---|---|
| R1 Tonset | R1 Tpeak | ||||
| LiAlH4-20 wt% TiCl3-MWCNTs | 75 | — | TGA, 5 °C min−1 | 3.6 wt% at 110 °C | 32 |
| LAH-confined-Ni-MCS | 66 | 154 | TPD, 3 °C min−1 | 3.82 wt% at 150 °C | 45 |
| TiCl3·1/3AlCl3-doped LiAlH4 | 100 | — | TG, 2 °C min−1 | <4 wt% in 60 min at 125 °C | 7 |
| 2% TiN–LiAlH4 | 90 | 137.2 | TPD, 2 °C min−1 | <3 wt% in 1 h at 130 °C | 46 |
| Fe2O3-doped LiAlH4 | 80 | — | PCT, 5 °C min−1 | 4.7 wt% in 70 min at 90 °C | 47 |
| 5 wt% CAs/TiO2–LiAlH4 | 95 | — | TPD, 2 °C min−1 | — | 16 |
| 37LAH–25TiO2/38HPC | 64 | 115 | TPD, 5 °C min−1 | 4.3 wt% in 40 min at 130 °C | This work |
As 37LAH–25TiO2/38HPC shows the best thermodynamics, we further study its kinetic properties at different holding temperatures. As shown in Fig. 4a, the quantity of released hydrogen (calculated from pure LiAlH4 without containing TiO2 or HPC) as well as the rate of dehydrogenation rises as the holding temperature increases. At a temperature of 100 °C, it can release 2.3 wt% H2 within 100 min. When the temperature increases to 130 °C, 4.3 wt% H2 can be released within 40 min, exhibiting a fast and efficient hydrogen release process. The capacity of released hydrogen is competitive to previous reports (Table 1).
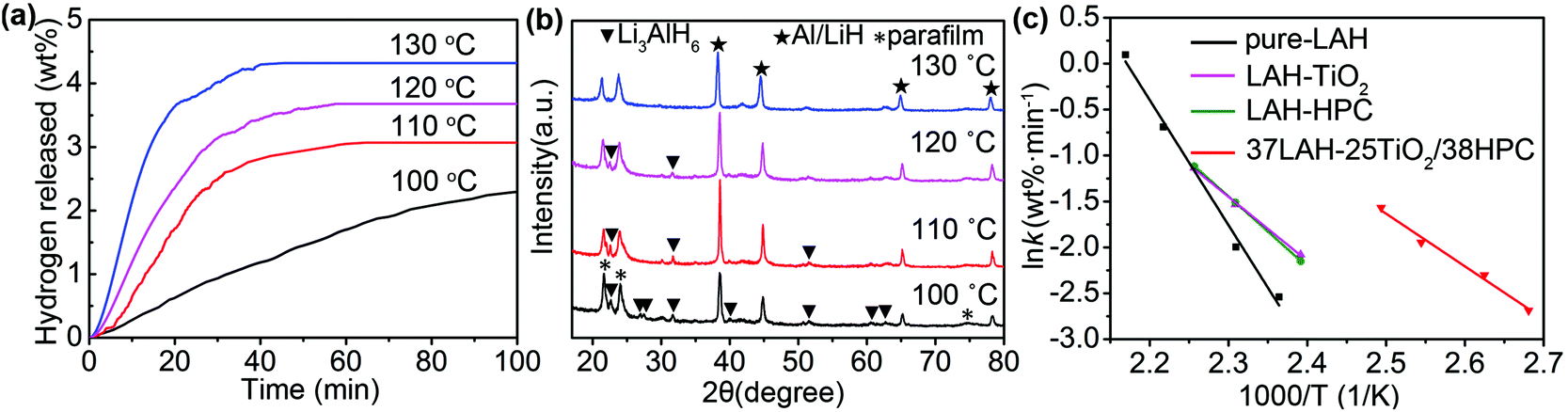 | ||
| Fig. 4 (a) Hydrogen desorption kinetic curves of 37LAH–25TiO2/38HPC at different temperatures, (b) XRD patterns of 37LAH–25TiO2/38HPC dehydrogenated at 100 °C, 110 °C, 120 °C and 130 °C. (c) The Arrhenius plots of pure-LAH, LAH–HPC, LAH–TiO2 and 37LAH–25TiO2/38HPC. | ||
XRD was implied to detect the dehydrogenated products at different holding temperatures in Fig. 4b. When dehydrogenated at 100 °C, distinct diffraction peaks of Li3AlH6 and Al/LiH are detected, while no peaks of LiAlH4 have appeared, confirming the reaction of reaction (1). Moreover, with the increase of the holding temperature, the intensity of Li3AlH6 peaks gradually gets weaker and Al/LiH becomes the main product. This demonstrates that the decomposition degree of LiAlH4 was related to various temperatures.
Furthermore, the isothermal dehydrogenation measurements of pure-LAH, LAH–HPC and LAH–TiO2 were also conducted (Fig. S8 and Table S2†). Compared to pure-LAH, the amount of released hydrogen and the rate of hydrogen release have obviously improved after adding TiO2 or HPC. The composite LAH–HPC has a higher hydrogen releasing capacity, while the dehydrogenation rate of LAH–TiO2 is slightly faster. This suggests that the TiO2 nanoparticles in TiO2/HPC enhance the desorption kinetics of 37LAH–25TiO2/38HPC, while HPC in TiO2/HPC could improve the hydrogen desorption capacity. Thus, the synergistic effect of TiO2/HPC enables 37LAH–25TiO2/38HPC to display a better desorption performance.25
The activation energy has been calculated from various isotherm curves through the Arrhenius equation:7
| lnk = lnA − Ea/RT | (4) |
k − 1/T in Fig. 4c, the activation energy is obtained (Table S3,† the error bars for Ea calculation are presented in Fig. S9†). The activation energy of pure LiAlH4 is calculated to be 113.2 ± 6.4 kJ mol−1, which is consistent with the value of 116.2 kJ mol−1 reported.13 With the additives of HPC or TiO2, the activation energies decrease to 63.5 ± 0.4 or 57.8 ± 2.1 kJ mol−1, respectively. While, the Ea for 37LAH–25TiO2/38HPC is 47.1 ± 3.5 kJ mol−1, which is about 66.1 kJ mol−1 lower than that of pure LiAlH4. This confirms that the high dispersion of TiO2 nanoparticles on the surface of HPC as a mixed catalyst plays a critical role in enhancing the kinetic properties of 37LAH–25TiO2/38HPC.
The composite of 37LAH–25TiO2/38HPC with 37 wt% LiAlH4 is further studied after full dehydrogenation. Fig. 5a displays the SEM image, in which HPC maintains its hierarchically porous structure. The EDS elemental mapping images (Fig. S10†) indicate that the Al, Ti, and O are uniformly distributed on the matrix of C, suggesting no aggregation of dehydrogenated products and TiO2 nanoparticles. This confirms that the HPC is a stable nanoscaffold material for dispersing TiO2 nanoparticles and preventing the dehydrogenated products from aggregation. In Fig. 5b, the dehydrogenated products of 37LAH–25TiO2/38HPC are composed of Al and LiH without LiAlH4, Li3AlH6 or other additional phases. It is noteworthy that there is no diffraction peak of TiO2 or the Ti-containing phase, which may be in the form of an amorphous state.
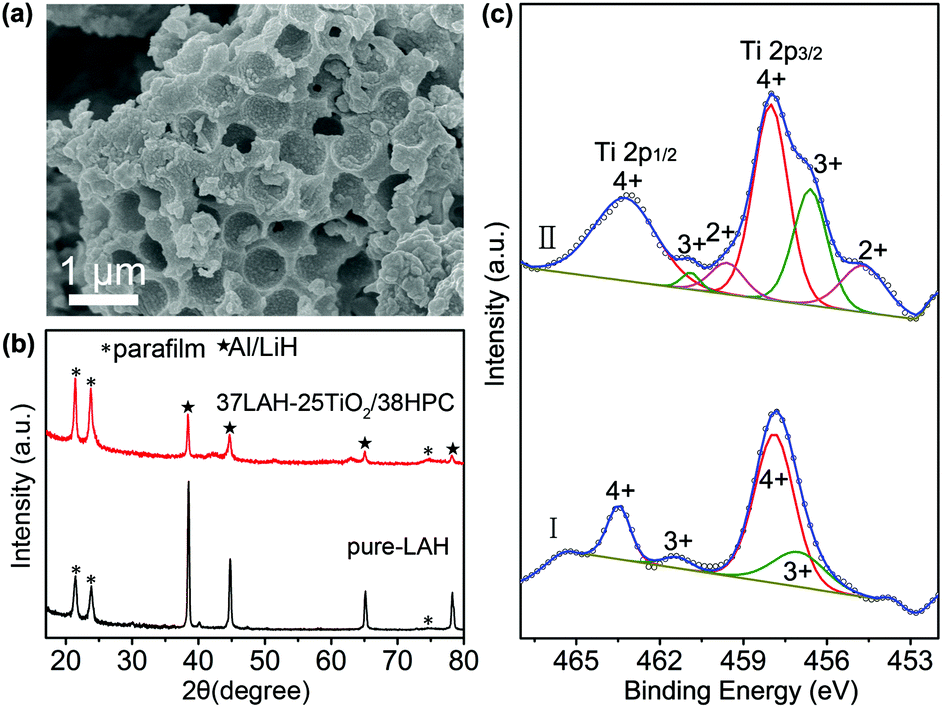 | ||
| Fig. 5 (a) SEM image of 37LAH–25TiO2/38HPC after dehydrogenation, (b) XRD patterns of pure-LAH and 37LAH–25TiO2/38HPC after dehydrogenation, (c) Ti 2p XPS spectra of 37LAH–25TiO2/38HPC before dehydrogenation (I) and after dehydrogenation (II). | ||
To understand the nature of the Ti-catalyst, 37LAH–25TiO2/38HPC was further studied by XPS before and after dehydrogenation in Fig. 5c. Compared to TiO2/HPC (Fig. 1f), it is apparent that the shape of the Ti 2p spectra has greatly changed. The broadening of XPS spectra suggests the presence of multiple oxidation states of Ti. Before dehydrogenation (I) (a.c. after impregnation), in addition to two main peaks at 463.5 eV and 457.9 eV for Ti4+, another pair of spin–orbit doublets at 461.4 eV and 456.6 eV could be corresponded to Ti3+. The lower binding energy values indicate the partial reduction of TiO2 resulting from the reaction between TiO2 and LiAlH4 during the impregnation process. After full dehydrogenation (II), the spectra became more complicated. Peaks located at 460.0 and 455.1 eV, attributed to Ti2+, appeared,41 illustrating that a deep reduction of TiO2 took place with the increase of the temperature. From Gaussian fitting of the peak area, the Ti(4+)/Ti(3+)/Ti(2+) atomic ratio is 0.62:0.22:0.16, demonstrating the multiple valence on the oxide surface with dominant Ti4+. Since a microstructured composite with Ti0/Ti2+/Ti3+ defect sites could enhance the dehydriding kinetics,7,24 the reduction of Ti4+ and the in situ formation of Ti-containing lower oxidation valence species have played a significant role in improving the dehydrogenation properties of LiAlH4.
In order to examine the reversibility for hydrogen storage, the dehydrogenated product of 37LAH–25TiO2/38HPC (LiH–Al–TiO2/HPC) was rehydrogenated at 300 °C for 24 h under a hydrogen pressure of 4 MPa. Fig. 6 shows the FTIR spectra of the dehydrogenated product LiH–Al–TiO2/HPC and the rehydrogenated product. Fig. 6a shows no peak of [AlH4]− or [AlH6]3−, suggesting the complete dehydrogenation of 37LAH–25TiO2/38HPC. Fig. 6b displays the infrared vibrations of the Al–H bond in [AlH6]3− between 1600–1400 cm−1 in the rehydrogenated products, indicating the partial reversibility of reaction (2). Additionally, the SEM image and TEM image (Fig. S11†) show no aggregation or growth of the TiO2 nanoparticles after re-adsorbing H2. In comparison, little H2 could be re-adsorbed for the dehydrogenated products of LAH–HPC and LAH–TiO2 since a higher pressure was necessary.48 These results further confirm the important effect of the TiO2/HPC nanocomposites in the reversibility of the 37LAH–25TiO2/38HPC.
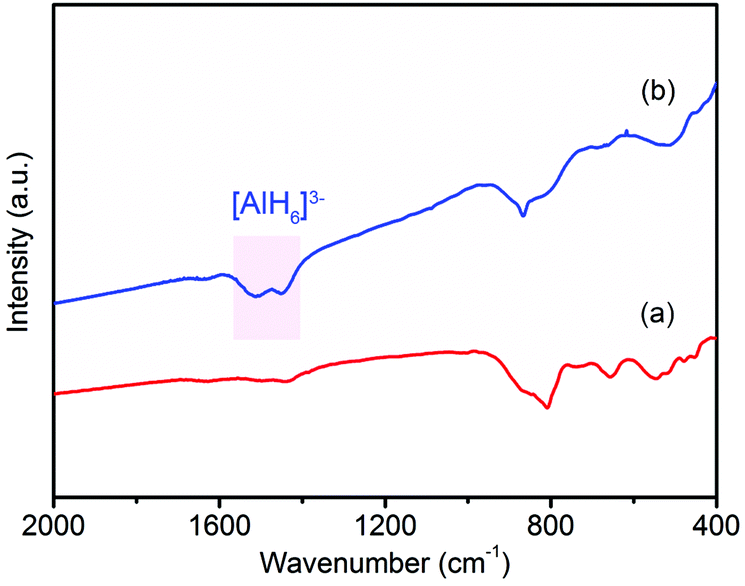 | ||
| Fig. 6 FTIR spectra of (a) dehydrogenated product LiH–Al and (b) rehydrogenated product of 37LAH–25TiO2/38HPC. | ||
From the above investigation, the as-synthesized TiO2/HPC nanocomposites exhibit excellent catalytic activity in the de/rehydrogenation of LiAlH4. The interpretations from two aspects are summarized to comprehend the catalytic function.49–51 On one hand, the high distribution of TiO2 nanoparticles is correlated with abundant active sites. The unique structure of TiO2/HPC provides both the inner and the outer surface contact with LiAlH4. Thus, a great deal of active sites is favorable to the kinetic enhancement of LiAlH4. Meanwhile, the hierarchically porous morphology and structure of the obtained carbon material offers a stable scaffold to highly disperse LiAlH4 and provide a hydrogen transporting pathway. This not only decreases the particle size of LiAlH4 but also prevents the dehydrogenated products from agglomerating. On the other hand, the reduction of Ti4+ during the impregnation and dehydriding process is accompanied by the formation of Ti(4+)/Ti(3+)/Ti(2+) defect sites. As a result, the decomposition pathway of LiAlH4 in LAH–TiO2/HPC was altered and the reaction energy barrier significantly decreased. For the rehydrogenation process, LiH–Al on TiO2/HPC is much easier to re-adsorb H2 at a relatively low hydrogen pressure.
Conclusions
LAH–TiO2/HPC nanocomposites with LiAlH4 supporting on TiO2/HPC were prepared. The optimized nanocomposite of 37LAH–25TiO2/38HPC with 37 wt% LiAlH4 started to release H2 at 64 °C, which is about 100 °C lower than that of pure LiAlH4. Furthermore, it could release 4.3 wt% H2 within 40 min at 130 °C and 6.2 wt% H2 within 60 min at 160 °C. The activation energy of the dehydrogenation process is 47.1 ± 3.5 kJ mol−1. In addition, we demonstrated the partial reversibility of this nanocomposite at 300 °C under a hydrogen pressure of 4 MPa. The enhanced hydrogen storage properties of LAH–TiO2/HPC are attributed to the synergistic effect of TiO2/HPC nanocomposites. HPC acted as a stable nanoscaffold to disperse LiAlH4 on the surface of TiO2/HPC. TiO2 nanoparticles with high distribution provide numerous active sites and Ti-containing defect sites, decreasing the energy barrier of the de/rehydrogenation of LiAlH4. This catalyst is useful to promote the de/rehydrogenation performance of complex hydrides.Acknowledgements
This work was supported by National NSFC (51371100 and 51271094), and MOE (B12015 and IRT13R30).Notes and references
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- B. Peng and J. Chen, Coord. Chem. Rev., 2009, 253, 2805–2813 CrossRef CAS.
- Y. Pang, Y. Liu, M. Gao, L. Ouyang, J. Liu, H. Wang, M. Zhu and H. Pan, Nat. Commun., 2014, 5, 3519 Search PubMed.
- M. Ismail, Y. Zhao, X. B. Yu, I. P. Nevirkovets and S. X. Dou, Int. J. Hydrogen Energy, 2011, 36, 8327–8334 CrossRef CAS.
- M. Ahmed and X. Guo, Inorg. Chem. Front., 2016, 3, 578–590 RSC.
- X. Ma, K. Zhao, H. Tang, Y. Chen, C. Lu, W. Liu, Y. Gao, H. Zhao and Z. Tang, Small, 2014, 10, 4664–4670 CrossRef CAS PubMed.
- J. Chen, N. Kuriyama, Q. Xu, H. T. Takeshita and T. Sakai, J. Phys. Chem. B, 2001, 105, 11214–11220 CrossRef CAS.
- W. Li, C. Li, H. Ma and J. Chen, J. Am. Chem. Soc., 2007, 129, 6710–6711 CrossRef CAS PubMed.
- B. Peng and J. Chen, Energy Environ. Sci., 2008, 1, 479–483 CAS.
- Q. L. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478–512 CAS.
- H. Reardon, J. M. Hanlon, R. W. Hughes, A. Godula-Jopek, T. K. Mandal and D. H. Gregory, Energy Environ. Sci., 2012, 5, 5951–5979 CAS.
- X. B. Yu, D. M. Grant and G. S. Walker, J. Phys. Chem. C, 2009, 113, 17945–17949 CAS.
- L. Ouyang, J. Tang, Y. Zhao, H. Wang, X. Yao, J. Liu, J. Zou and M. Zhu, Sci. Rep., 2015, 5, 10776 CrossRef CAS PubMed.
- X. Liu, H. W. Langmi, S. D. Beattie, F. F. Azenwi, G. S. McGrady and C. M. Jensen, J. Am. Chem. Soc., 2011, 133, 15593–15597 CrossRef CAS PubMed.
- L. Li, Y. Xu, Y. Wang, Y. Wang, F. Qiu, C. An, L. Jiao and H. Yuan, Dalton Trans., 2014, 43, 1806–1813 RSC.
- P. Rangsunvigit, P. Purasaka, T. Chaisuwan, B. Kitiyanan and S. Kulprathipanja, Chem. Lett., 2012, 41, 1368–1370 CrossRef CAS.
- A. Andreasen, J. Alloys Compd., 2006, 419, 40–44 CrossRef CAS.
- F. Zhai, P. Li, A. Sun, S. Wu, Q. Wan, W. Zhang, Y. Li, L. Cui and X. Qu, J. Phys. Chem. C, 2012, 116, 11939–11945 CAS.
- M. A. Wahab, Y. Jia, D. Yang, H. Zhao and X. Yao, J. Mater. Chem. A, 2013, 1, 3471–3478 Search PubMed.
- Z. B. Li, S. S. Liu, X. L. Si, J. Zhang, C. L. Jiao, S. Wang, S. Liu, Y. J. Zou, L. X. Sun and F. Xu, Int. J. Hydrogen Energy, 2012, 37, 3261–3267 CrossRef CAS.
- J. Du, J. Qi, D. Wang and Z. Tang, Energy Environ. Sci., 2012, 5, 6914–6918 CAS.
- T. K. Nielsen, M. Polanski, D. Zasada, P. Javadian, F. Besenbacher, J. Bystrzycki, J. Skibsted and T. R. Jensen, ACS Nano, 2011, 5, 4056–4064 CrossRef CAS PubMed.
- K. Zhao, S. Zhao, J. Qi, H. Yin, C. Gao, A. M. Khattak, Y. Wu, A. Iqbal, L. Wu, Y. Gao, R. Yu and Z. Tang, Inorg. Chem. Front., 2016, 3, 488–493 RSC.
- P. B. Amama, J. T. Grant, P. J. Shamberger, A. A. Voevodin and T. S. Fisher, J. Phys. Chem. C, 2012, 116, 21886–21894 CAS.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283–1315 CrossRef CAS PubMed.
- K. Zhao, J. Qi, H. Yin, Z. Wang, S. Zhao, X. Ma, J. Wan, L. Chang, Y. Gao, R. Yu and Z. Tang, J. Mater. Chem. A, 2015, 3, 20465–20470 CAS.
- J. Qi, J. Chen, G. Li, S. Li, Y. Gao and Z. Tang, Energy Environ. Sci., 2012, 5, 8937–8941 CAS.
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He, Q. Li, H. Zhao, X. Huang, Y. Gao and Z. Tang, Adv. Mater., 2016, 28, 6485–6490 CrossRef CAS PubMed.
- R. J. Xiong, G. Sang, X. Y. Yan, G. H. Zhang and X. Q. Ye, J. Mater. Chem., 2012, 22, 17183–17189 RSC.
- G. Liu, Y. J. Wang, F. Y. Qiu, L. Li, L. F. Jiao and H. T. Yuan, J. Mater. Chem., 2012, 22, 22542–22549 RSC.
- C. H. Yang, C. P. Hsu, S. L. Lee, K. W. Wang and J. K. Chang, ChemSusChem, 2015, 8, 2713–2718 CrossRef CAS PubMed.
- C. Y. Tan and W. T. Tsai, Int. J. Hydrogen Energy, 2014, 39, 20038–20044 CrossRef CAS.
- M. Christian and K. F. Aguey-Zinsou, Nanoscale, 2010, 2, 2587–2590 RSC.
- T. Mueller and G. Ceder, ACS Nano, 2010, 4, 5647–5656 CrossRef CAS PubMed.
- M. Han, Q. Zhao, Z. Zhu, Y. Hu, Z. Tao and J. Chen, Nanoscale, 2015, 7, 18305–18311 RSC.
- T. K. Nielsen, P. Javadian, M. Polanski, F. Besenbacher, J. Bystrzycki and T. R. Jensen, J. Phys. Chem. C, 2012, 116, 21046–21051 CAS.
- S. S. Y. Lin, J. Yang, H. H. Kung and M. C. Kung, Top. Catal., 2013, 56, 1937–1943 CrossRef CAS.
- J. Gao, P. Adelhelm, M. H. W. Verkuijlen, C. Rongeat, M. Herrich, P. J. M. van Bentum, O. Gutfleisch, A. P. M. Kentgens, K. P. de Jong and P. E. de Jongh, J. Phys. Chem. C, 2010, 114, 4675–4682 CAS.
- J. Z. Zhao, F. Y. Cheng, C. H. Yi, J. Liang, Z. L. Tao and J. Chen, J. Mater. Chem., 2009, 19, 4108–4116 RSC.
- A. W. Xu, Y. Gao and H. Q. Liu, J. Catal., 2002, 207, 151–157 CrossRef CAS.
- N. Li, X. Zhang, W. Zhou, Z. Liu, G. Xie, Y. Wang and Y. Du, Inorg. Chem. Front., 2014, 1, 521–525 RSC.
- L. Deng, Y. Chen, M. Yao, S. Wang, B. Zhu, W. Huang and S. Zhang, J. Sol-Gel Sci. Technol., 2009, 53, 535–541 CrossRef.
- T. Zhang, X. Yang, S. Yang, D. Li, F. Cheng, Z. Tao and J. Chen, Phys. Chem. Chem. Phys., 2011, 13, 18592–18599 RSC.
- J. Zhao, J. Shi, X. Zhang, F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2010, 22, 394–397 CrossRef CAS PubMed.
- M. A. Wahab and J. N. Beltramini, Int. J. Hydrogen Energy, 2014, 39, 18280–18290 CrossRef CAS.
- L. Li, F. Y. Qiu, Y. J. Wang, Y. N. Xu, C. H. An, G. Liu, L. F. Jiao and H. T. Yuan, Int. J. Hydrogen Energy, 2013, 38, 3695–3701 CrossRef CAS.
- M. Ismail, A. M. Sinin, C. K. Sheng and W. B. W. Nik, Int. J. Electrochem. Sci., 2014, 9, 4959–4973 Search PubMed.
- J. Graetz, J. Wegrzyn and J. J. Reilly, J. Am. Chem. Soc., 2008, 130, 17790–17794 CrossRef CAS PubMed.
- F. Cheng and J. Chen, J. Mater. Res., 2006, 21, 2744–2757 CrossRef CAS.
- J. Chen and F. Wu, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 989–994 CrossRef CAS.
- J. Chen, S. L. Li and Z. L. Tao, J. Alloys Compd., 2003, 356, 413–417 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis procedures of HPC and dehydrogenation performances of LAH–HPC and LAH–TiO2, and additional XRD patterns, SEM images, FTIR, BET, EDS, and N2 adsorption/desorption curves. See DOI: 10.1039/c6qi00200e |
| This journal is © the Partner Organisations 2016 |