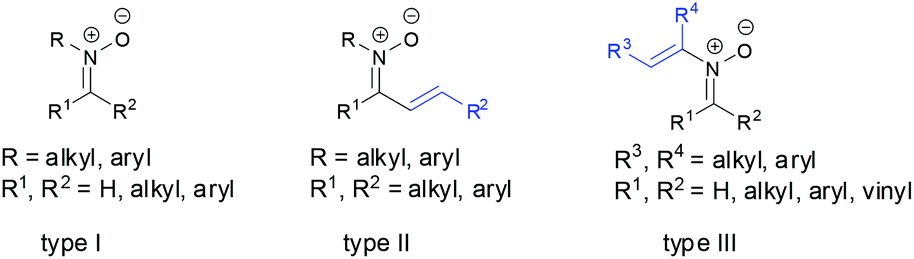DOI:
10.1039/C5QO00122F
(Review Article)
Org. Chem. Front., 2016,
3, 116-130
New developments of ketonitrones in organic synthesis†
Received
13th April 2015
, Accepted 2nd November 2015
First published on 2nd November 2015
Abstract
Nitrone chemistry has been widely developed for a long time and many reviews have outlined its rich transformation chemistry. However, most of these reviews involve aldonitrones due to their easy preparation and high reactivity. In recent years, N-substituents α,β-unsaturated ketonitrones and N-vinyl nitrones have attracted much attention of synthetic chemists to develop novel methodologies for their preparation and transformation. Various properties and new transformations of these nitrones have been discovered and applied into the synthesis of important heterocycle scaffolds. This review describes some recent breakthroughs in the development of new transformation of these nitrones. These new reactions include novel strategies for preparation of N-substituents α,β-unsaturated ketonitrones or N-vinyl nitrones, new rearrangement of nitrones, and reaction of nitrones with dipolarophiles such as alkynes, allenes and isocyanates.
 Wei-Min Shi | Wei-Min Shi was born in Guangxi in 1991. He gained his BS degree from Yulin Normal University in 2014. Then, he joined professor Dong-Liang Mo's group in Guangxi Normal University, and is currently a second-year graduate. His research interests focus on developing new transformation via N–O bond rearrangement. |
 Xiao-Pan Ma | Xiao-Pan Ma was born in Henan in 1989. She received her BS degree from Anyang Normal University in 2014. Then, she joined professor Dong-Liang Mo's group in Guangxi Normal University and is currently a second-year graduate. Her current research interests include preparation of nitrones and transition metal catalysis. |
 Gui-Fa Su | Gui-Fa Su was born in Guangxi in 1963. He earned his PhD degree in organic chemistry from Sun Yat-Sen University. In 1987, he joined the faculty at Guangxi Normal University, and is currently a full professor. His current research interests include design, synthesis and their biological evaluation of heterocyclic compounds; and design, synthesis and the structural study of conformationally constrained amino acids and peptides. |
 Dong-Liang Mo | Dong-Liang Mo was born in Guangxi in 1982. He gained his PhD in organic chemistry from Shanghai Institute of Organic Chemistry (SIOC) in 2011 under the supervision of Professors Li-Xin Dai and Xue-Long Hou. He spent 2011–2014 with Professor Laura Anderson at the University of Illinois at Chicago as a postdoctoral fellow. In 2014, he joined the faculty at Guangxi Normal University as a full professor, where he has started his independent research career. His current research interests are synthetic versatility of N–O bond rearrangement and transition metal catalysis in organic synthesis. |
1 Introduction
The chemistry of nitrones has been well developed in the literature since their discovery more than a century ago.1 Nitrones are not only widely recognized as important building blocks in the synthesis of various natural and biologically active compounds,2 but also used as both spin-trap reagents3 and therapeutic agents.4 Most of these early studies have focused on exploring the 1,3-dipole character of nitrones in [3 + 2] dipolar cycloadditions with dipolarophiles to construct functionalized five-membered heterocycles which are important intermediates in organic synthesis.5 Dipolarophiles include alkenes,6 alkynes,7 allenes8 and other cumulated double bonds.9 Some of other applications involving nitrones are also studied well, such as the synthesis of β-lactams by copper-catalyzed [2 + 2] cycloaddition with terminal alkynes,10 [3 + 2] cycloaddition with methylenecyclopropane,11 [3 + 3] cycloaddition with cyclopropanes,12 [3 + 3] cycloaddition with vinylcarbenes,13 [4 + 3] cycloaddition with cyclobutanes,14 and [2 + 2 + 3] cycloaddition with 1,6-enynes to give tricyclic 1,2-oxazepanes,15 umpolung of nitrones by SmI2 for C–C bond formation with electrophiles.16 However, most of these transformations are applied to N-alkyl/N-aryl aldonitrones, N-alkyl/N-aryl ketonitrones (Fig. 1, type I),17a or N-alkyl/N-aryl α,β-unsaturated aldonitrones17b,c due to their easy preparation and high reactivity. There are only a few applications found for N-substituents α,β-unsaturated ketonitrones (Fig. 1, type II)18 or N-vinyl nitrones (Fig. 1, type III)19 in the past few decades because of the challenges of their synthesis. Hence it is not surprising that the properties of these nitrones are unclear and desirable to be studied. Recently, not only some new methods for synthesis of N-substituents α,β-unsaturated ketonitrones (type II) and N-vinyl nitrones (type III) have been developed but also novel transformations of these nitrones have been demonstrated in organic synthesis. Various transformations have shown that the nature of these nitrones is distinguished and interesting in the reaction. In this review, we focus on describing several recent developments of preparation of N-substituents α,β-unsaturated ketonitrones (type II) or N-vinyl nitrones (type III) as well as novel transformations involving these nitrones, which include: (1) synthesis of N-substituents α,β-unsaturated ketonitrones and N-vinyl nitrones, (2) the new rearrangement of ketonitrones, (3) the reaction of ketonitrones with alkynes, (4) the reaction of ketonitrones with allenes, and (5) the reaction of ketonitrones with isocyanates.
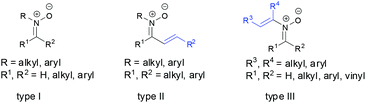 |
| | Fig. 1 Different types of nitrones. | |
1.1 Novel strategies in synthesis of ketonitrones
Various methodologies have been reported for preparation of nitrones. But the most convenient and commonly used methods are by condensing N-monosubstituted hydroxylamines and carbonyl compounds,20 or by the oxidation of the corresponding hydroxylamines,21 imines,22 amines23 or alkylation of aldoxime.24 The reaction of N-substituted hydroxylamines and aldehydes is typically facile and gives N-substituted aldonitrones in high yields under mild conditions. However, except for the intra-molecular variant, condensation of N-substituted hydroxylamines with corresponding ketones usually requires harsh conditions and the scope of the reaction is limited. Up to now, no direct condensation of N-aryl hydroxylamines with α,β-unsaturated ketones has been developed except for the successful condensation of N-alkyl hydroxylamine with α,β-unsaturated ketones.25
1.1.1 Preparation of type II nitrones.
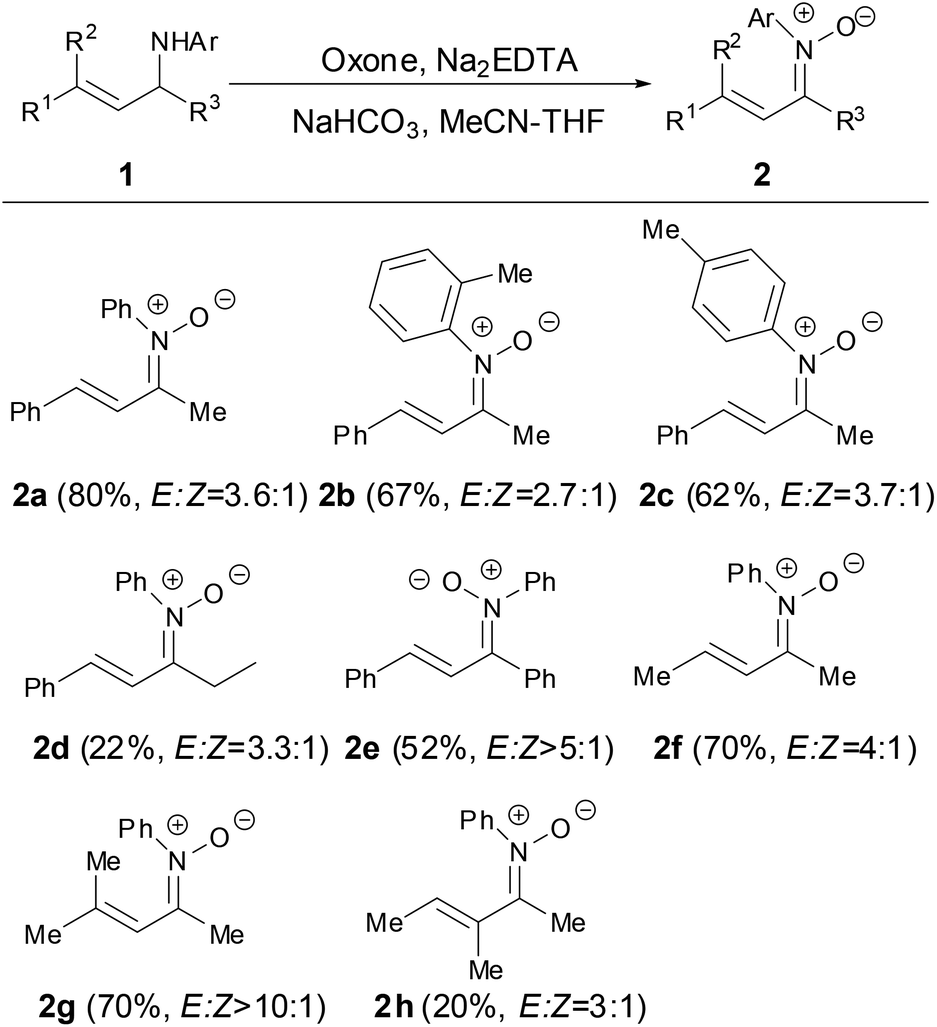
In 2012, Yang and coworkers developed a modular approach to N-aryl α,β-unsaturated ketonitrones by the oxidation of N-allyl anilines with oxone which was prepared by the condensation of anilines and enals followed by the alkylation of the resulting α,β-unsaturated imines.26 This method was general and provided efficient access to diversely substituted N-aryl α,β-unsaturated ketonitrones in good yields. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of nitrones 2 appeared to be generated in the E-geometry while the CN double bonds of the nitrone 2 were formed as E,Z-mixtures in ratios ranging from 2
C bonds of nitrones 2 appeared to be generated in the E-geometry while the CN double bonds of the nitrone 2 were formed as E,Z-mixtures in ratios ranging from 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to >10:1. The N-substituted groups in nitrones tolerated para-, meta- or ortho-substituted on the aryl ring. It is worth noting that the α- and β-positions of N-aryl α,β-unsaturated ketonitrones could be further substituted with the methyl group (2g and 2h) (Scheme 1).
:1 to >10:1. The N-substituted groups in nitrones tolerated para-, meta- or ortho-substituted on the aryl ring. It is worth noting that the α- and β-positions of N-aryl α,β-unsaturated ketonitrones could be further substituted with the methyl group (2g and 2h) (Scheme 1).
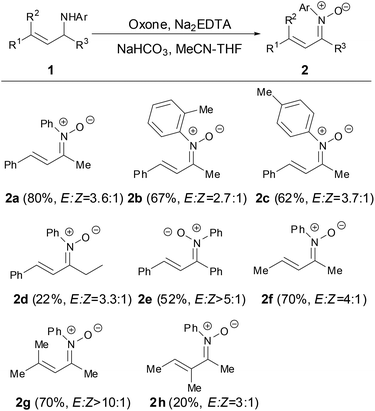 |
| | Scheme 1 Synthesis of N-aryl α,β-unsaturated ketonitrones by oxidation of aniline. | |
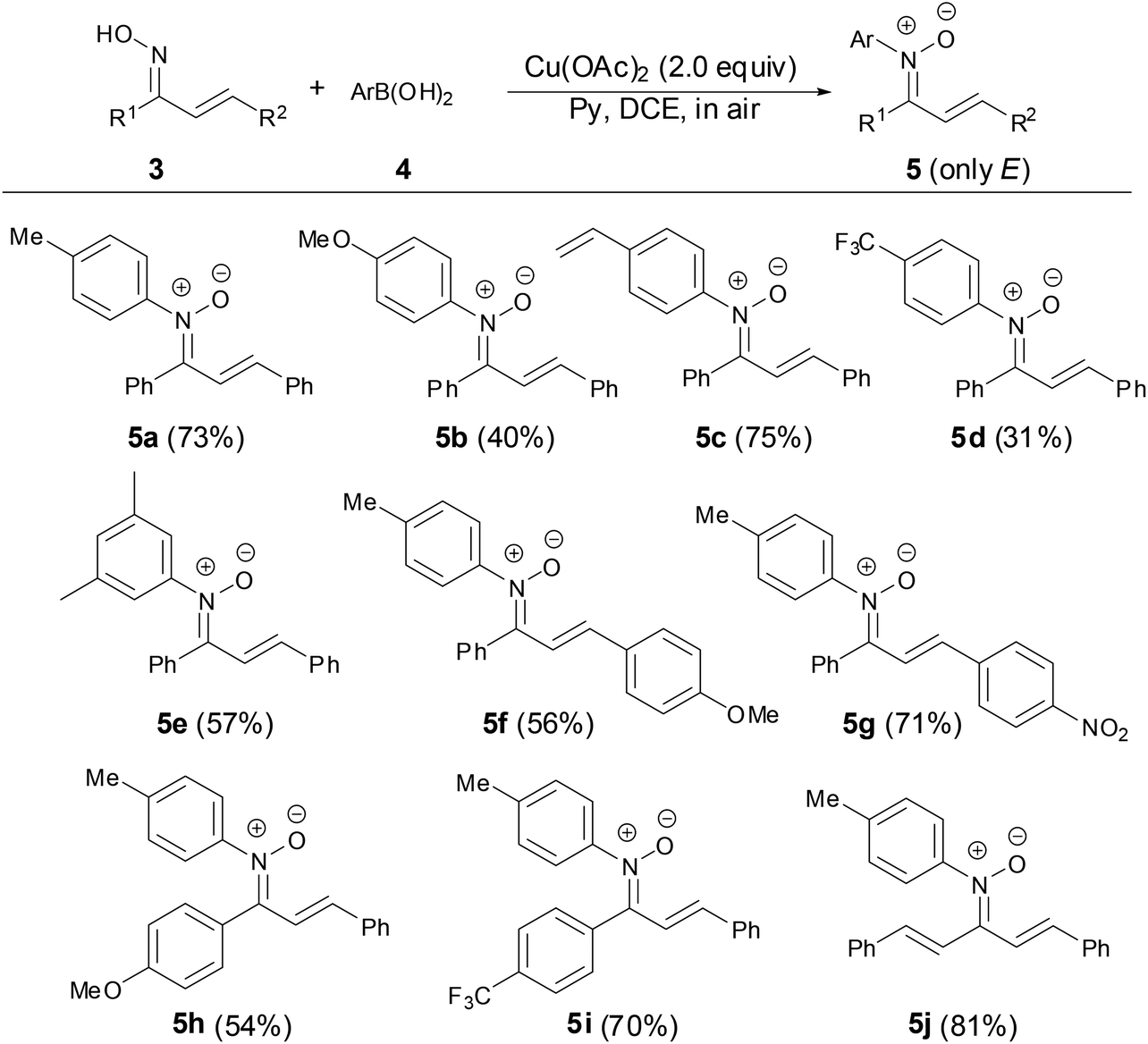
In 2013, Anderson and coworkers found that N-aryl α,β-unsaturated ketonitrones can be prepared by the copper-mediated cross-coupling reaction of chalcone oximes with various arylboronic acids in good yields (Scheme 2).27 The scope of nitrones tolerated aryl, vinyl, bromide, nitro groups. The geometry of chalcone oxime 3 has no effect on the formation of N-aryl α,β-unsaturated ketonitrones with E-isomer as major products. It is a very useful transformation that only the E-isomer nitrones were obtained after fast column chromatography. However, this copper-mediated coupling reaction was not suitable for ortho-substituted arylboronic acids.
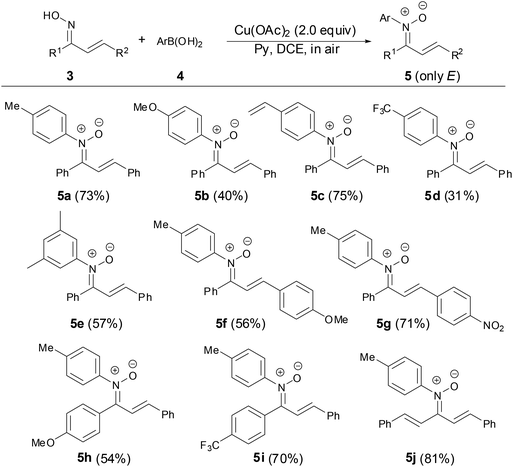 |
| | Scheme 2 Synthesis of N-aryl ketonitrones by copper-mediated reaction. | |
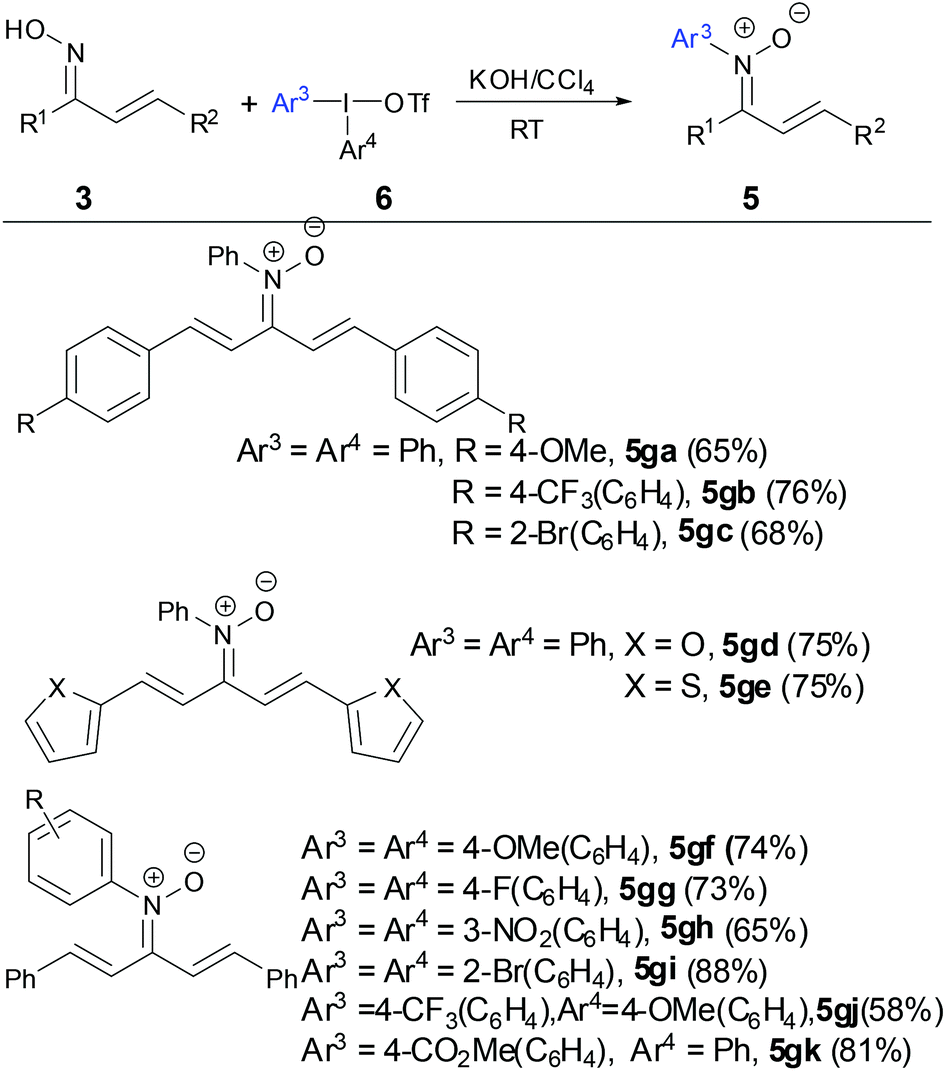
In order to resolve the aforementioned drawbacks of requirement of copper salts and synthesis of ortho-substituted N-aryl α,β-unsaturated ketonitrones, a simple and efficient transition-metal free method to prepare N-aryl α,β-unsaturated ketonitrones was developed by Mo and coworkers in 2015 (Scheme 3).28 The reaction showed good functional group tolerance for both electron-rich and electron-deficient substituents on both oximes and diaryliodonium salts. When unsymmetrical diaryliodonium salts were tested, the N-arylation reaction proceeded with high chemoselectivity and electron-deficient aryl moieties were preferentially transferred to the nitrones. The nitrones can be easily prepared on a gram scale in good yields. A DFT calculation revealed a rational formation of nitrone via O-attack diaryliodonium salts and [1,3]-phenyl migration of the O-coordinated oximate complex.
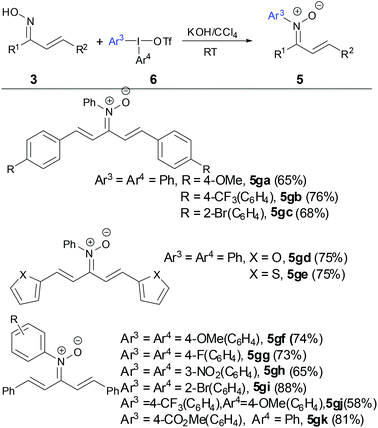 |
| | Scheme 3 Synthesis of N-aryl ketonitrones by diaryliodonium salts. | |
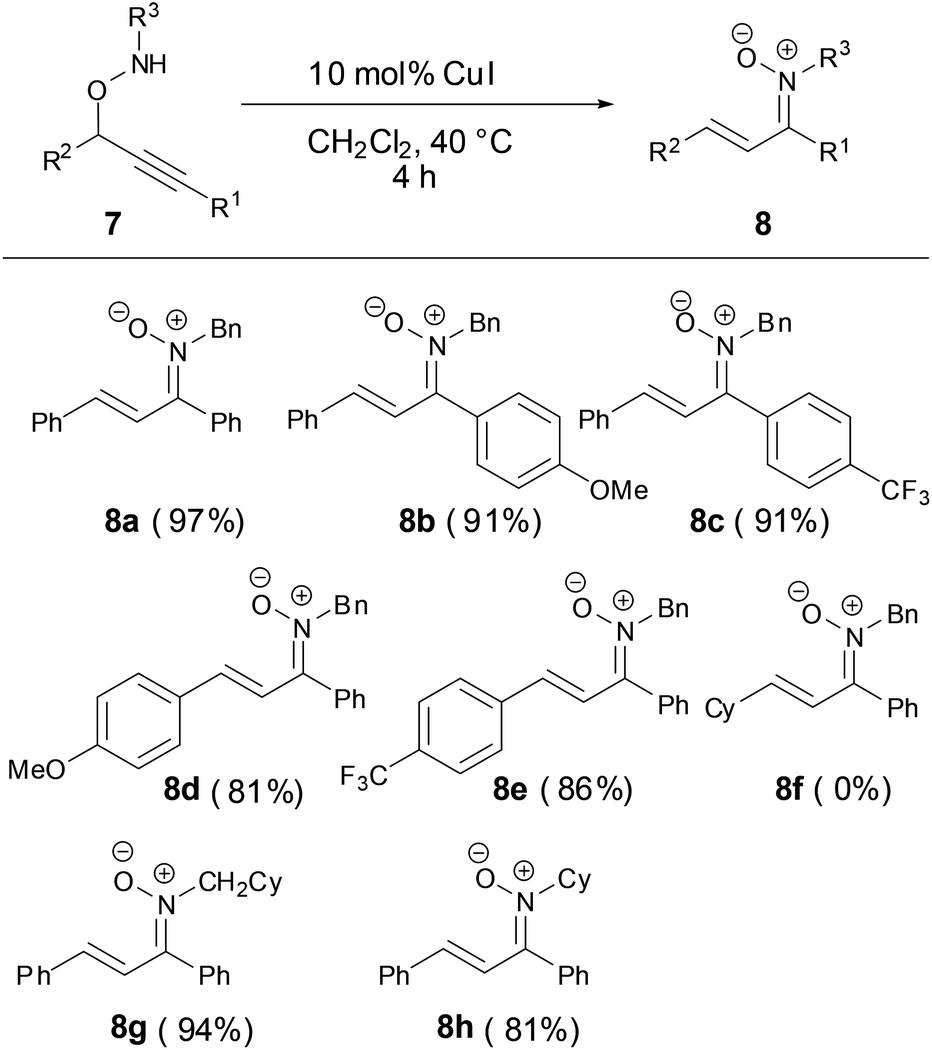
In 2014, a Cu-catalyzed rearrangement of propargyloxyamines to synthesize the N-alkyl α,β-unsaturated ketonitrones with excellent yields was developed by Nakamura and coworkers (Scheme 4).29 The R1 and R2 substituted groups in propargyloxyamines 7 tolerated various aryl substituted groups. However, the reaction failed when the R2 group was an alkyl substituent in substrate 7f.
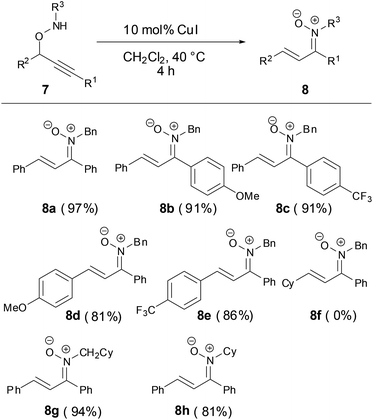 |
| | Scheme 4 Synthesis of N-alkyl ketonitrones by copper-catalyzed reaction. | |
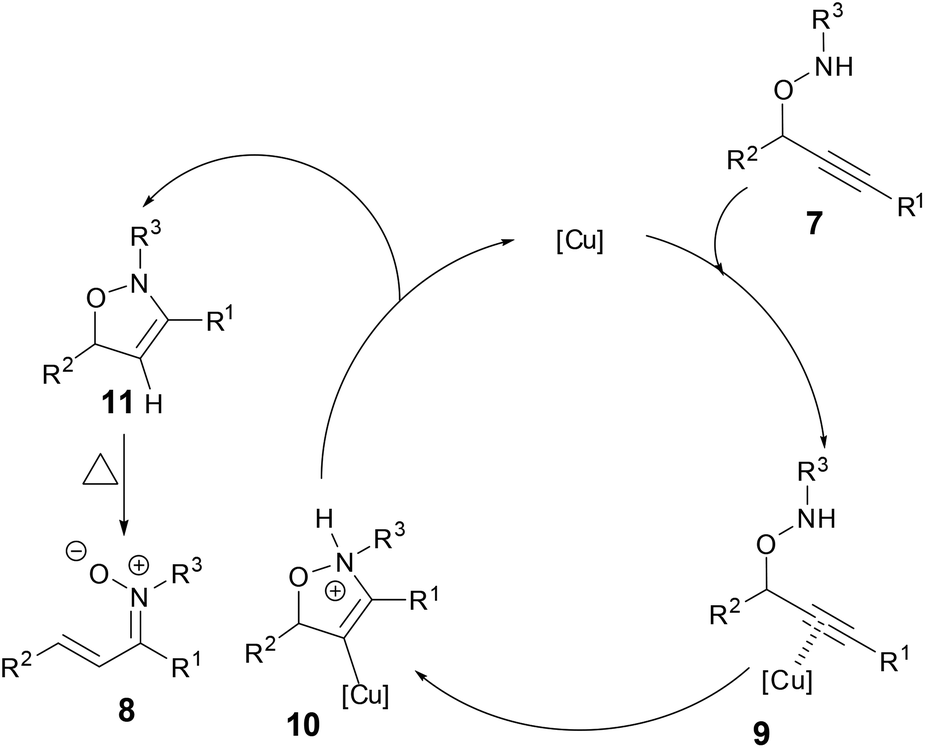
A plausible mechanism was proposed based on the isolation of the intermediate 11 (Scheme 5). The π-acidic Cu catalyst coordinated to the alkyne 7 to form intermediate 9. Nucleophilic attack of the nitrogen atom onto the electrophilically activated C–C triple bond occurred in a 5-endo manner, leading to the cyclized Cu-vinyl intermediate 10. Subsequent protonolysis of the Cu-vinyl intermediate afforded 3-isoxazoline 11 while regenerating a Cu catalyst. An aryl substituent at the propargylic position would promote the electrocyclic ring opening of 11 to provide N-alkyl α,β-unsaturated ketonitrones 8.
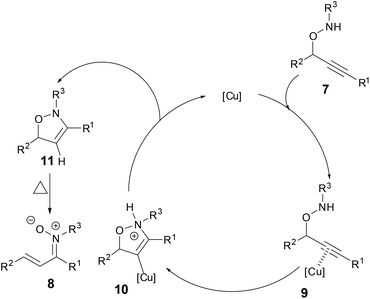 |
| | Scheme 5 A plausible mechanism of formation of N-alkyl α,β-unsaturated ketonitrones. | |
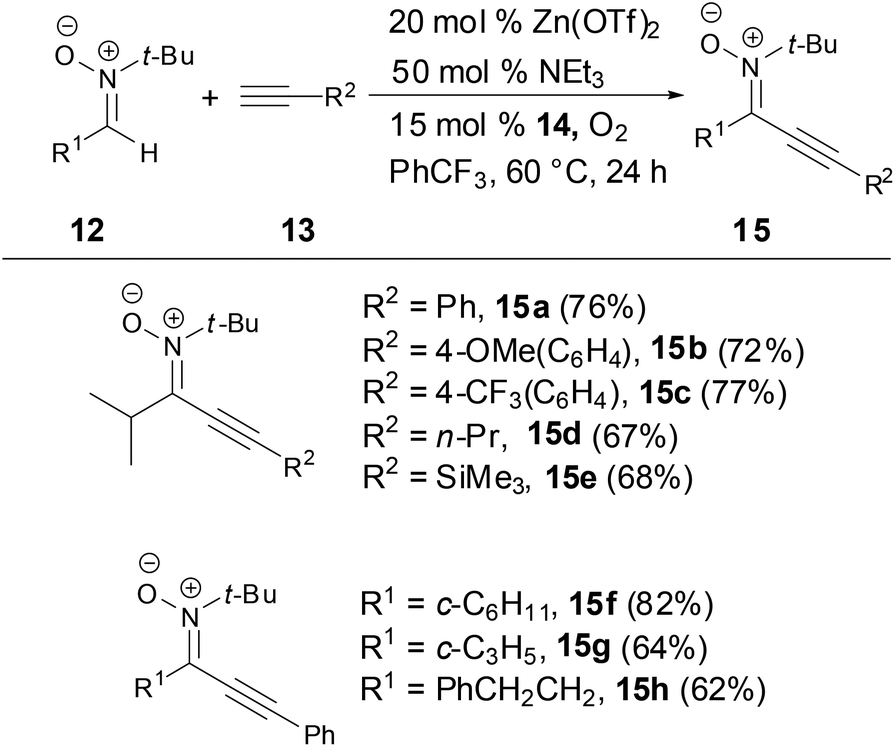
Recently, Studer and coworkers developed a mild, simple zinc triflate catalyzed cross-dehydrogenative coupling (CDC) between aldonitrones and terminal alkynes to synthesize alkynylnitrones in good yields by using cheap, readily available 3,3′-tetra-tert-butyldipheno-quinone (14) and oxygen as oxidants (Scheme 6).30 The nitrones were easily converted to important 3,5-disubstituted isoxazoles.
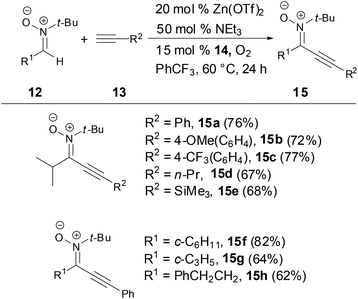 |
| | Scheme 6 Synthesis of N-alkyl alkynylnitrones. | |
1.1.2 Preparation of type III nitrones.
N-Vinyl nitrones are unusual compounds that have rarely been targeted as synthetic intermediates due to the challenges for their preparation by traditional methods. Most previous examples about N-vinyl nitrones were reported as side products or proposed intermediates.31 No general practical synthesis of N-vinyl nitrones has been reported. In 2006, Denmark and Montgomery revealed that N-vinyl aldonitrones could be prepared from nitroalkenes and aldehydes in four steps with good yields.19 The Sammakia group developed a two-step method to synthesize the N-vinyl aldonitrones via condensation of an α-chloroaldehyde with a substituted benzylhydroxylamine and sequence of a 1,4-elimination.32 However, it is not suitable for synthesis of N-vinyl ketonitrones.
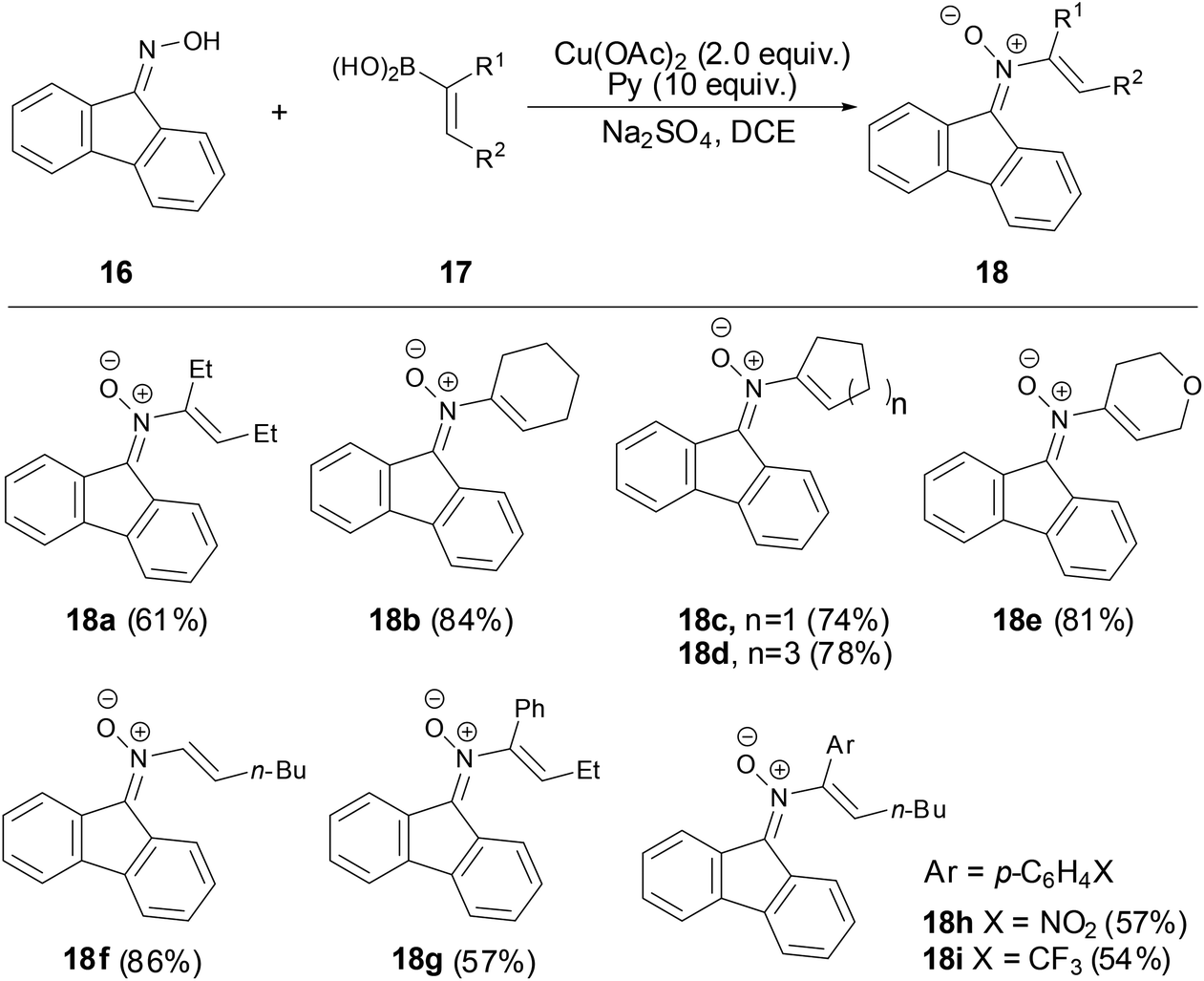
In 2012, Anderson and coworkers developed an efficient and simple methodology to prepare N-vinyl ketonitrones via a copper-mediated cross-coupling reaction of 9-fluorenone oxime with various vinylboronic acids.33 The exact structure of N-vinyl ketonitrone 18 was determined by X-ray crystallography. A variety of vinylboronic acids were applied in preparing N-vinyl nitrones with moderate to good yields (54%–86%). As shown in Scheme 7, the best results were obtained using cyclic vinyl boronic acids and trans-monosubstituted vinylboronic acids. This reaction provided a practical method for the synthesis of N-vinyl ketonitrones which allowed further facile investigation of N-vinyl ketonitrones.
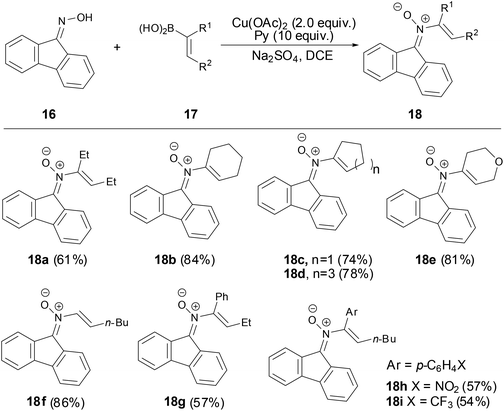 |
| | Scheme 7 Synthesis of N-vinyl ketonitrones by copper-mediated reaction. | |
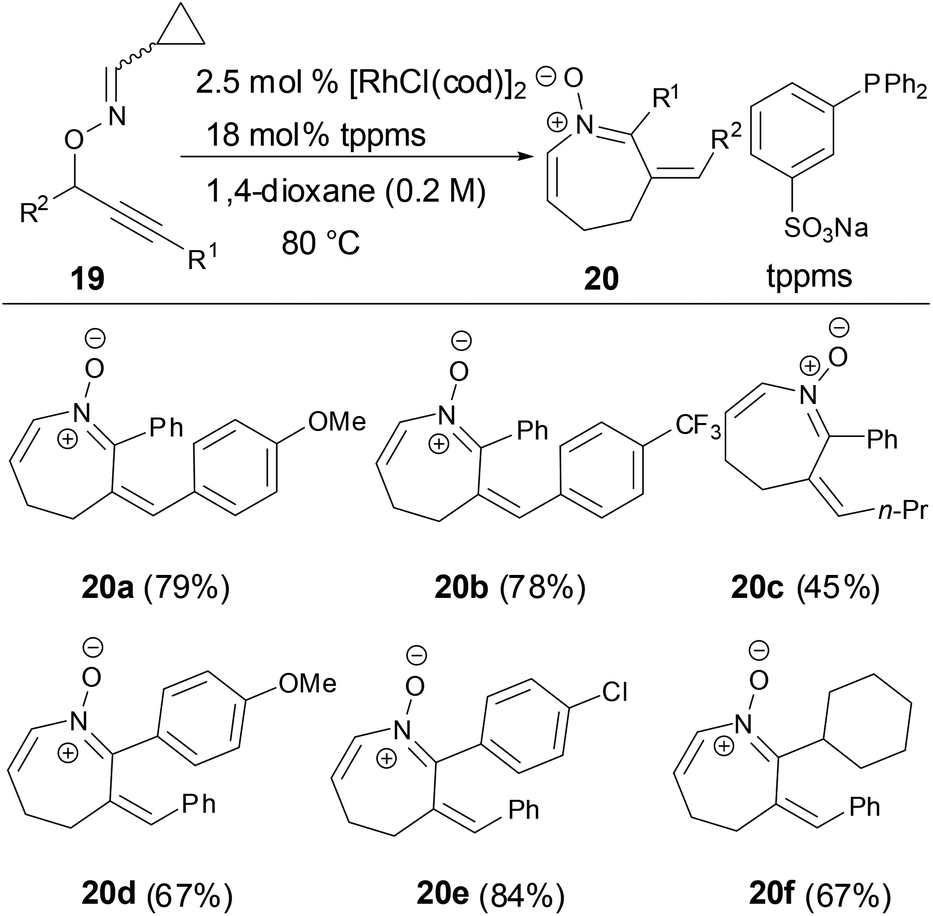
Nakamura and coworkers reported a reaction of O-propargylic cyclopropyl cabaldoximes in the presence of rhodium catalysts to provide azepine N-vinyl nitrones from good to high yields through a tandem 2,3-rearrangement and heterocyclization (Scheme 8).34 The scope of substrates tolerated different R1 and R2 substituents in substrate 19. The geometry of the imines has no effect on the formation of azepine N-vinyl nitrones.
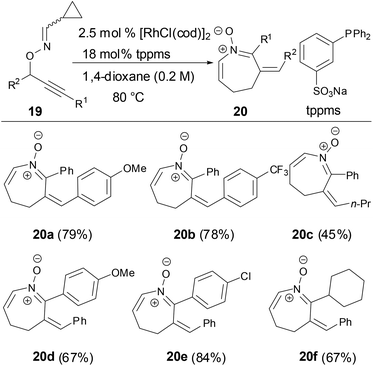 |
| | Scheme 8 Synthesis of N-vinyl ketonitrones by Rh-catalyzed reaction. | |
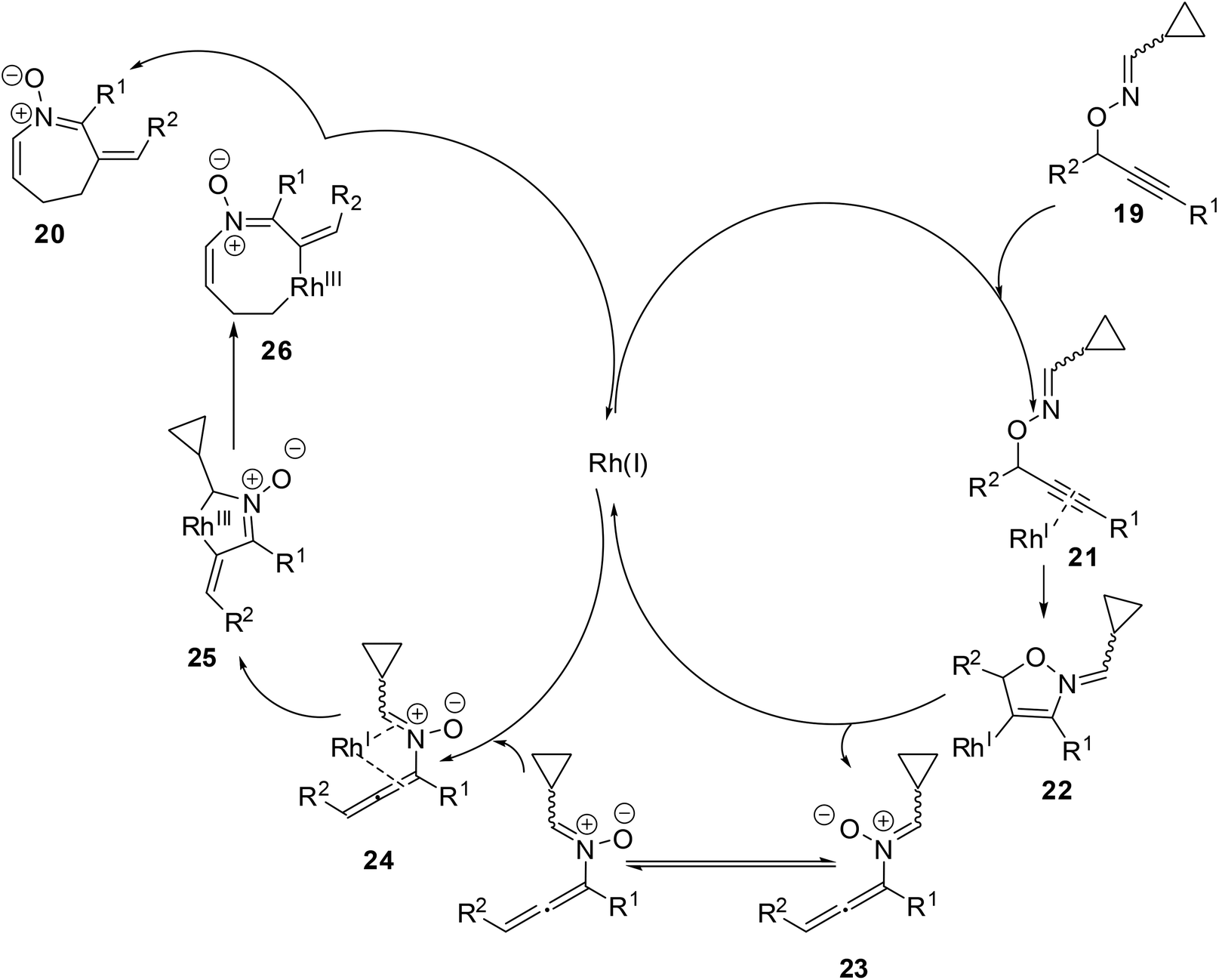
A plausible mechanism was proposed for the formation of product 16 from O-propargylic cyclopropyl cabaldoximes 19 (Scheme 9). The Rh catalyst coordinated to the triple bond of alkyne 20 to form π-complex 21. Nucleophilic attack by the nitrogen atom on the activated π-complex 21 would lead to five-membered cyclic vinylrhodium intermediate 22. Cleavage of the carbon–oxygen bond and elimination of the Rh-catalyst afforded N-allenylnitrone intermediate 23. The formation of the s-cis isomer of N-allenylnitrone allows the η4-coordination to the Rh-catalyst from the opposite side of R2, as shown in the intermediate 24. The formation of aza-rhodacycle 25 and ring expansion involving selective cleavage of the less hindered carbon–carbon bond would give eight-membered aza-rhodacycle 26. Finally, the reductive elimination of the Rh-catalyst produced azepine N-vinyl nitrone 20.
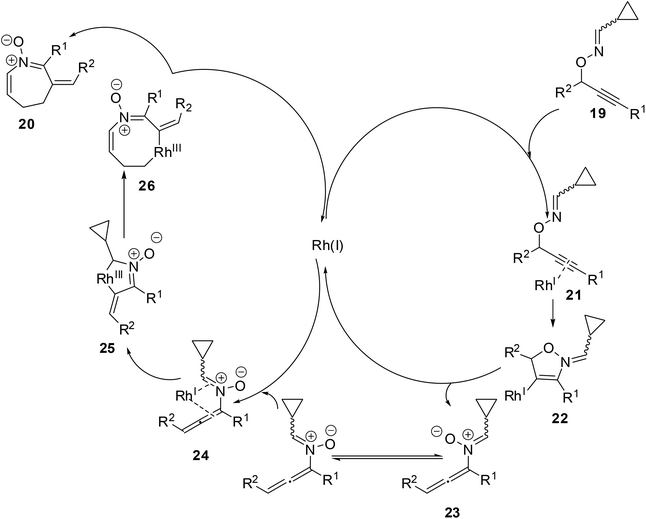 |
| | Scheme 9 A plausible mechanism of formation of azepine N-vinyl nitrone 20. | |
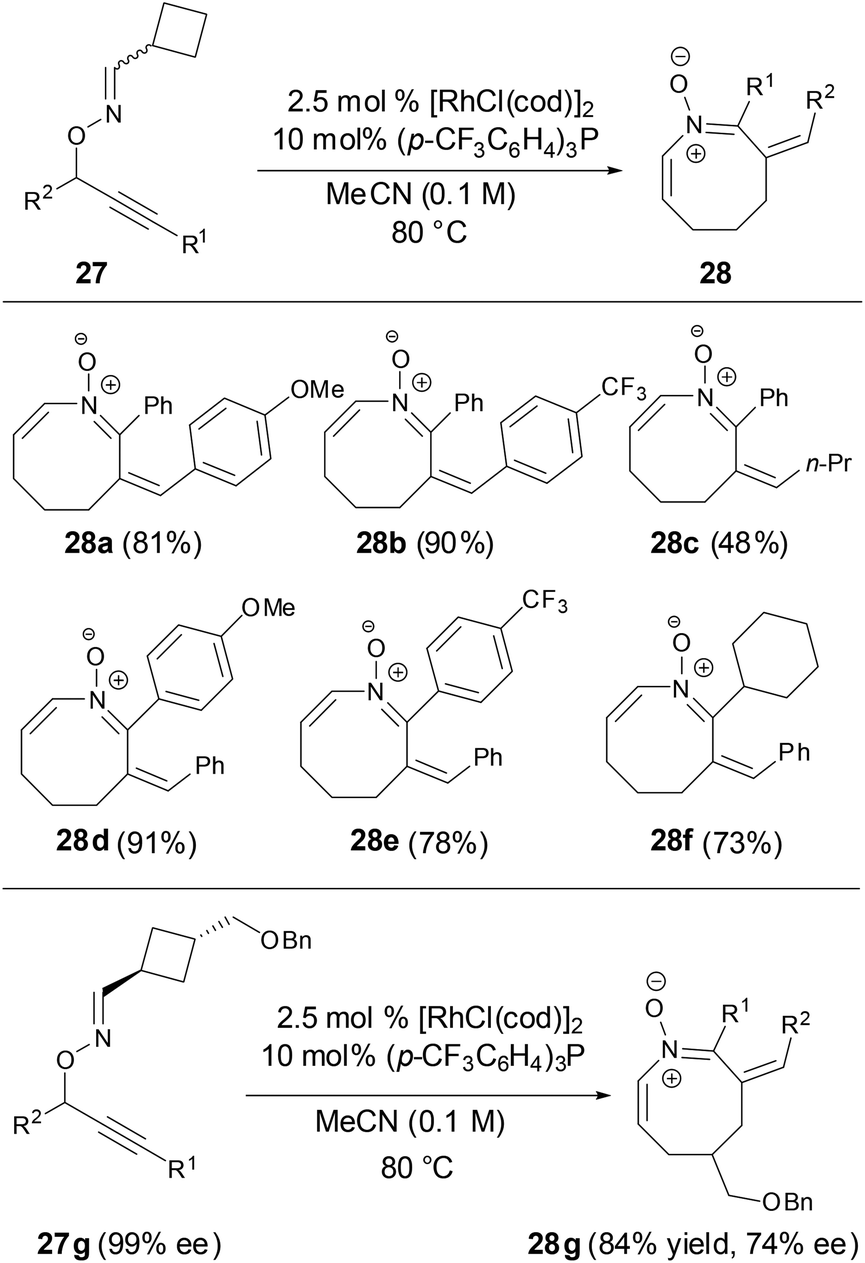
More recently, Nakamura and coworkers revealed that azocine derivatives can be prepared from O-propargylic oximes via a Rh-catalyzed 2,3-rearrangement and heterocyclization cascade reaction (Scheme 10).35 More importantly, the chirality of the substrate was maintained throughout the cascade process to provide the corresponding optically active azocines.
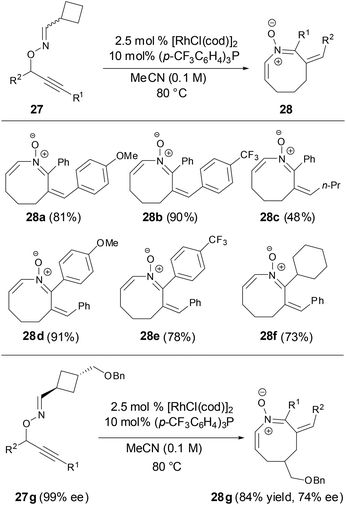 |
| | Scheme 10 Synthesis of azocines by Rh-catalyzed reaction. | |
1.2 The new rearrangement of ketonitrones
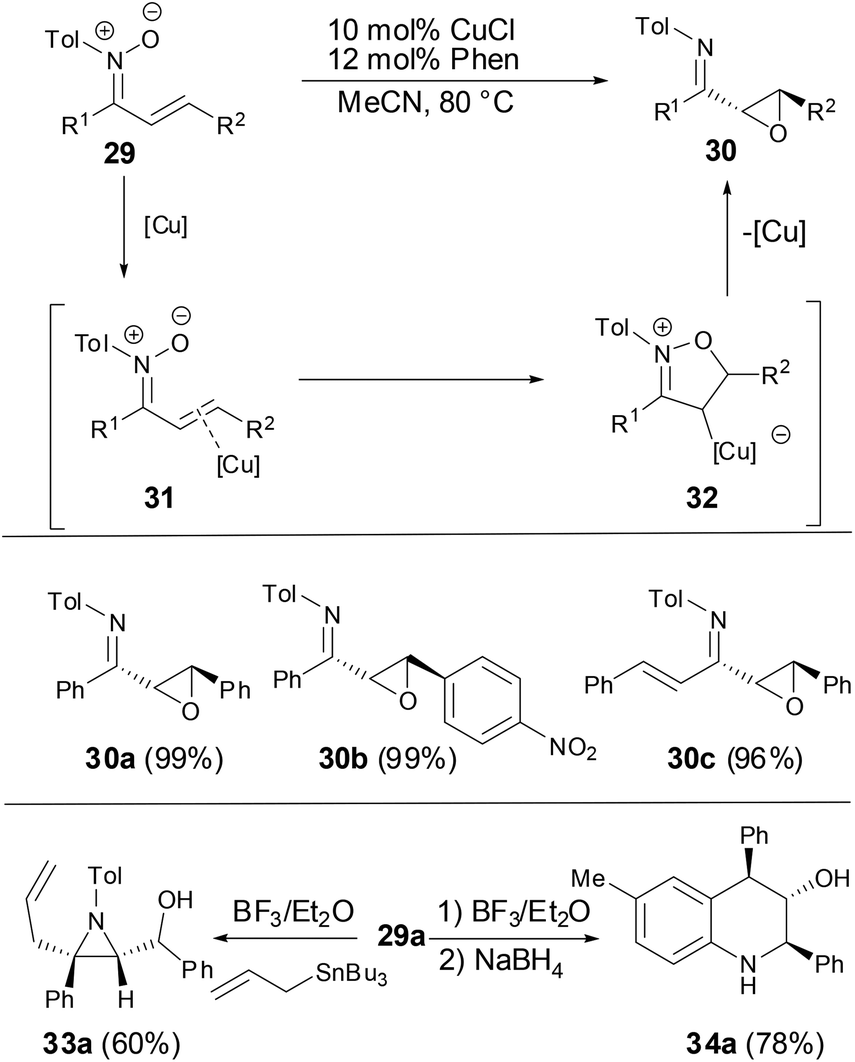
Recently, Anderson and coworkers found that the rearrangement of N-aryl α,β-unsaturated nitrones can form epoxyketimines in the presence of catalytic amounts of CuCl and 1,10-phenanthroline (Phen) (Scheme 11).27 The reaction proceeded via a nucleophilic attack of the nitrone oxygen atom on the electrophilically activated carbon–carbon double bond. Subsequent N–O bond cleavage of 31 resulting in cyclized intermediate 32 took place via an oxygen-atom transfer reaction to form α,β-epoxyketimines 30 in high yields and excellent diastereoselectivity. Treatment of epoxyketimine 30a with BF3/Et2O followed by reduction with NaBH4 efficiently afforded tetrahydroqinolines 34a in 78% yield as a single diastereomer. Exposure of epoxyketimine 30a to an allylic tin reagent in the presence of BF3/Et2O provided trisubstituted aziridine 33a in 60% yield as a single diastereomer.
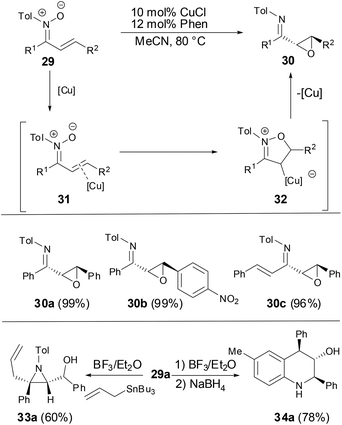 |
| | Scheme 11 Copper-catalyzed oxygen-atom transfer reaction. | |
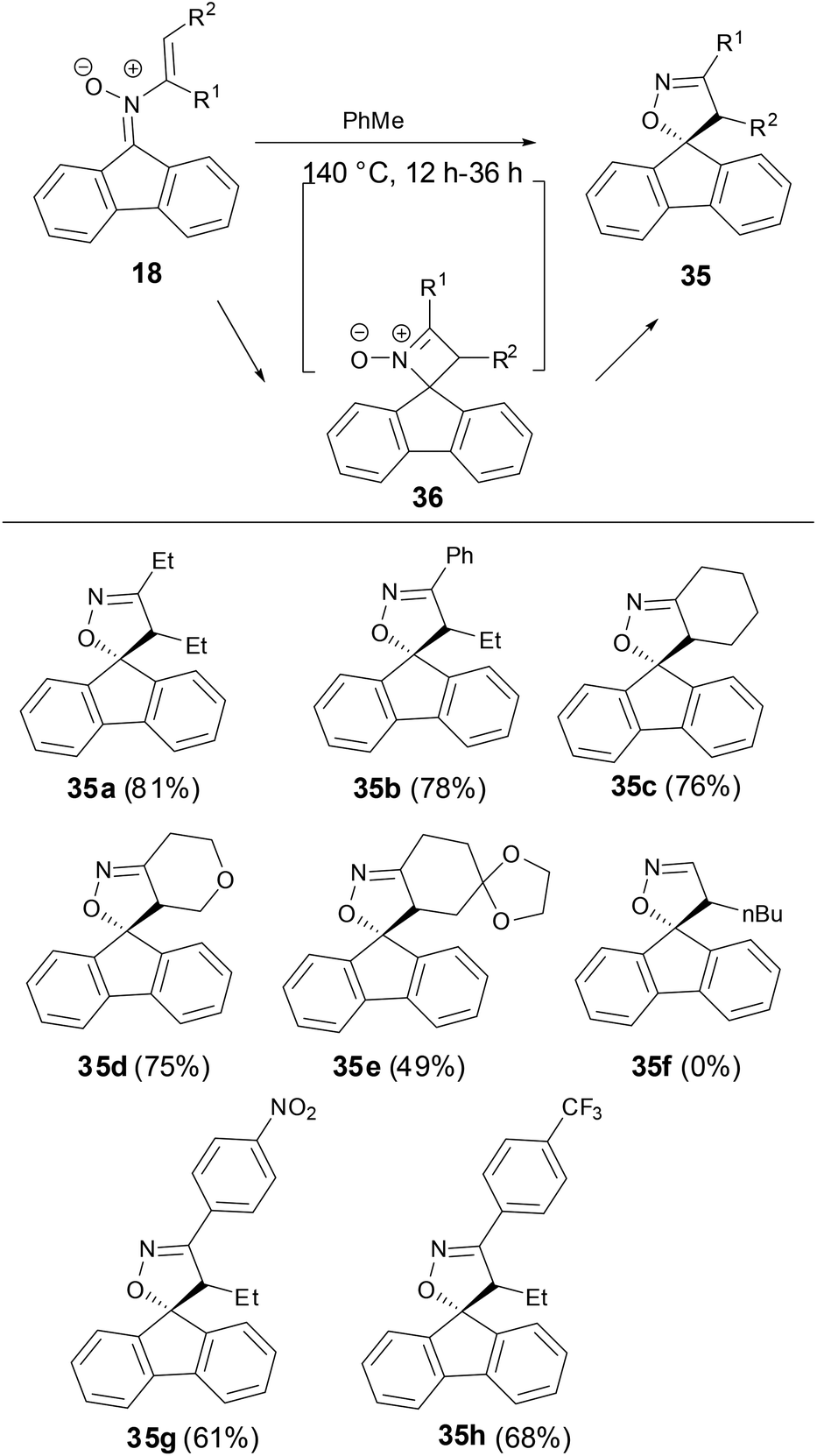
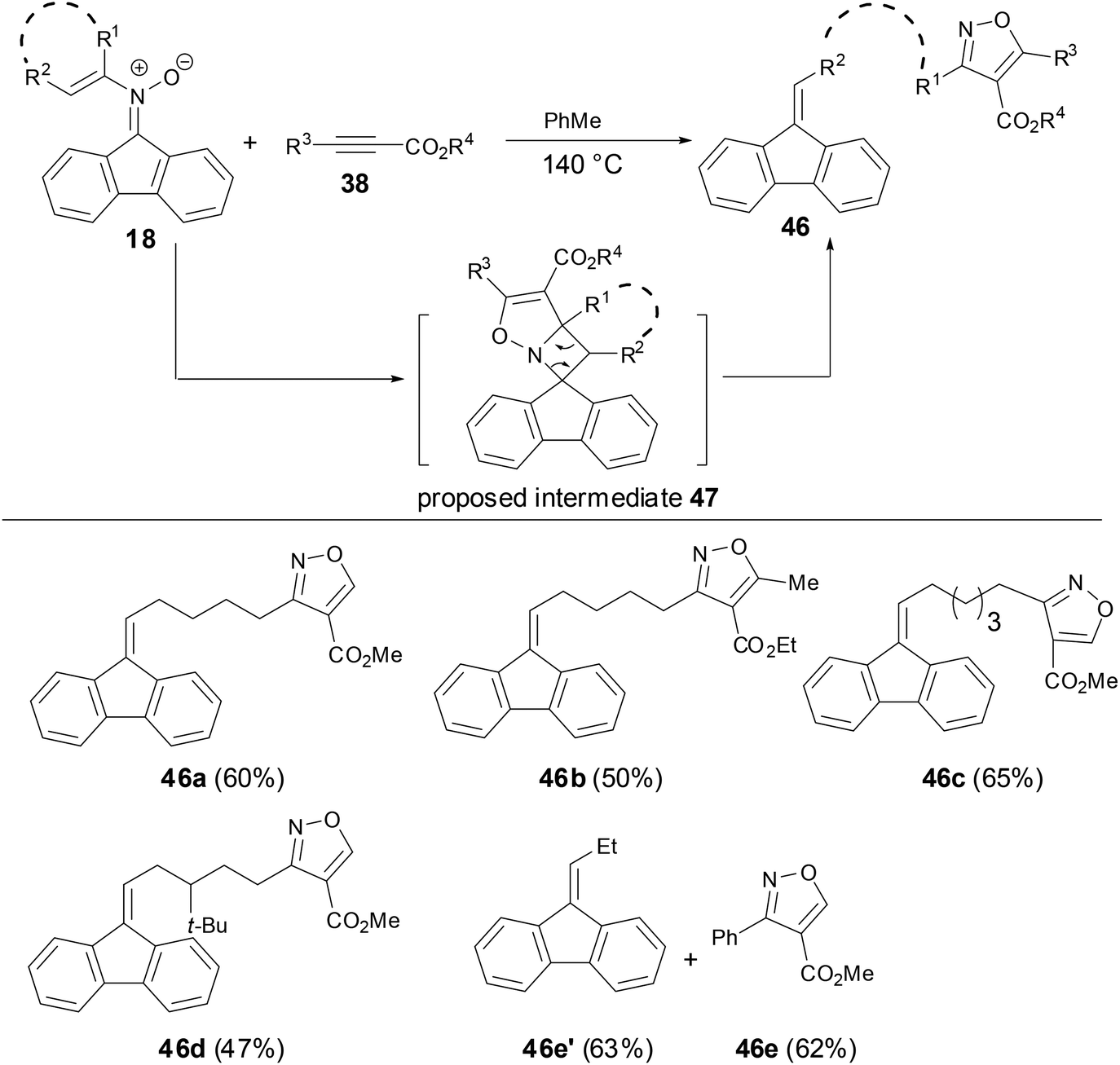
A thermal rearrangement of fluorenone N-vinyl nitrones 18 to provide spiroisoxazolines 35 has been observed by Anderson and coworkers (Scheme 12).33 The reaction proceeded well above 120 °C, however, no conversion of the N-vinyl nitrone to spiroisoxazoline was observed below 100 °C and only the starting material was recovered. The scope of this rearrangement was broad with both disubstituted and cyclic vinyl nitrones tolerated as substrates. However, monosubstituted N-vinyl nitrone decomposed when the reaction ran above 120 °C. Although there was no definite evidence for the mechanism of this transformation now, the authors proposed an optional intermediate 36 by a 4π electron cyclization or perhaps from the isomerization of nitrone to oxaziridines.
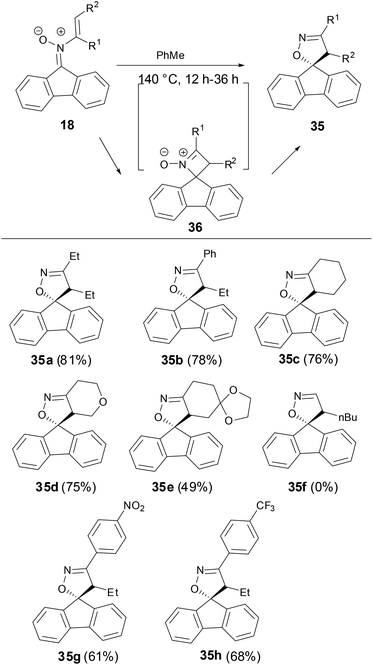 |
| | Scheme 12 Thermal rearrangement of N-vinyl nitrones. | |
1.3 The reaction of ketonitrones with alkynes
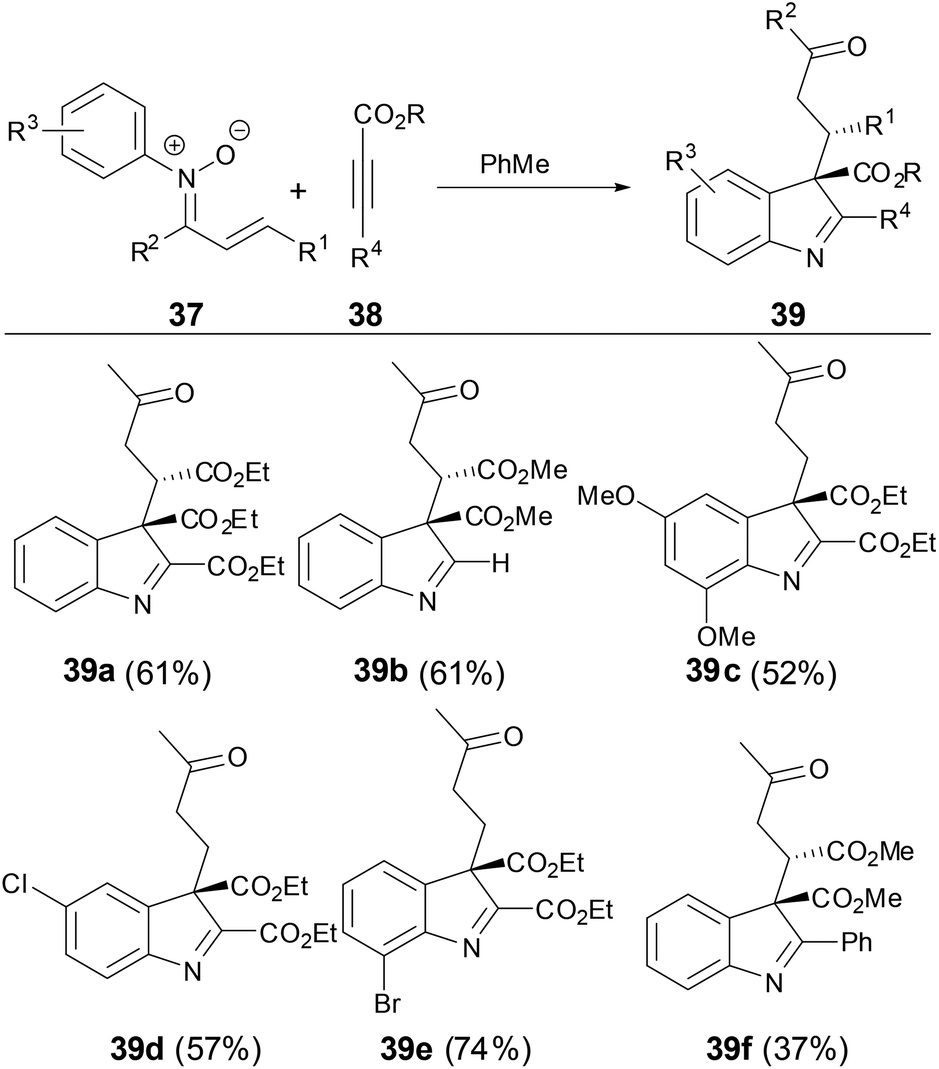
In 2012, Yang and coworkers36 found that when N-aryl α,β-unsaturated ketonitrones were treated with activated alkynes, C3-quaternary indolenines were obtained with concomitant formation of two adjacent quaternary and tertiary chiral centers in high diastereoselectivity, which are versatile intermediates in the synthesis of indole-containing structural motifs in natural products and pharmaceutical compounds (Scheme 13). This transformation provided an effective approach to synthesis of complex indole-containing compounds. The reaction likely started from the stepwise [5 + 2] cycloaddition of N-aryl α,β-unsaturated ketonitrones with activated alkynes, which further underwent a series of transformations including [3,3]-sigmatropic rearrangement, tautomerization and intra-molecular condensation to afford C3-quaternary indolenine.
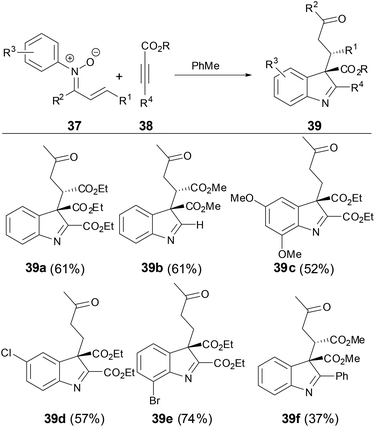 |
| | Scheme 13 [5 + 2] Cycloaddition of N-aryl nitrones with activated alkynes. | |
Based on the above results, in the presence of an appending N-nucleophilic center at the activated alkyne, the initial formation of C3-quaternary indolenines might be followed by spontaneous N-cyclization to form the additional heterocycle of pyrrolindoline in one step.37 However, pyrrolindoline products 42 were isolated with low yields (Scheme 14).
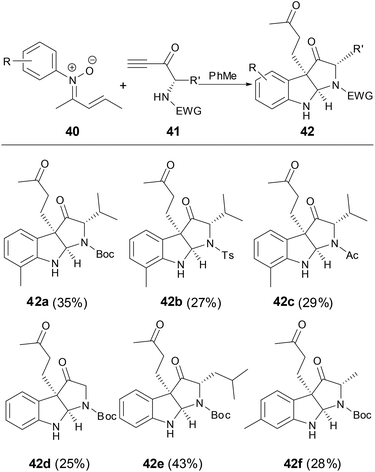 |
| | Scheme 14 [5 + 2] Cycloaddition of N-aryl nitrones with activated N-nucleophilic center alkynes. | |
Recently, Nakamura and coworkers found that N-alkyl α,β-unsaturated ketonitrones underwent [3 + 2] cycloaddition reactions to construct the corresponding 2-isoxazoline in excellent yields under mild conditions when treated with electron-deficient alkynes (Scheme 15).29
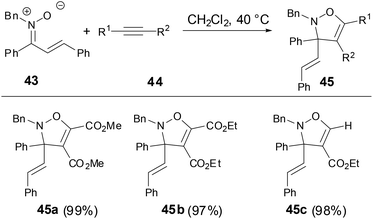 |
| | Scheme 15 [3 + 2] Cycloaddition of N-alkyl nitrones with activated alkynes. | |
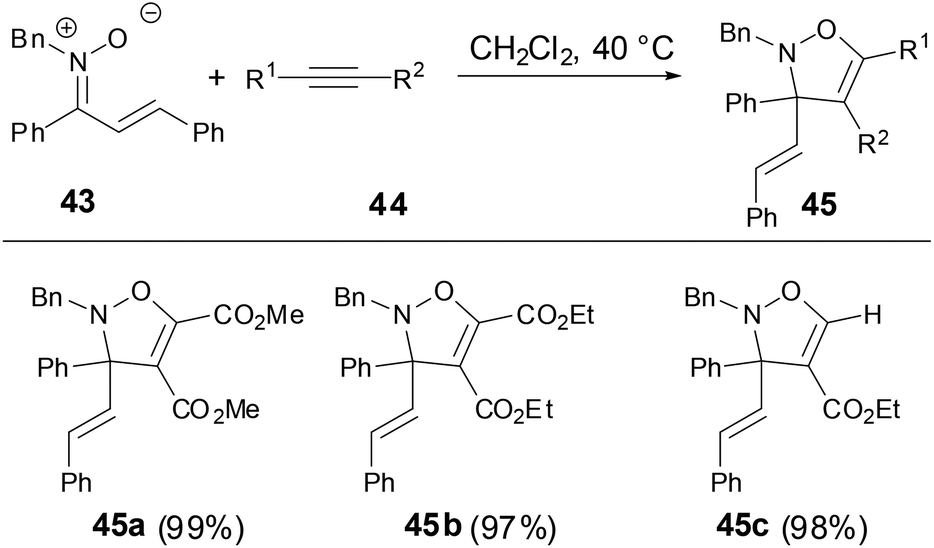
Anderson and coworkers have shown that during the studies of thermal rearrangements of fluorenone N-vinyl nitrones 18, 18 provided fluorine-tethered isoxazoles 46 in good yields on treatment with electron-deficient alkynes to test for [3 + 2] cycloaddition reactivity of N-vinyl nitrones (Scheme 16).33 Both the linear alkenyl and cyclo-alkenyl N-substituent nitrones underwent an analogous transformation with either terminal or internal alkynes 38 to afford alkyne addition and C–C bond cleavage products 46. An alternative mechanism for the preparation of tethered-isoxazoles or mixtures of fluorenes and isoxazoles might involve an initial interaction between the electron-deficient alkyne and the N-vinyl nitrone prior to cycloreversion. This result has revealed that the reactivity of fluorenone-based N-vinyl nitrones differed significantly from the intra-molecular [4 + 2] cycloaddition chemistry and provided a new access to [3 + 2] cycloaddition chemistry.
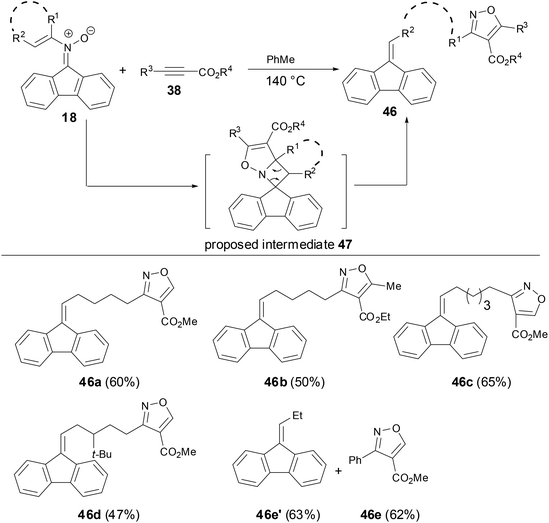 |
| | Scheme 16 Cycloaddition of N-vinyl nitrones with activated alkynes. | |
1.4 The reaction of ketonitrones with allenes
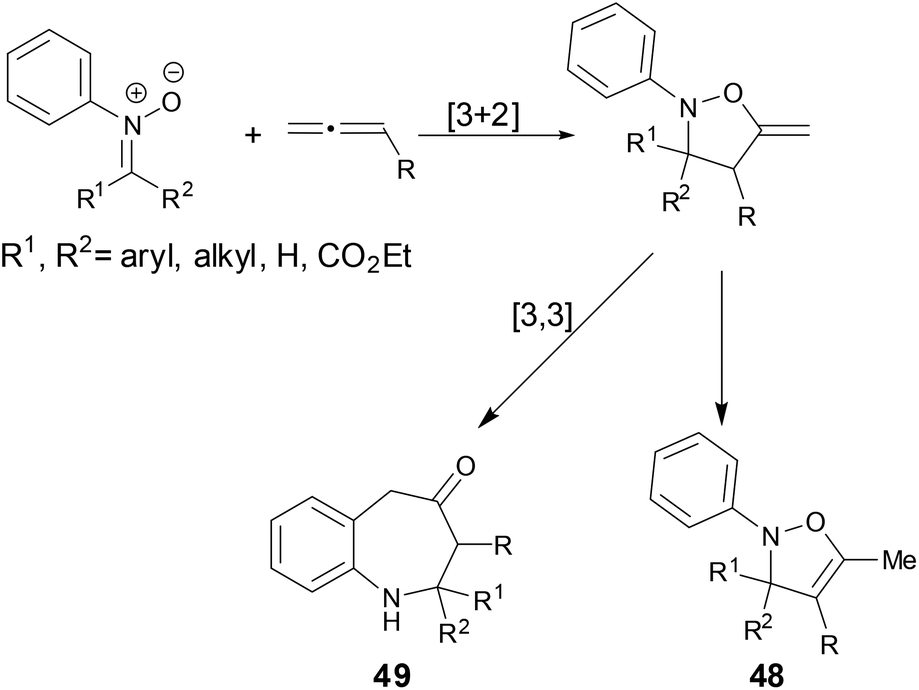
Cycloaddition reactions are powerful methods for the synthesis of cyclic compounds. The cycloaddition reactions of N-aryl nitrones with allenes have been studied in the past.38 In these cases, the [3 + 2] cycloaddition products 48 were obtained or the [3 + 2] cycloaddition intermediates went through a [3,3]-rearrangement to form benzazepine compounds 49 (Scheme 17).
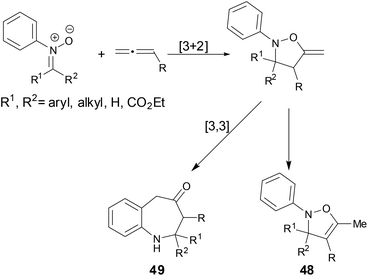 |
| | Scheme 17 Cycloaddition of N-aryl nitrones with allenes. | |
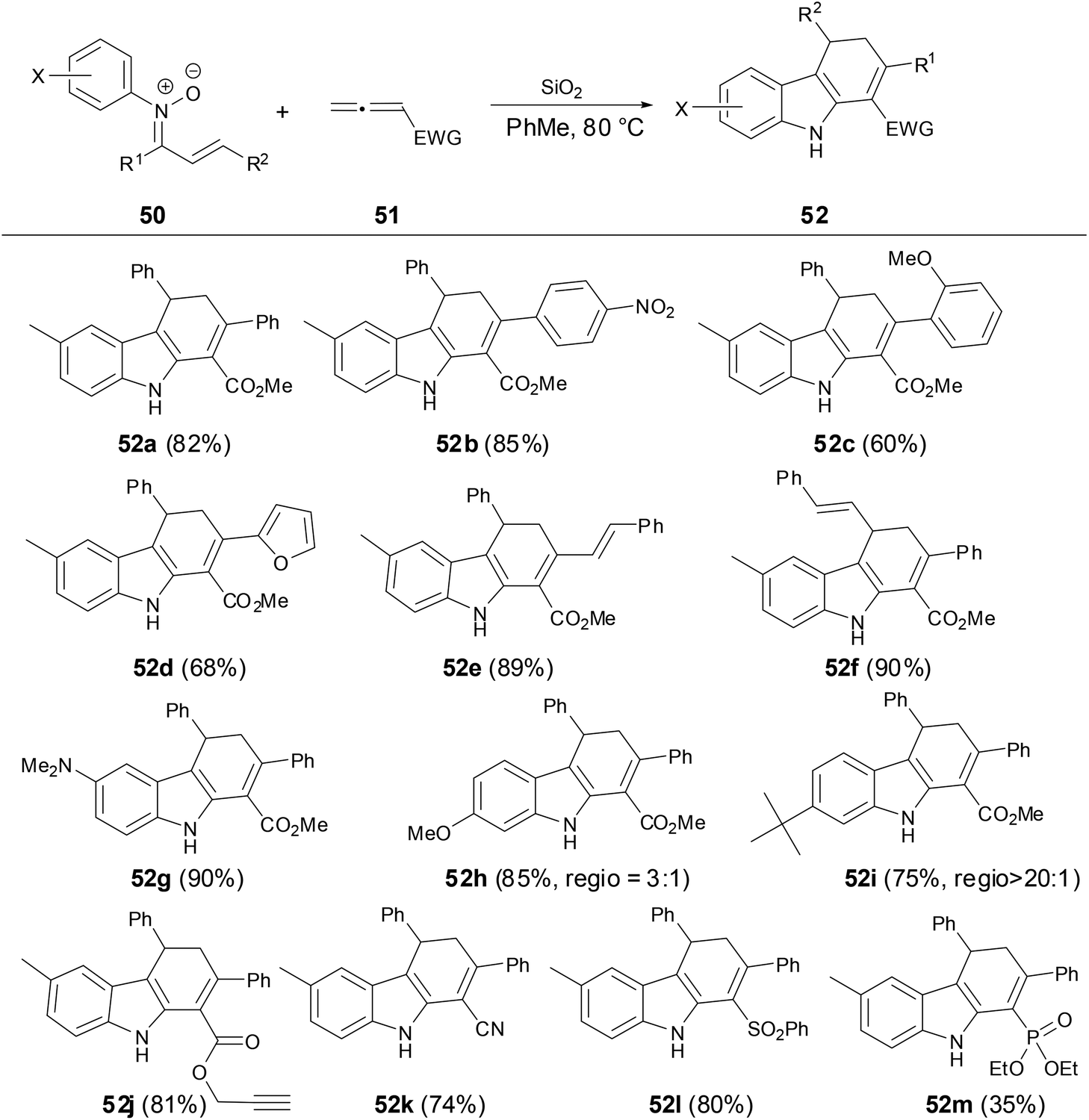
In 2014, Anderson and coworkers reported a new solvent-controlled cascade process of N-aryl α,β-unsaturated nitrones with electron-deficient allenes to prepare dihydrocarbazoles or dihydropyridoindoles (Scheme 18).39 The reactions proceeded smoothly under metal-free conditions in high yields and excellent diastereoselectivity. Firstly, the author found that N-aryl α,β-unsaturated nitrones were treated with allenoates under SiO2 as an additive in toluene, and dihydrocarbazoles were obtained in high yields and high regioselectivity of the C–C bond formation which was controlled by steric hindrance on N-substituents.
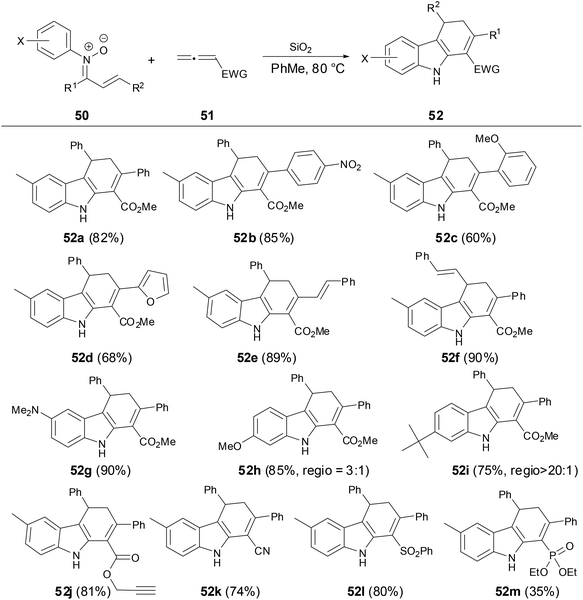 |
| | Scheme 18 Reaction of N-aryl nitrones with allenes in PhMe. | |
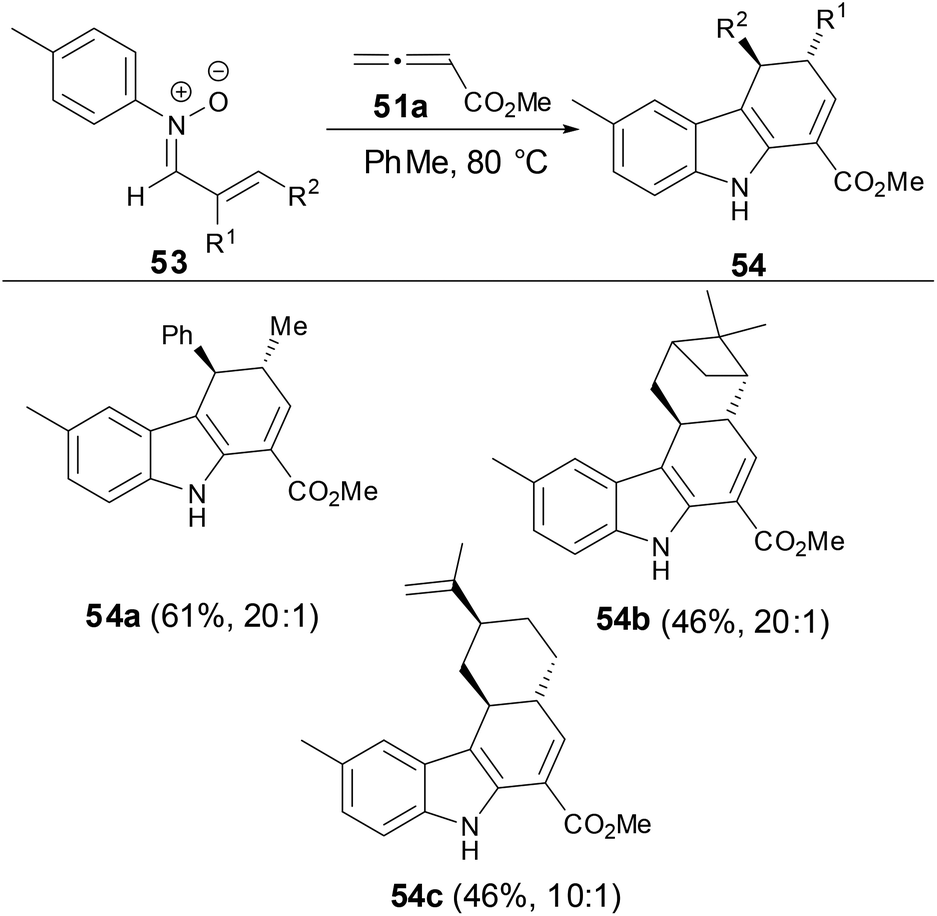
In order to investigate the relative stereoselectivity for preparation of dihydrocarbazoles, the reaction of N-aryl α,β-disubstituted nitrones with allenes was examined. As shown in Scheme 19, the dihydrocarbazoles favored the trans-isomers in moderate yields. These results suggested that the relative stereochemistry of dihydrocarbazole products 54 can be controlled by N-aryl α,β-disubstituted nitrones.
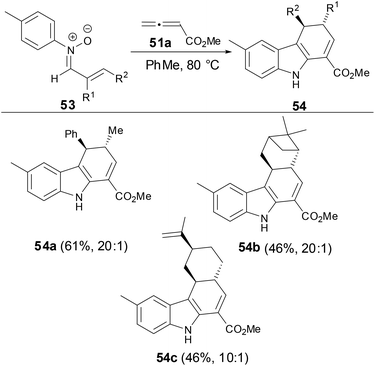 |
| | Scheme 19 The studies of diastereoselectivity on reaction of N-aryl nitrones with allenes. | |
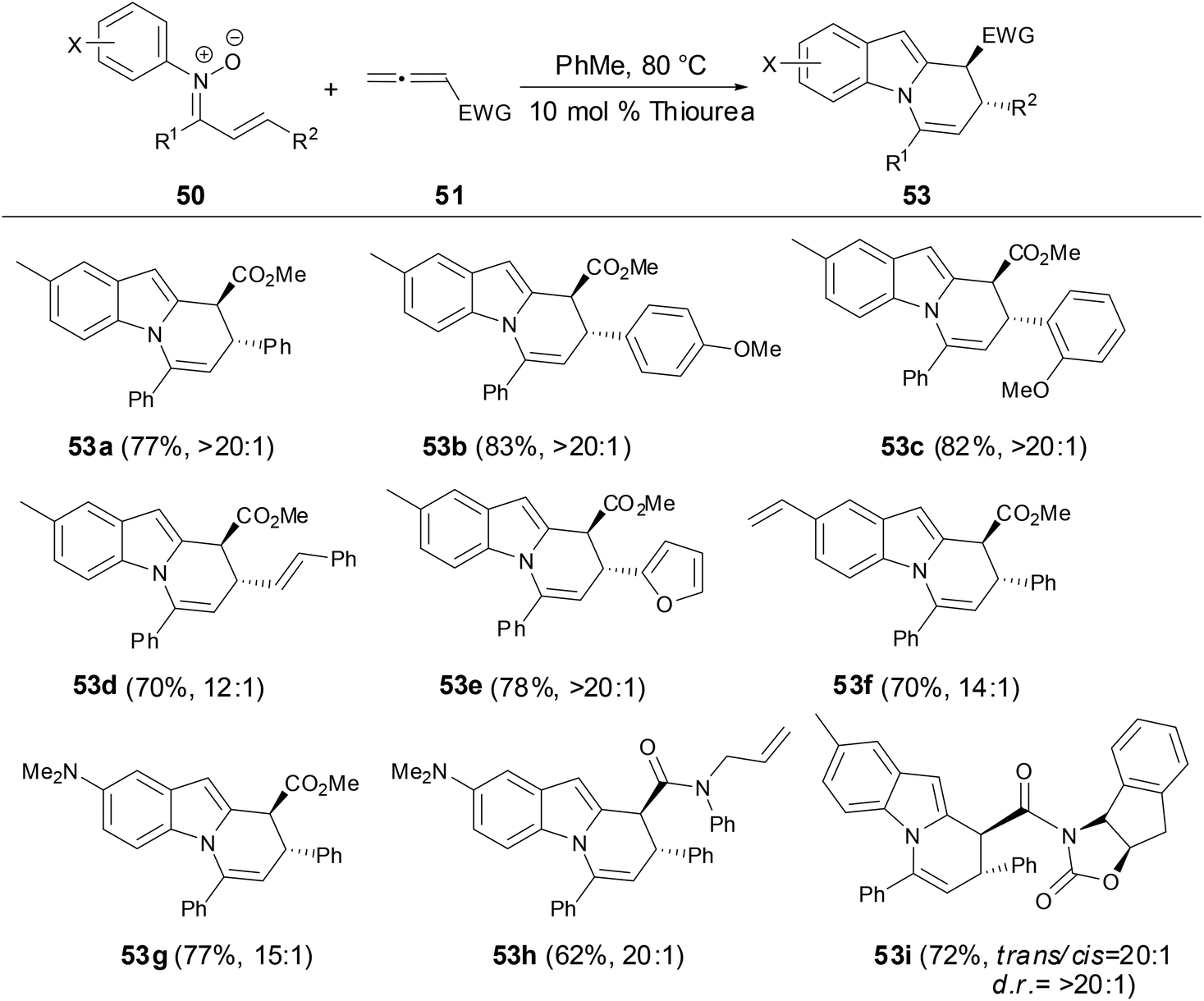
During the screening of solvents, the authors found that the formation of dihydropyridoindole was strongly favored in EtOAc and i-PrOAc. The yields and diastereoselectivity of dihydropyridoindoles were significantly increased by the addition of 10 mol% of thiourea catalyst and molecular sieves. The scope of the cascade process for preparation of dihydropyridoindoles tolerated not only a variety of electron-rich and electron-deficient arene substituent nitrones but also a range of N-aryl substituents (Scheme 20). Moreover, the relative and absolute stereochemistry of dihydropyridoindole formation can be controlled through the cascade process by using oxazolidinone chiral auxiliaries (53i).
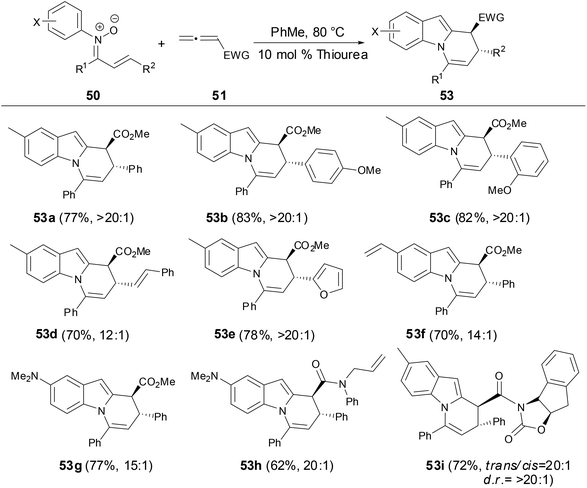 |
| | Scheme 20 Synthesis of dihydropyridoindole by N-aryl nitrones with allenes. | |
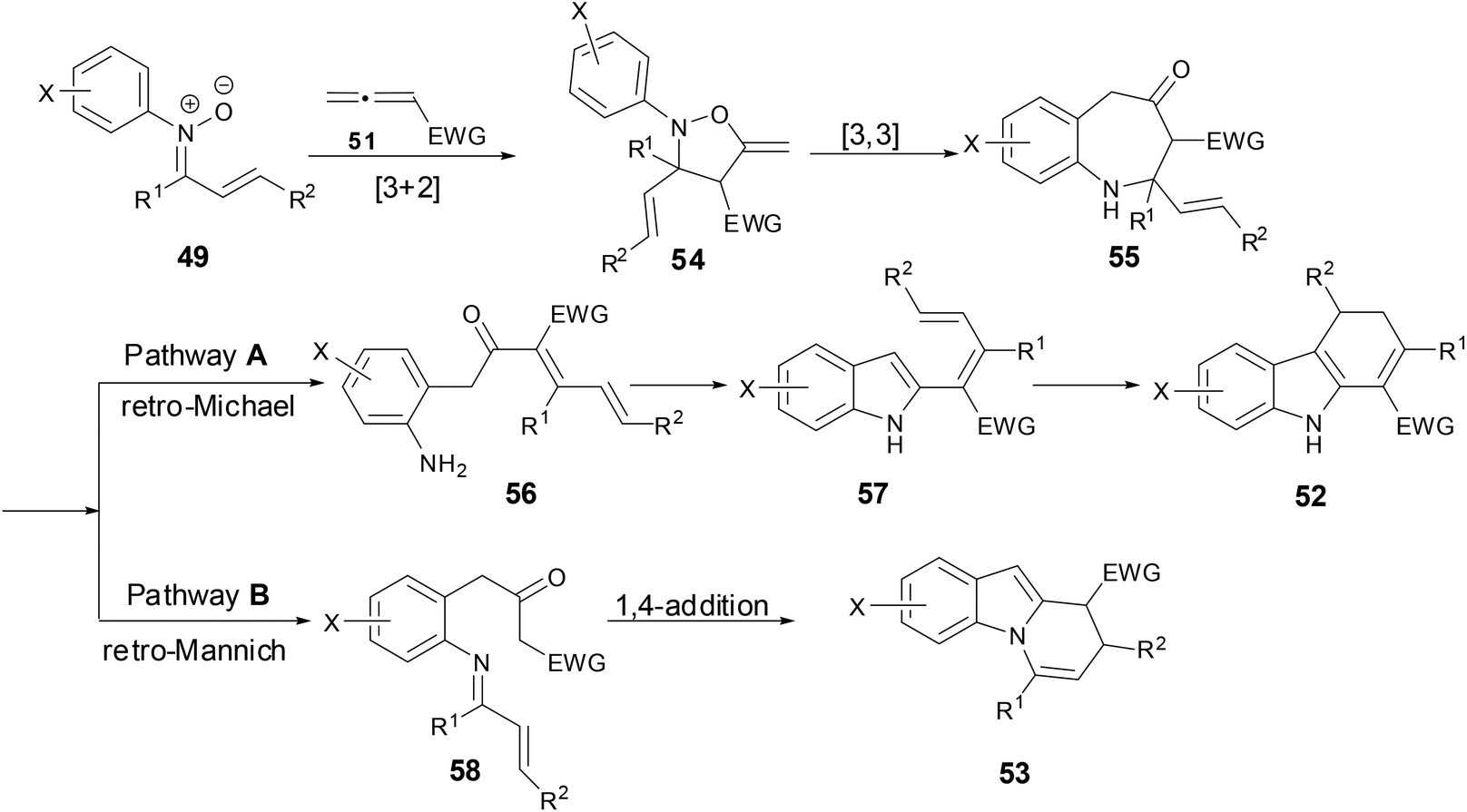
Based on the literature and experimental data, the authors proposed a rational mechanism (Scheme 21). An initial [3 + 2]-cycloaddition of nitrone and allene regioselectively provided exomethylene isoxazolidine 54, which then underwent a [3,3]-rearrangement to give benzazapine 55. For pathway A, a retro-aza-Michael addition followed by intra-molecular condensation of the resulting aniline then afforded dienyl indole 57. The dienyl indole went through a subsequent conjugate addition or 6π-electrocycliczation to provide dihydrocarbazoles after proton transfer. As for pathway B, a retro-Mannich reaction formed imine 58 which then underwent a [4 + 2] cycloaddition or a stepwise 1,4-addition of the β-ketoester enolate to the α,β-unsaturated imine to provide dihydropyridoindole after a subsequent elimination.
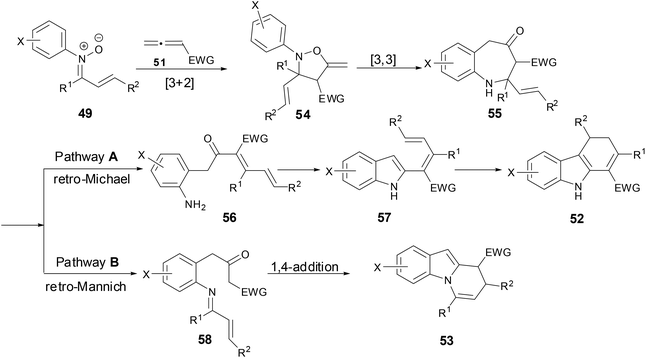 |
| | Scheme 21 A plausible mechanism for reaction of N-aryl nitrones with allenes. | |
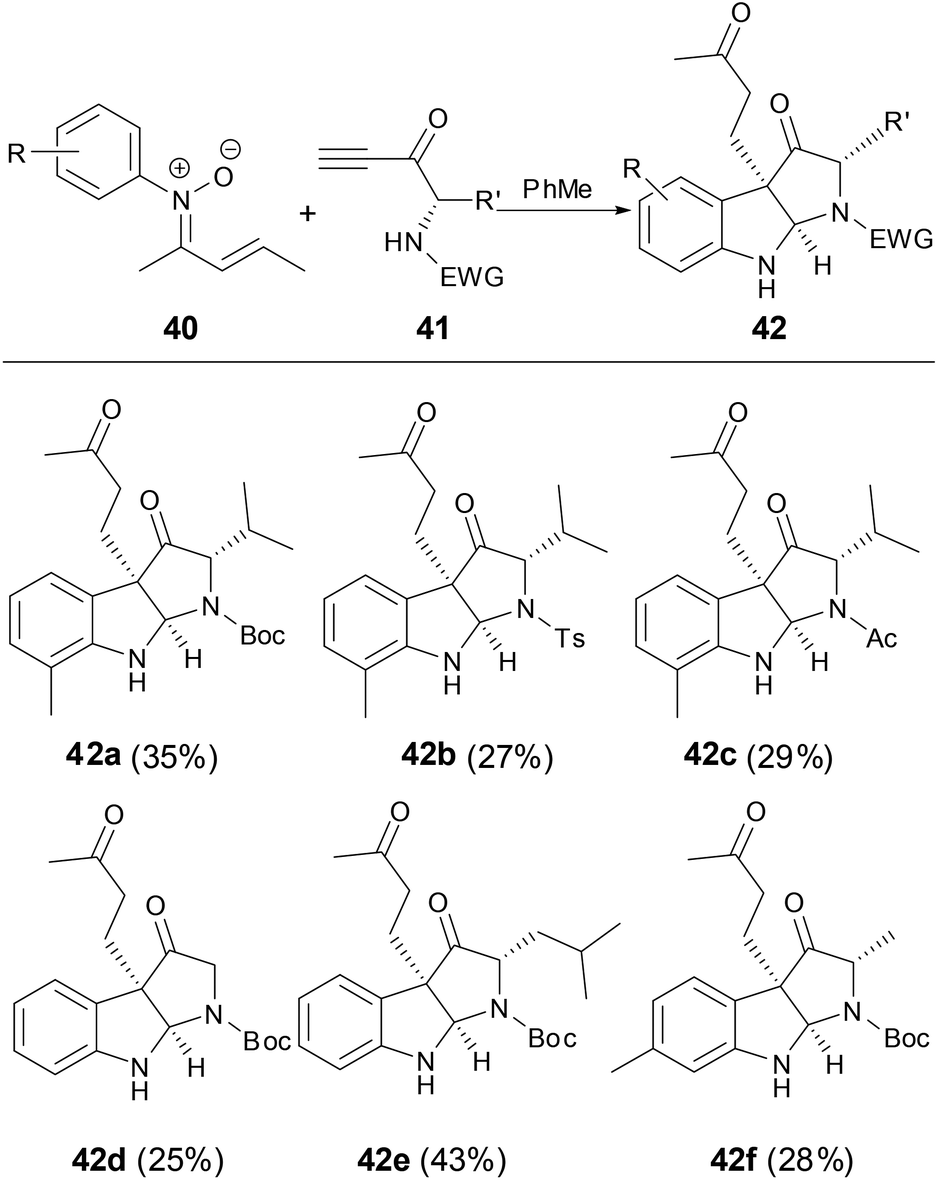
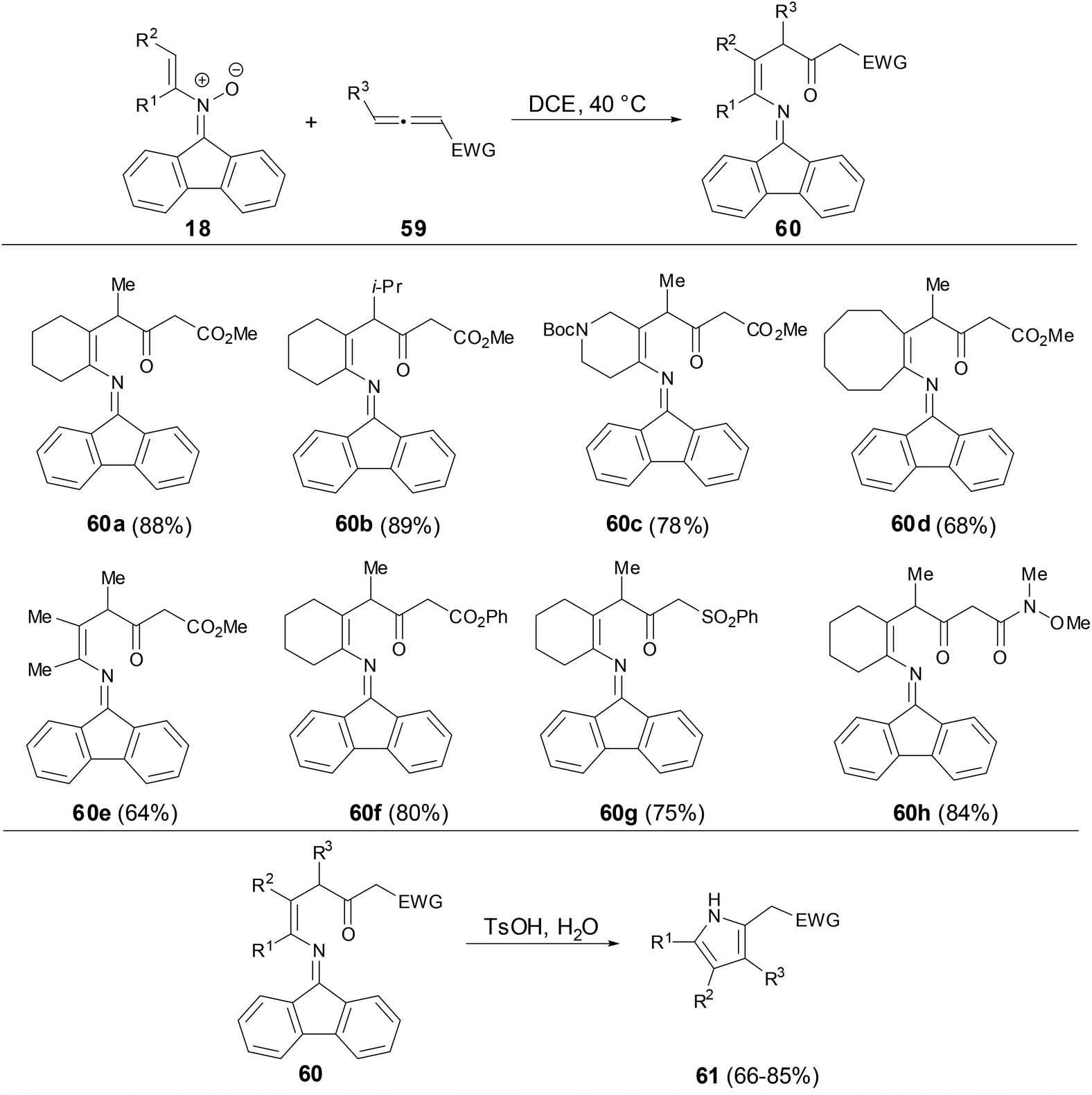
Recently, Anderson and coworkers found that fluorenonylimine protected 1,4-enamino ketones can be prepared through the [3,3]-rearrangement of dialkenylhydroxylamines which were generated from the addition of N-alkenylnitrones to electron-deficient allenes (Scheme 22).40 This reaction particularly well tolerated cycloalkenyl nitrone substituents and electron-deficient allenes. The fluorenonylimine protected 1,4-enamino ketones could be effectively transferred to pyrroles, 1,4-dinones and furans.
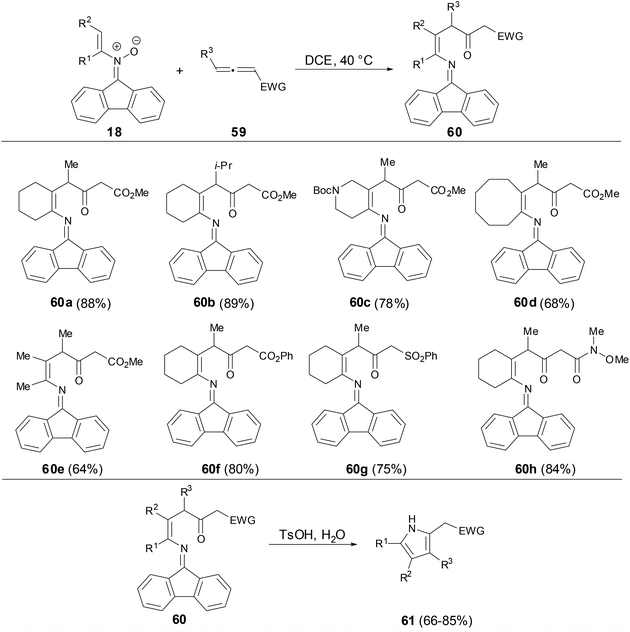 |
| | Scheme 22 Reaction of N-vinyl nitrones with allenes. | |
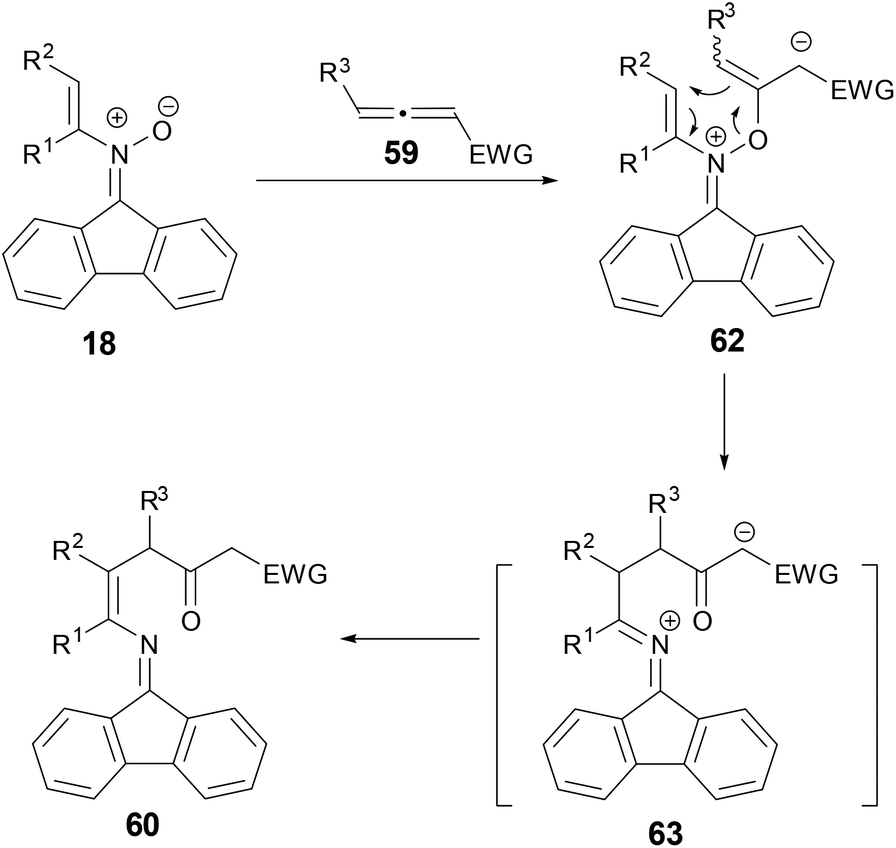
A plausible mechanism was proposed for the conversion of N-alkenylnitrones and allenes to 1,4-enamino ketones by either 1H or 13C NMR spectroscopy monitoring during the conversion of a mixture of 18 and 59 to 60 in DCE-d4 (Scheme 23). Addition of nitrone to electron-deficient allene could form intermediate 61, which could go through a [3,3]-rearrangement to form azallenium 62 and a subsequent proton transfer to form 58.
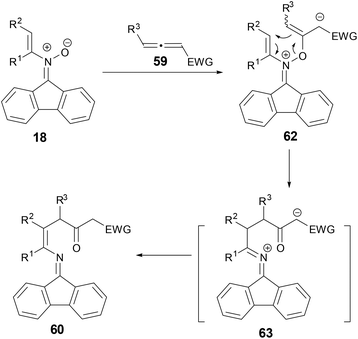 |
| | Scheme 23 Plausible mechanism for reaction of N-vinyl nitrones with allenes. | |
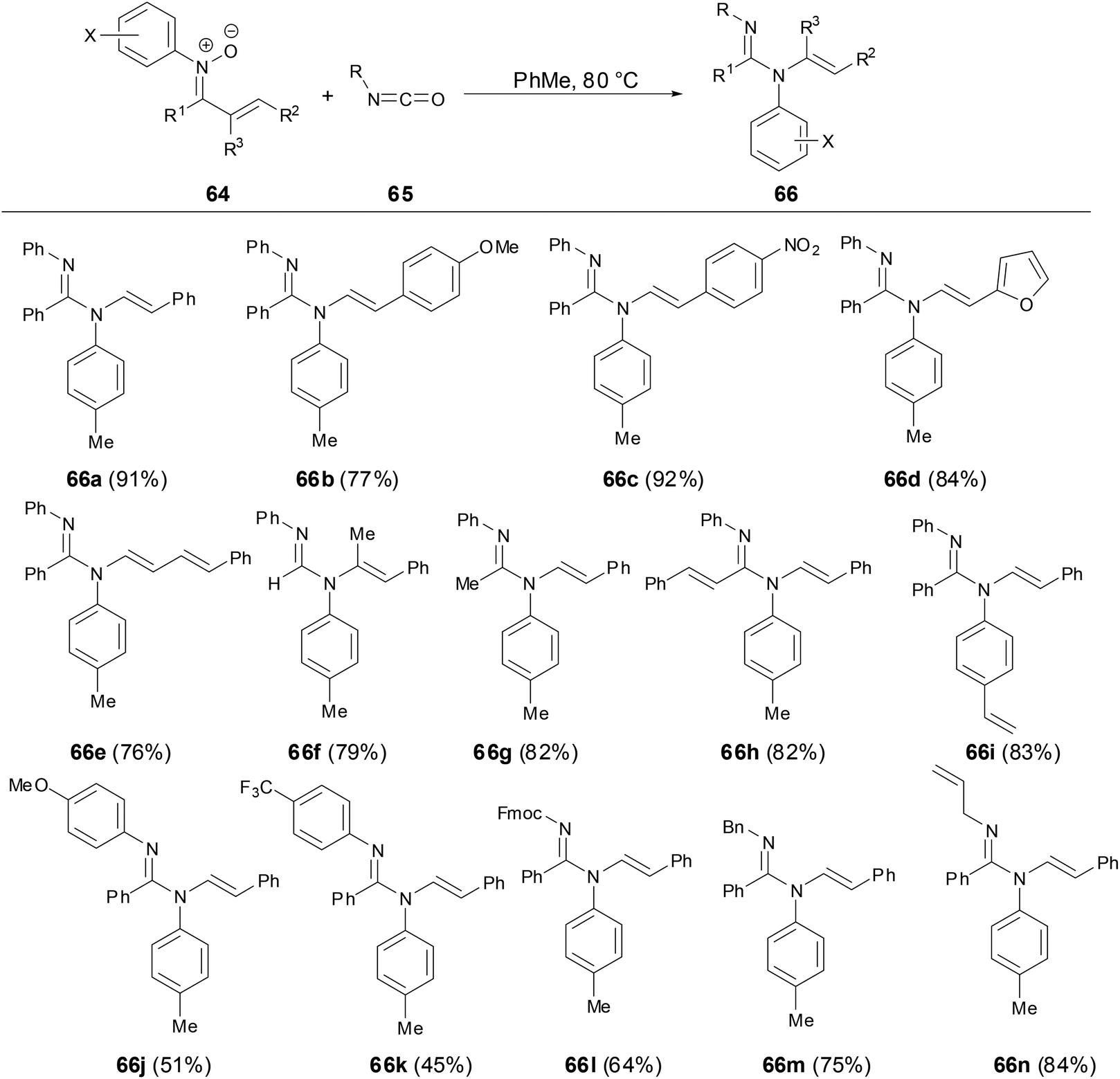
1.5 The reaction of ketonitrones with isocyanates
The cycloaddition reaction of N-aryl nitrones with isocyanates has been developed in the past few decades.41 However, Anderson and coworkers have revealed that N-styrenyl amidines can be prepared from N-aryl α,β-unsaturated nitrones and isocyanates (Scheme 24).42 The scope of nitrones tolerated not only a variety of different substitution patterns on the nitrone component but also different N-aryl substituent nitrones including halogen, vinyl and naphthyl substitutions. The mechanism studies showed that the transformation proceeded through an initial cycloaddition of a nitrone to an isocyanate to form an oxadiazolidinone followed by the subsequent CO2 elimination and styrenyl migration. This transformation provided a new route to N-alkenyl amidines which are challenging to access by traditional methods for amidine synthesis.
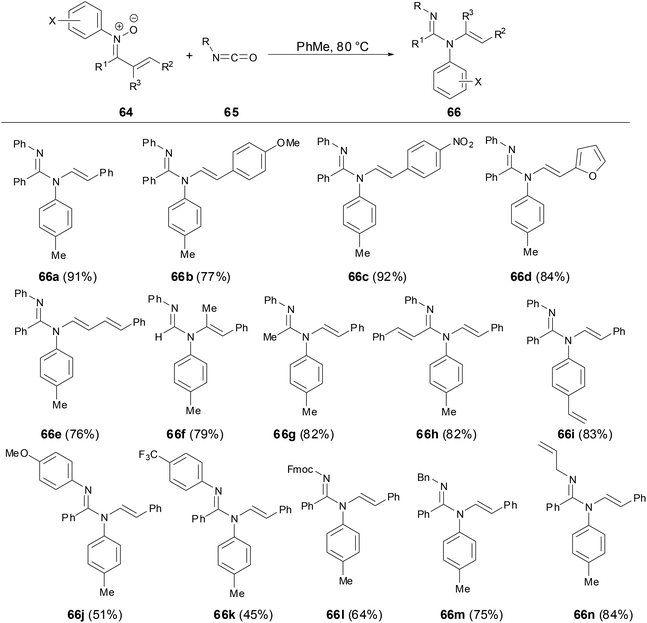 |
| | Scheme 24 Reaction of N-aryl nitrones with isocyanates. | |
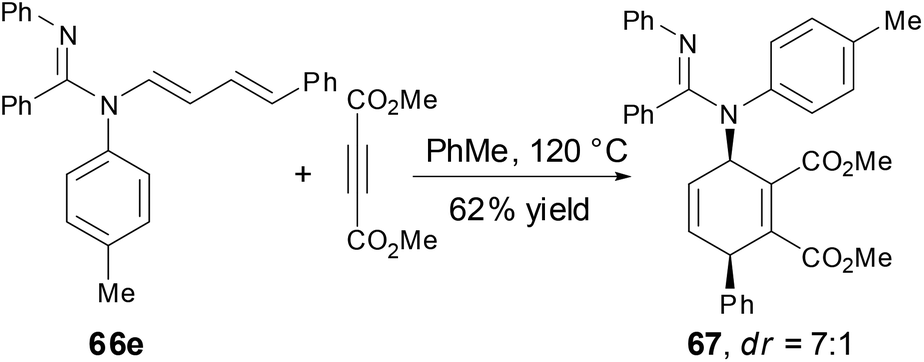
The N-alkenyl amidines 66e underwent a [4 + 2] cycloaddition reaction with dimethylacetylene dicarboxylate (DMAD) to provide the cycloadduct 67 with good yield and good diastereoselectivity, which demonstrated the synthetic utility of enamine functionality (Scheme 25).
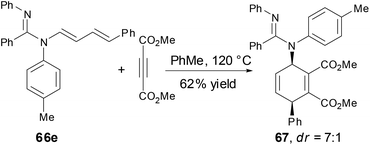 |
| | Scheme 25 [4 + 2] Reaction of N-alkenyl amidine with DMAD. | |
2 Conclusion
A copper-mediated cross coupling reaction from readily available oximes with aryl- or vinyl boronic acids under mild conditions, Rh- or Cu-catalyzed rearrangement of propargyloxyamines to prepare N-substitutents α,β-unsaturated ketonitrones or N-vinyl nitrones has been developed. Using different dipolarophiles such as alkynes, allenes, and isocyanates to test the reactivity of these nitrones has revealed that these nitrones involved new reactive intermediates and provided a variety of useful organic transformations. Although these contributions and significant progress have been made by researchers, many challenges still remain in nitrone chemistry. More efficient, simple and practical methodologies for preparing N-substituents α,β-unsaturated ketonitrones or N-vinyl nitrones will easily shed more light on the potential transformations of nitrones. The better understanding of transformations of these nitrones can provide more inspiration on new reaction design. To explore further the transformation of nitrones in organic synthesis remains a great challenge, especially, the synthesis and transformation of N-vinyl nitrones are highly demanded. We hope that our review will inspire more explorations in the field of nitrone chemistry.
Conflict of interest
The authors declare no competing financial interest.
Acknowledgements
Financial support from Overseas 100 Talents Program of Guangxi Education, Basic Research Program of Guangxi Normal University (grant no. 2014ZD005), National Natural Science Foundation of China (21562005, 21262007, 21462008), Ministry of Education of China (IRT1225), and Guilin Science and Technology Development Fund (20110331) is greatly appreciated.
References
- For some early reviews, see:
(a) L. I. Smith, Chem. Rev., 1938, 38, 193 CrossRef;
(b) J. Hamer and A. Macaluso, Chem. Rev., 1964, 64, 473 CrossRef CAS.
- For some reviews, see:
(a) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS PubMed;
(b) K. Rück-Braun, T. H. E. Freysoldt and F. Wierschem, Chem. Soc. Rev., 2005, 34, 507 RSC;
(c) H. Pellissier, Tetrahedron, 2007, 63, 3235 CrossRef CAS;
(d) V. Nair and T. D. Suja, Tetrahedron, 2007, 63, 12247 CrossRef CAS;
(e) A. Brandi, F. Cardona, S. Cicchi, F. M. Cordero and A. Goti, Chem. – Eur. J., 2009, 15, 7808 CrossRef CAS PubMed;
(f) C. Nájera and J. M. Sansano, Org. Biomol. Chem., 2009, 7, 4567 RSC;
(g) M. Kissane and A. R. Maguire, Chem. Soc. Rev., 2010, 39, 845 RSC;
(h) A. J. M. Burrell and I. Coldham, Curr. Org. Synth., 2010, 7, 312 CrossRef CAS.
-
(a) H. Zhang, J. Joseph, J. Vasquez-Vivar, H. Karoui, C. Nsanzumuhire, P. Martasek, P. Tordo and B. Kalyanaraman, FEBS Lett., 2000, 473, 58 CrossRef CAS PubMed;
(b) F. A. Villamena, S. Xia, J. K. Merle, R. Lauricella, B. Tuccio, C. M. Hadad and J. L. Zweier, J. Am. Chem. Soc., 2007, 129, 8177 CrossRef CAS.
-
(a) R. A. Floyd, Ageing Cell, 2006, 5, 51 CrossRef CAS PubMed;
(b) J. E. Slemmer, J. J. Shacka, M. Sweeney and J. T. Weber, Curr. Med. Chem., 2008, 15, 404 CrossRef CAS PubMed;
(c) A. Piperno, S. V. Giofre, D. Iannazzo, R. Romeo, G. Romeo, U. Chiacchio, A. Rescifina and D. G. Piotrowska, J. Org. Chem., 2010, 75, 2798 CrossRef CAS PubMed.
- For some early reviews, see:
(a) R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565 CrossRef;
(b) D. S. C. Black, R. F. Crozier and V. C. Davis, Synthesis, 1975, 205 CrossRef CAS;
(c) A. Padwa, Angew. Chem., Int. Ed., 1976, 15, 123 CrossRef;
(d) W. Opplzer, Angew. Chem., Int. Ed., 1977, 16, 10 CrossRef;
(e) C. P. Dell, J. Chem. Soc., Perkin Trans. 1, 1998, 3873 RSC.
- For some examples with alkenes, see:
(a) P. N. Confalone and E. M. Huie, Org. React., 2004, 36, 1 Search PubMed;
(b) N. Morita, K. Fukui, J. Irikuchi, H. Sato, Y. Takano, I. Okamoto, H. Ishibashi and O. Tamura, J. Org. Chem., 2008, 73, 7164 CrossRef CAS PubMed;
(c) T. B. Nguyen, A. Martel, R. Dhal and G. Dujardin, Org. Lett., 2008, 10, 4493 CrossRef CAS PubMed;
(d) C. M. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 14516 CrossRef CAS PubMed;
(e) R. Lahiri, A. Palanivel, S. A. Kulkarni and Y. D. Vankar, J. Org. Chem., 2014, 79, 10786 CrossRef CAS PubMed.
- For some examples with alkynes, see:
(a) C. Lu, A. V. Dubrovskiy and R. C. Larock, J. Org. Chem., 2012, 77, 2279 CrossRef CAS PubMed;
(b) S. Bhunia, C. J. Chang and R. S. Liu, Org. Lett., 2012, 14, 5522 CrossRef CAS PubMed;
(c) C. S. Mckay, M. Chigrinova, J. A. Blake and J. P. Pezaccki, Org. Biomol. Chem., 2012, 10, 3066 RSC;
(d) Y. Q. Zhang and J. L. Zhang, Chem. Commun., 2012, 48, 4710 RSC;
(e) Z. S. Qi, M. Wang and X. W. Li, Org. Lett., 2013, 15, 5440 CrossRef CAS PubMed;
(f) R. P. Temming, L. Eggermont, M. B. van Eldijk, J. C. M. van Hest and F. L. van Delft, Org. Biomol. Chem., 2013, 11, 2772 RSC;
(g) L. Wozniak, O. Staszewska-Krajewska and M. Michalak, Chem. Commun., 2015, 51, 1933 RSC.
- For some examples with allenes, see:
(a) J. J. Tufariello, S. A. Ali and H. O. Klingele, J. Org. Chem., 1979, 44, 4213 CrossRef CAS;
(b) A. Padwa, S. P. Carter, Y. Chiacchio and D. N. Kline, Tetrahedron Lett., 1986, 27, 2683 CrossRef CAS;
(c) A. Padwa, M. Matzinger, Y. Tomioka and M. K. Venkatramanan, J. Org. Chem., 1988, 53, 955 CrossRef CAS;
(d) T. Wirth and S. Blechert, Synlett, 1994, 717 CrossRef CAS;
(e) X. Wu, R. S. Na, H. L. Liu, J. Liu, M. Wang, J. C. Zhong and H. C. Guo, Tetrahedron Lett., 2012, 53, 342 CrossRef CAS;
(f) G. H. Li, W. Zhou, X. X. Li, Q. W. Bi, Z. Wang, Z. G. Zhao, W. X. Hu and Z. L. Chen, Chem. Commun., 2013, 49, 4770 RSC;
(g) J. Malinina, T. Q. Tran, A. V. Stepakov, V. V. Gurzhiy, G. L. Starova, R. R. Kostikov and A. P. Molchanov, Tetrahedron Lett., 2014, 55, 3663 CrossRef CAS.
- For some examples with isocyanates, ketenes, ketenimines, see:
(a) S. P. Joseph and D. N. Dhar, Tetrahedron, 1988, 44, 5209 CrossRef CAS;
(b) N. Murai, M. Komatsu, Y. Ohshiro and T. Agawa, J. Org. Chem., 1977, 42, 448 CrossRef CAS;
(c) K. Namitharan and K. Pitchumani, Org. Lett., 2011, 13, 5728 CrossRef CAS PubMed.
-
(a) M. Kinugasa and S. Hashimoto, J. Chem. Soc., Chem. Commun., 1972, 466 RSC;
(b) M. M. C. Lo and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 4572 CrossRef CAS PubMed;
(c) R. Shintani and G. C. Fu, Angew. Chem., Int. Ed., 2003, 42, 4082 CrossRef CAS PubMed;
(d) M. C. Ye, J. Zhou, Z. Z. Huang and Y. Tang, Chem. Commun., 2003, 2554 RSC;
(e) S. Stecko, A. Mames, B. Furman and M. Chmielewski, J. Org. Chem., 2009, 74, 3094 CrossRef CAS PubMed;
(f) J. H. Chen, S. H. Liao, X. L. Sun, Q. Shen and Y. Tang, Tetrahedron Lett., 2012, 68, 5042 CrossRef CAS;
(g) S. Santoro, R. Z. Liao, T. Marcelli, P. Hammar and F. Himo, J. Org. Chem., 2015, 80, 2649 CrossRef CAS PubMed.
-
(a) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213 CrossRef CAS PubMed;
(b) T. Q. Tran, V. V. Diev, G. L. Starova, V. V. Gurzhiy and A. P. Molchanov, Eur. J. Org. Chem., 2012, 2054 CrossRef CAS;
(c) D. L. Mo, T. Yuan, C. H. Ding, L. X. Dai and X. L. Hou, J. Org. Chem., 2013, 78, 11470 CrossRef CAS PubMed.
-
(a) I. S. Young and M. A. Kerr, Angew. Chem., Int. Ed., 2003, 42, 3023 CrossRef CAS PubMed;
(b) I. S. Young, J. L. Williams and M. A. Kerr, Org. Lett., 2005, 7, 953 CrossRef CAS PubMed;
(c) C. A. Carson and M. A. Kerr, Angew. Chem., Int. Ed., 2006, 45, 6560 CrossRef CAS;
(d) M. K. Sibi, Z. Mz and C. P. Jasperse, J. Am. Chem. Soc., 2005, 127, 5764 CrossRef CAS PubMed;
(e) Y. B. Kang, X. L. Sun and Y. Tang, Angew. Chem., Int. Ed., 2007, 46, 3918 CrossRef CAS PubMed;
(f) H. B. Yang and M. Shi, Org. Biomol. Chem., 2012, 10, 8236 RSC;
(g) C. M. Braun, E. A. Congdon and K. A. Nolin, J. Org. Chem., 2015, 80, 1979 CrossRef CAS PubMed.
-
(a) X. Wang, X. Xu, P. Y. Zavalij and M. P. Doyle, J. Am. Chem. Soc., 2011, 133, 16402 CrossRef CAS PubMed;
(b) X. Xu, M. O. Ratnikov, P. Y. Zavalij and M. P. Doyle, Org. Lett., 2011, 13, 6122 CrossRef CAS PubMed;
(c) Y. Qian, X. Xu, X. Wang, P. Y. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2012, 51, 5900 CrossRef CAS PubMed;
(d) X. Wang, Q. M. Abrahams, P. Y. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2012, 51, 5907 CrossRef CAS PubMed;
(e) X. F. Xu, P. J. Zavalij and M. P. Doyle, Chem. Commun., 2013, 49, 10287 RSC.
-
(a) R. Shintani, S. Park, W. L. Duan and T. Hayashi, Angew. Chem., Int. Ed., 2007, 46, 5901 CrossRef CAS PubMed;
(b) R. Shintani, M. Murakami and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 12356 CrossRef CAS PubMed;
(c) A. C. Stevens, C. Palmer and B. L. Pagenkopf, Org. Lett., 2011, 13, 1528 CrossRef CAS PubMed.
- S. A. Gawade, S. Bhunia and R. S. Liu, Angew. Chem., Int. Ed., 2012, 51, 7835 CrossRef CAS PubMed.
-
(a) G. Masson, P. Cividino, S. Py and Y. Vallée, Angew. Chem., Int. Ed., 2003, 42, 2265 CrossRef CAS;
(b) D. Riber and T. Skrydstrup, Org. Lett., 2003, 5, 229 CrossRef CAS PubMed;
(c) J. Marco-Contelles, Angew. Chem., Int. Ed., 2004, 43, 2198 CrossRef CAS PubMed;
(d) P. Cividino, S. Py, P. Delair and A. E. Greene, J. Org. Chem., 2007, 72, 485 CrossRef CAS PubMed.
-
(a) J. Yang, Synlett, 2012, 2293 CrossRef CAS;
(b) M. P. S. Ishar, K. Kumar and R. Singh, Tetrahedron Lett., 1998, 39, 6547 CrossRef CAS;
(c) T. Ghosh and C. Bandyopadhyay, Tetrahedron Lett., 2004, 45, 6169 CrossRef CAS;
(d) K. Wittstein, A. B. García, M. Schürmann and K. Kumar, Synlett, 2012, 227 CAS.
- G. Bartoli, E. Marcantoni and M. Petrini, J. Org. Chem., 1990, 55, 4456 CrossRef CAS.
- S. E. Denmark and J. I. Montgomery, J. Org. Chem., 2006, 71, 6211 CrossRef CAS PubMed.
-
(a) P. N. Confalone and E. M. Huie, Org. React., 1988, 36, 1 CAS;
(b)
K. B. G. Torssell, Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis, ed. H. Feuer, VCH Publishers, New York, 1988 Search PubMed.
-
(a) S. Cicchi, M. Corsi and A. Goti, J. Org. Chem., 1999, 64, 7243 CrossRef CAS;
(b) S. Cicchi, M. Marradi, A. Goti and A. Brandi, Tetrahedron Lett., 2001, 42, 6503 CrossRef CAS;
(c) R. Saladino, V. Neri, F. Cardona and A. Goti, Adv. Synth. Catal., 2004, 346, 639 CrossRef CAS.
-
(a) F. Busqué, P. de March, M. Figueredo, J. Font, T. Gallagher and S. Milán, Tetrahedron: Asymmetry, 2002, 13, 437 CrossRef;
(b) G. Soldaini, F. Cardona and A. Goti, Org. Lett., 2007, 9, 473 CrossRef CAS PubMed.
-
(a) M. Forcato, W. A. Nugent and G. Licini, Tetrahedron Lett., 2003, 44, 49 CrossRef CAS;
(b) C. Gella, E. Ferrer, R. Alibes, F. Busque, P. de March, M. Figueredo and J. Font, J. Org. Chem., 2009, 74, 6365 CrossRef CAS PubMed;
(c) M. Forcato, M. Mba, W. A. Nugent and G. Licini, Eur. J. Org. Chem., 2010, 740 CrossRef CAS;
(d) M. Abrantes, I. S. Goncalves, M. Pillinger, C. Vurchio, F. M. Cordero and A. Brandi, Tetrahedron Lett., 2011, 52, 7079 CrossRef CAS.
-
(a) K. Nakama, S. Seki and S. Kanemasa, Tetrahedron Lett., 2002, 43, 829 CrossRef CAS;
(b) S. K. Hota, A. Chatterjee, P. K. Bhattacharya and P. Chattopadhyay, Green Chem., 2009, 11, 169 RSC.
-
(a) S. Franco, F. L. Merchan, P. Merino and T. Tejero, Synth. Commun., 1995, 25, 2272 CrossRef;
(b) J. Y. Pfeiffer and A. M. Beauchemin, J. Org. Chem., 2009, 74, 8381 CrossRef CAS PubMed.
- T. S. Hood, C. B. Huehls and J. Yang, Tetrahedron Lett., 2012, 53, 4679 CrossRef CAS.
- D. L. Mo and L. L. Anderson, Angew. Chem., Int. Ed., 2013, 52, 6722 CrossRef CAS PubMed.
- X. P. Ma, W. M. Shi, X. L. Mo, X. H. Li, L. G. Li, C. X. Pan, B. Chen, G. F. Su and D. L. Mo, J. Org. Chem., 2015, 80, 10098 CrossRef CAS PubMed.
- I. Nakamura, T. Onuma, R. Kanazawa, Y. Nishigai and M. Terada, Org. Lett., 2014, 16, 4198 CrossRef CAS PubMed.
- S. Murarka and A. Studer, Org. Lett., 2011, 13, 2746 CrossRef CAS PubMed.
-
(a) M. L. M. Pennings, D. N. Reinhoudt, S. Harkema and G. J. Van Hummel, J. Am. Chem. Soc., 1980, 102, 7570 CrossRef CAS;
(b) M. L. M. Pennings and D. N. Reinhoudt, J. Org. Chem., 1982, 47, 1816 CrossRef CAS;
(c) R. Grigg, T. R. Perrior, G. J. Sexton, S. Surendrakumar and T. Suzuki, J. Chem. Soc., Chem. Commun., 1993, 372 RSC;
(d) F. Heaney, B. M. Kelly, S. Bourke, O. Rooney, D. Cunningham and P. McArdle, J. Chem. Soc., Perkin Trans. 1, 1999, 143 RSC;
(e) H. S. Yeom, E. So and S. Shin, Chem. – Eur. J., 2011, 17, 1764 CrossRef CAS PubMed.
- R. E. Michael, K. M. Chando and T. Sammakia, J. Org. Chem., 2015, 80, 6930 CrossRef CAS PubMed.
- D. L. Mo, D. A. Wink and L. L. Anderson, Org. Lett., 2012, 14, 5187 Search PubMed.
- I. Nakamura, M. Okamoto, Y. Sato and M. Terada, Angew. Chem., Int. Ed., 2012, 51, 10816 CrossRef CAS PubMed.
- I. Nakamura, Y. Sato, K. Takeda and M. Terada, Chem. – Eur. J., 2014, 20, 10214 CrossRef CAS PubMed.
- C. B. Huehls, T. S. Hood and J. Yang, Angew. Chem., Int. Ed., 2012, 51, 5110 CrossRef CAS PubMed.
- C. B. Huehls, J. H. Huang and J. Yang, Tetrahedron, 2015, 71, 3593 CrossRef CAS.
-
(a) A. Padwa, D. N. Kline, K. F. Koehler, M. Matzinger and M. K. Venkatramanan, J. Org. Chem., 1987, 52, 3909 CrossRef CAS;
(b) A. Padwa, W. H. Bullock, D. N. Kline and J. Perumattam, J. Org. Chem., 1989, 54, 2862 CrossRef CAS.
- D. L. Mo, D. A. Wink and L. L. Anderson, Chem. – Eur. J., 2014, 20, 13217 CrossRef CAS PubMed.
- W. H. Pecak, J. W. Son, A. J. Burnstine and L. L. Anderson, Org. Lett., 2014, 16, 3440 CrossRef CAS PubMed.
-
(a) A. M. T. Bell, J. Bridges, R. Cross, C. P. Falshaw, B. F. Taylor, G. A. Taylor, I. C. Whittakeer and M. J. Begley, J. Chem. Soc., Perkin Trans. 1, 1987, 2593 RSC;
(b) T. Ritter and E. M. Carreira, Angew. Chem., Int. Ed., 2005, 44, 936 CrossRef CAS PubMed.
- D. L. Mo, W. H. Pecak, M. Zhao, D. A. Wink and L. L. Anderson, Org. Lett., 2014, 16, 3696 CrossRef CAS PubMed.
Footnote |
| † This work is dedicated to Professor Xue-Long Hou (from Shanghai Institute of Organic Chemistry) on the occasion of his 60th birthday. |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.