
Triphenylphosphine promoted regio and stereoselective α-halogenation of ynamides†‡
B.
Prabagar
,
Sanatan
Nayak
,
Rajendra K.
Mallick
,
Rangu
Prasad
and
Akhila K.
Sahoo
*
School of Chemistry, University of Hyderabad, Hyderabad-500046, India. E-mail: akhilchemistry12@gmail.com; akssc@uohyd.ac.in
First published on 30th November 2015
Abstract
In this study, we demonstrate metal free regio- and stereoselective α-halogenation (Cl, Br and I) of ynamides. The halogenation of ynamides in the presence of PPh3 and CCl4, CBr4 or CHI3 in moist CH2Cl2 at room temperature provides good-to-excellent yields of a wide variety of stable (E)-α-haloenamides; thus, the reaction has a broad scope. Chlorination of ynamides at room temperature is particularly notable. The sequential one pot α-iodination and alkynylation of ynamides provide synthetically useful amidoenynes. The chlorination of 3-acetoxy ynamide delivers 3-acetoxy-α-chloro enamide without the migratory-assistance of the acetoxy moiety.
Introduction
Enamides are versatile motifs, and are widely present in various natural products and complex molecular entities.1 In general, enamides are more stable than enamines; thus, enamide scaffolds are synthetically viable and broadly useful.2 Notably, delocalization of the nitrogen lone-pair readily assists the functionalization of the β-position of enamides.3 In contrast, modification of the α-position of enamides is challenging. α-Haloenamides are particularly notable in this context because functionalization of their α- and β-carbons is easy.4 The β-carbon of α-haloenamides is nucleophilic and is therefore useful for stereoselective C–C/X bond formation, while their α-halo group can effectively be replaced by organometallic reagents through transition-metal-catalyzed cross-coupling reactions.5Hsung et al. first reported the stereoselective synthesis of α-haloenamides through the hydrohalogenation of ynamides with MgX2 and TMSBr in moist CH2Cl2; ynamide chlorination was possible at an elevated temperature (Scheme 1).6 The Kazmaier group showed Mo-catalyzed hydrostannylation of ynamides followed by Sn–iodine exchange to afford α-iodoenamide (Scheme 1).7 The Skrydstrup group exhibited a novel method for the synthesis of α-iodo/bromo acrylamides/acrylimides through a NIS/NBS promoted reaction on 3-acetoxy ynamide, while the identical reaction with NCS displayed poor conversion (Scheme 1).8 Stereoselective hydroflourination of ynamides was described by Evano and co-workers (Scheme 1).9 Iwasawa et al. demonstrated the regio- and stereospecific synthesis of (E)-α-haloenamide from ynamides in the presence of halotrimethylsilane and H2O (Scheme 1).10 Thus, the synthesis of α-haloenamides from ynamides involves step-wise conversion from Sn-species to iodo, regioselective addition of HX to ynamides, control of the double-bond geometry, or harsh conditions. Thus, the efficient introduction of common halo groups (Cl, Br or I) at the α-position of ynamides is considerably challenging. Therefore, an operationally simple and competent protocol for the regio- and stereoselective α-halogenation of ynamides is required. With our ongoing research on ynamides,11 we expect to trap halophosphonium salts generated from Ph3P and CCl4, CBr4 or CHI3 with the keteniminium ion species, the reactive resonance hybrid unit of ynamides,12 and finally produce stereoselective (E)-α-haloenamides (Scheme 1).
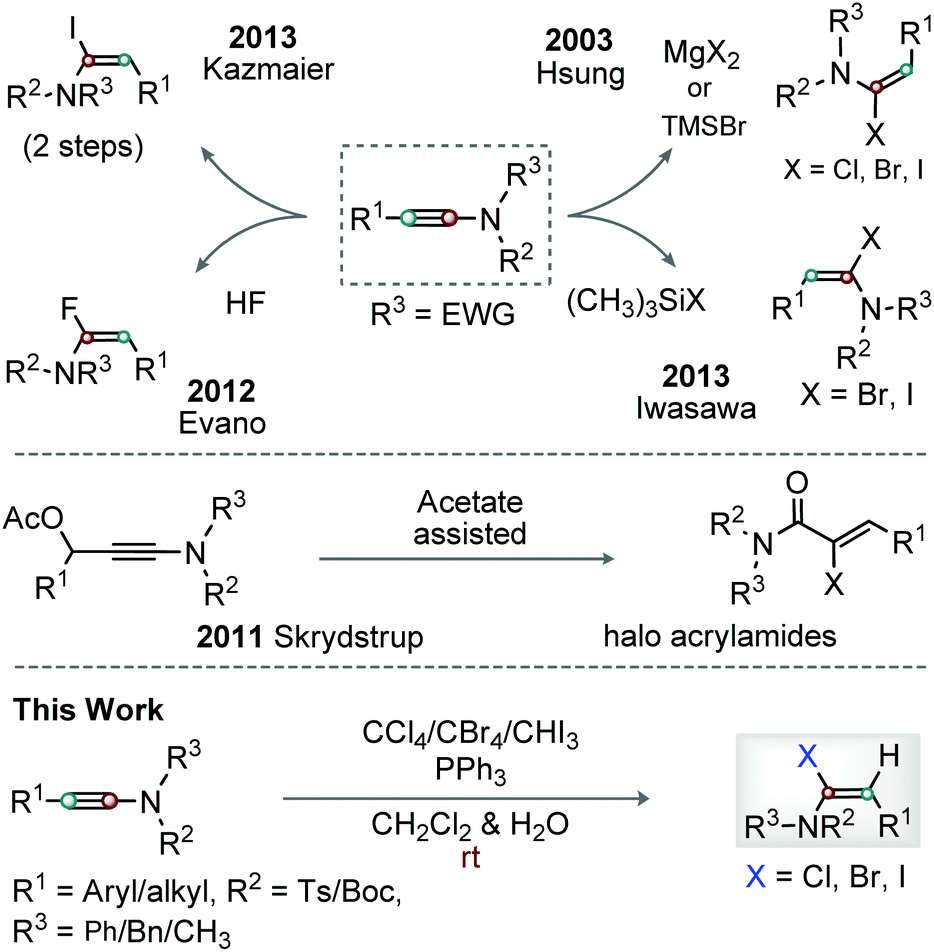 | ||
| Scheme 1 Synthetic strategies for α-haloenamide. | ||
We herein report a general, reliable, efficient, and gram-scale synthetic approach for the convergent and stereoselective synthesis of geometrically defined α-halo (Cl/Br/I)-enamides from ynamides in the presence of metal-free reagents [Ph3P and CCl4/CBr4/CHI3] in CH2Cl2 and H2O at rt; the halo moiety is successfully used for the synthesis of diverse molecular entities.
Results and discussion
Investigation was initiated by examining α-chlorination of 1a (Table 1). Thus, a mixture of 1a (0.5 mmol), PPh3 (1.2 equiv.), CCl4 (2.0 equiv.) and H2O (1.5 equiv.) was heated at 60 °C for 16 h; gratifyingly the desired (E)-α-chloroenamide 2a was isolated in 50% yield with the incomplete consumption of 1a (entry 1). Disappointingly, the reaction did not proceed at rt; the inadequate solubility of PPh3 in CCl4 at rt is presumably responsible for the poor outcome, an established fact reported by R. Apple (entry 2).13 Good to excellent yield of 2a was obtained, when the reaction was conducted at 80 °C (entries 3 and 4). The structure of 2a was confirmed by X-ray crystallographic analysis (Scheme 2).22 In the absence of PPh3, there was no consumption of the starting material (entry 5). Other phosphine reagents such as trialkylphosphine and trialkylphosphite provided a moderate yield of 2a (entries 6–9). Apparently the more nucleophilic trialkylphosphines should work better than triarylphosphines; however, the requirement of H2O in this transformation makes the pyrophoric trialkylphosphines ineffective, resulting in poor reaction outcome (entries 6 and 7), whereas the corresponding phosphonium salts of the phosphite esters are unstable and are readily transformed to dialkyl phosphonate esters, hampering the reaction productivity (entries 8 and 9).14 To our surprise, the reaction of 1a with PPh3 and CCl4 in CH2Cl2 (2.0 mL) was found to be efficient at rt, producing 2a in 98% yield in 2 h with excellent stereo-control (entry 10). Presumably, the solubility of PPh3 in CH2Cl2 enables faster interaction with CCl4 at rt to offer better results.15 Thus, stereoselective α-chlorination of ynamides at rt is truly notable. Despite the toxicity of CCl4, the current method requires only 2.0 equiv. of CCl4 rather than a solvent. The formation of [Ph3PCl]+ species from PPh3 and SOCl2 is known.16a,20 Accordingly, a reaction of 1a with [PPh3 and SOCl2] was conducted at rt; pleasingly, the desired α-chloroenamide 2a was obtained in 86% yield (entry 11). The use of CCl4 (1.2 equiv.) led to the decrease of product yield (entry 12). Furthermore, the reaction in the absence of water produced 2a in 26% yield (entry 13). The use of different amounts of H2O in the reaction provided 48–68% of 2a in 2 h (entries 14–16). The results from entries 12–16 suggest that H2O plays a critical role in this transformation. We believe that the attack of H2O on the phosphonium species terminates the reaction and liberates PPh3(O) as a by-product, which eventually controls the efficiency of the reaction. The isolation of PPh3(O) further supports the need of H2O in this study. The reaction of 1a with PPh3 and CHCl3 in CH2Cl2 did not proceed (entry 17, Table 1). The use of other solvents such as chloroform, dioxane and acetone instead of CH2Cl2 provided inferior yields of 2a.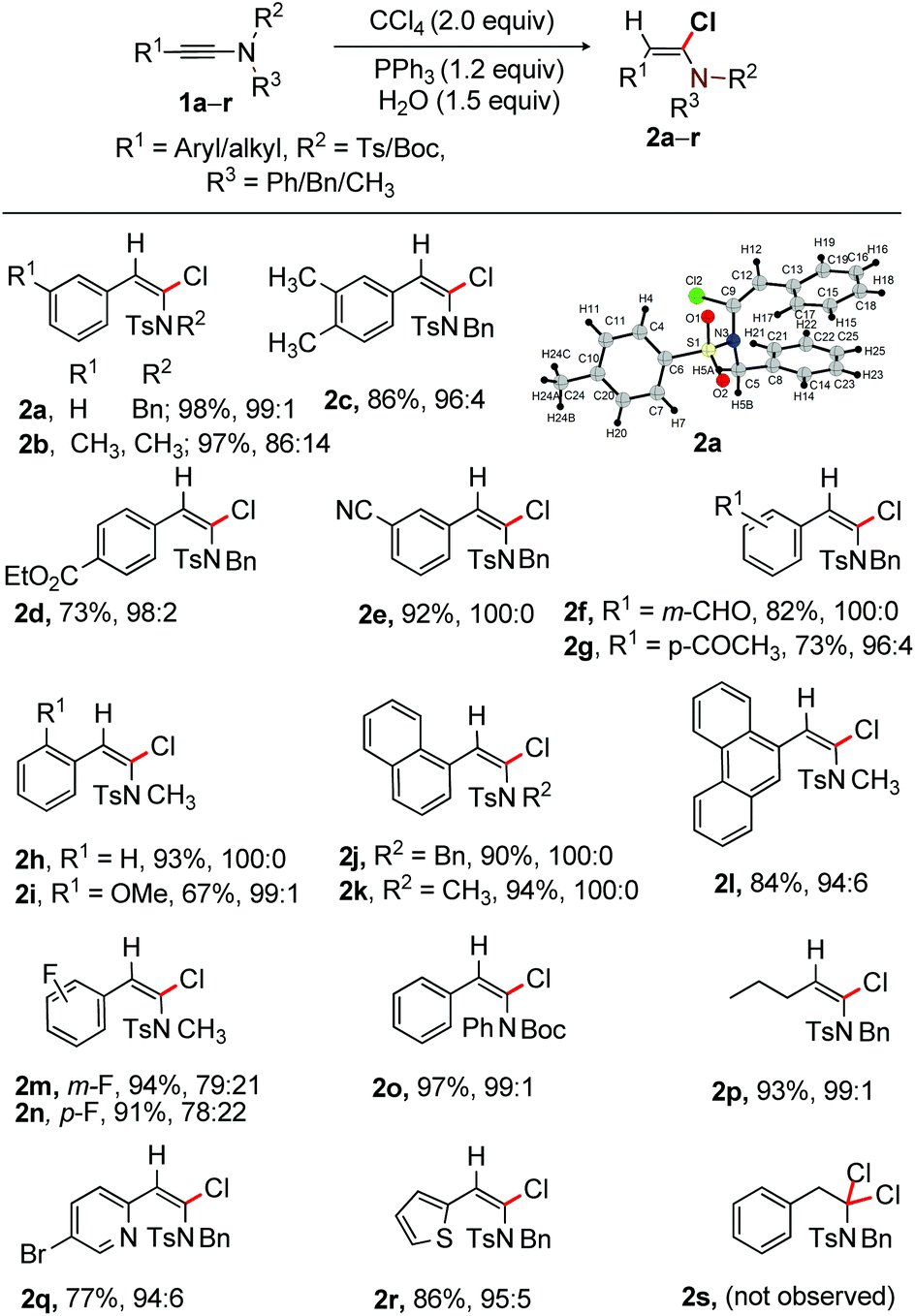 | ||
| Scheme 2 α-Chlorination of ynamides: reactions were carried out using 1 (0.5 mmol), CCl4 (1.0 mmol), PPh3 (0.6 mmol) in CH2Cl2 (2.0 mL) and H2O (0.75 mmol) at rt for 2 h; stereoisomers are mentioned in the E/Z ratio (determined by 1H NMR). Isolated yield. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Solvent | Temp. (°C) | Time (h) | Yield (%)b2a/3a/4a |
a Reactions were carried out using 1 (0.5 mmol), CCl4 (2.0 equiv.), PPh3 (1.2 equiv.) in solvent (2.0 mL) at rt.
b Isolated yield.
c CCl4 (1.2 equiv.) was used.
d Absence of water.
e 0.5 equiv. of H2O.
f 1.0 equiv. of H2O.
g 1.5 equiv. of H2O.
h Bromination of 1a.
i Iodination of 1a.
j Formation of 2a (60%) along with 40% monohydration.
k Formation of 4a [E/Z(1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)]. NR = no reaction. RT = 25–28 °C. :1)]. NR = no reaction. RT = 25–28 °C.
|
|||||
| 1 | PPh3 | CCl4 | 60 | 16 | 50 |
| 2 | PPh3 | CCl4 | RT | 24 | NR |
| 3 | PPh3 | CCl4 | 80 | 12 | 68 |
| 4 | PPh3 | CCl4 | 80 | 24 | 98 |
| 5 | — | CCl4 | 80 | 24 | NR |
| 6 | P(Et)3 | CCl4 | 80 | 24 | 50 |
| 7 | P(Me)3 | CCl4 | 80 | 24 | 25 |
| 8 | P(iPrO)3 | CCl4 | 80 | 24 | 10 |
| 9 | P(EtO)3 | CCl4 | 80 | 24 | 45 |
| 10 | PPh 3 /CCl 4 | CH 2 Cl 2 | RT | 02 | 98 |
| 11 | PPh3/SOCl2 | CH2Cl2 | RT | 02 | 86 |
| 12c | PPh3/CCl4 | CH2Cl2 | RT | 02 | 84 |
| 13d | PPh3/CCl4 | CH2Cl2 | RT | 02 | 26 |
| 14e | PPh3/CCl4 | CH2Cl2 | RT | 02 | 48 |
| 15f | PPh3/CCl4 | CH2Cl2 | RT | 02 | 59 |
| 16g | PPh3/CCl4 | CH2Cl2 | RT | 02 | 68 |
| 17 | PPh3/CHCl3 | CH2Cl2 | RT | 04 | NR |
| 18 | PPh 3 /CBr 4 | CH 2 Cl 2 | RT | 05 | 94 |
| 19 | PPh 3 /CHI 3 | CH 2 Cl 2 | RT | 05 | 96 |
| 20j | PPh3/ICl | CH2Cl2 | RT | 02 | 60 |
| 21k | PPh3/I2 | CH2Cl2 | RT | 02 | 83 |
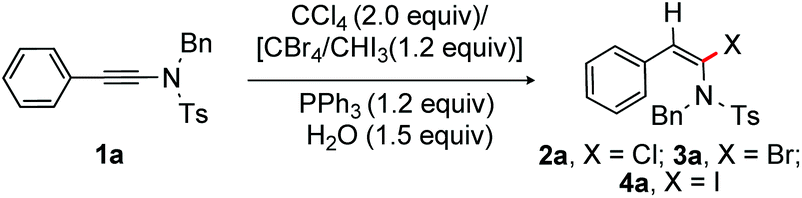
Next, α-bromination and iodination of ynamides were investigated (Table 1). As expected, the reaction of 1a with a mixture of PPh3, CBr4 and H2O (1.5 equiv.) in CH2Cl2 at rt led to α-bromoenamide 3a in 94% yield (entry 18). To accomplish the iodination of ynamides, 1a was exposed to PPh3, iodoform (CHI3) and H2O (1.5 equiv.) in CH2Cl2; gratifyingly, 96% of the desired α-iodoenamide 4a was obtained at rt (entry 19). The reaction of 1a with PPh3, ICl, and H2O (1.5 equiv.) in CH2Cl2 provided a mixture of chlorination product 2a along with the monohydration compound (60:40) (entry 20). In contrast, 83% of iodination product 4a (E/Z = 1:1) was obtained, when the reaction was performed with PPh3 and I2 (entry 21).
With the optimized conditions in hand, as shown in entry 10, Table 1, the scope of α-chlorination of various ynamides was explored; the results are summarized in Scheme 2. Wide ranges of N-(Ts/Boc)-phenyl/benzyl/methyl-ynamides having aryl, heteroaryl and alkyl substitution at the alkyne terminus were subjected to the optimized conditions (entry 10, Table 1); the corresponding (E)-α-chloroenamides 2a–r were obtained in good to excellent yields. The electro-neutral/-rich aryl-moiety bearing ynamides provided E-α-chloroenamides 2a–c in excellent yields. Easily modifiable ester, –CN, formyl, and –COMe functional groups did not show adverse effects, affording 2d–g in 73–92% yield. N-Methyl protected ynamides were no exception [2h (93%) and 2i (67%)]. The naphthalene and phenanthrene bearing α-chloroenamides 2j–l were readily synthesized. The fluoro group in ynamide did not affect the reaction outcome, providing 2m [E/Z = 79:21] and 2n [E/Z = 78:22] in 94% and 91% yield, respectively. The labile and easily removable N-Boc group survived, affording 97% of 2o. The alkyl-bearing (E)-α-chloroenamide 2p (93%) was smoothly accessed. The pyridyl and thiophenyl bearing ynamides were effectively reacted to provide 2q and 2r in good yields.
The β-position of α-chloroenamide is generally nucleophilic; the formation of the α,α′-gem-dichloroamide product is therefore anticipated from α-chloroenamide under the optimized conditions. To further assess the reaction feasibility of α-chloroenamide, compound 1a was subjected to excess amounts of Ph3P (2.5 equiv.) and CCl4 (4.0 equiv.) in CH2Cl2 at rt. Disappointingly, we did not observe the expected gem-dichloroamide product 2s (Scheme 2). We therefore believe that the α-position of ynamide 1a is more susceptible to chlorination with [Ph3PCl]+ than the enamide moiety in 2a.
Next, α-bromination of ynamide 1 under the optimized conditions (entry 18, Table 1) was examined at rt. The reaction proved to be general and to have a broad substrate scope; the results are depicted in Scheme 3. The phenyl substituted α-bromoenamide 3a (94%) was obtained with high selectivity, while 3b (E/Z = 88:12) was isolated in 94% yield. The 1-naphthyl bearing N-methyl protected bromoenamide 3c was produced in good yield. Phenanthrene substituted 3d (E/Z = 98:2) was formed in 87% yield. The heteroaryls such as 5-bromopyridinyl and thiophenyl bearing ynamides were compatible [3e (77%) and 3f (83%)]. The alkyl group bearing ynamide was no exception, providing 85% of 3g.
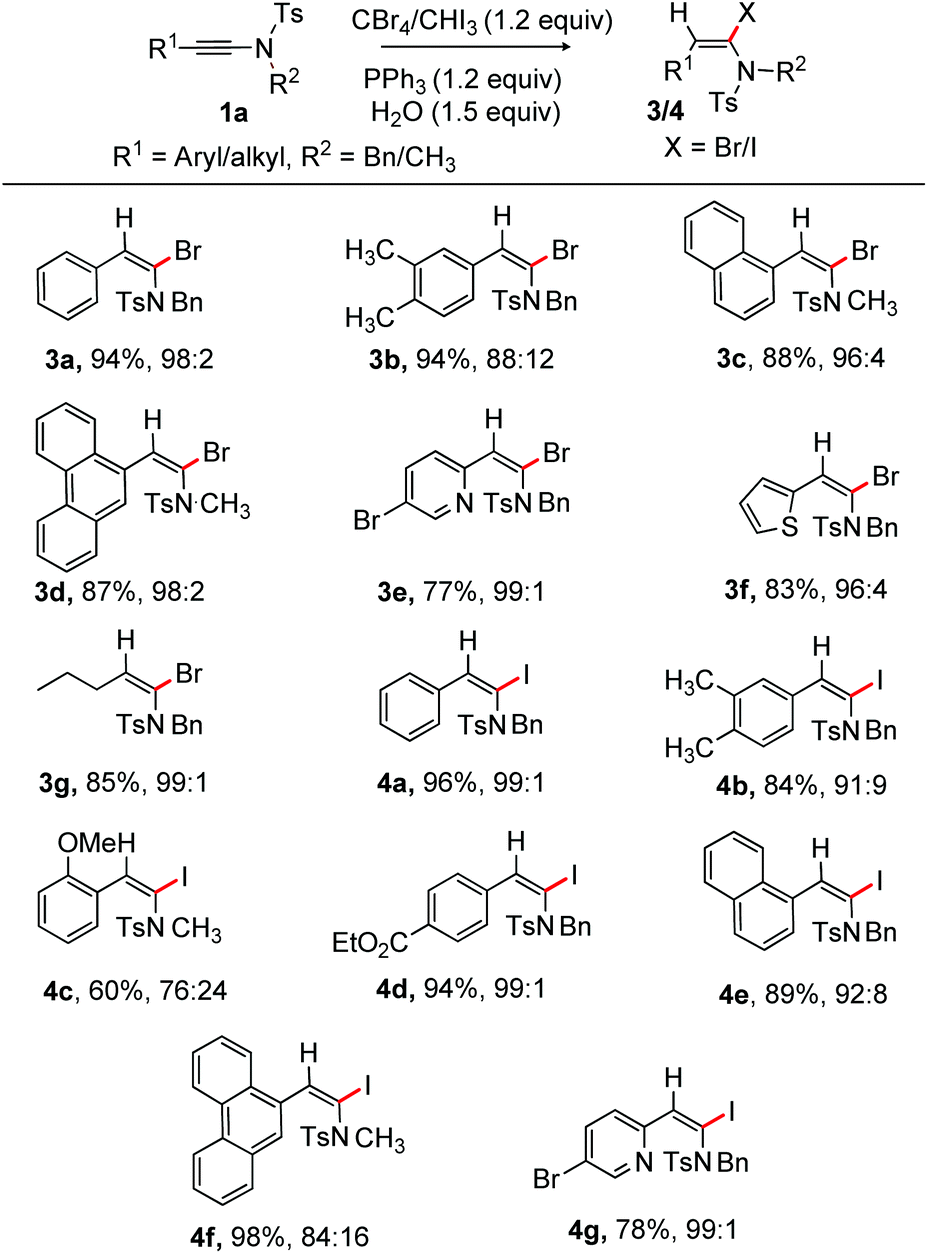 | ||
| Scheme 3 α-Bromo and α-iodination of ynamides: reactions were carried out using 1 (0.5 mmol), CBr4/CHI3 (0.6 mmol), PPh3 (0.6 mmol) in CH2Cl2 (2.0 mL) and H2O (0.75 mmol) at rt for 5 h; stereoisomers are mentioned in the E/Z ratio (determine by 1H NMR). Isolated yield. | ||
We next tested the α-iodination of ynamide under the conditions shown in entry 19, Table 1. The ynamide having electron-neutral and electron-rich aryl moieties on the alkyne terminus reacted efficiently to deliver 4a and 4b with high selectivity; whereas the o-substituted aryl ynamide yielded 4c [E/Z = 76:24] with moderate selectivity. The exact reason for the moderate selectivity is unknown; however, we believe that the participation of the o-OMe group in the reaction intermediate and the size of iodine play a crucial role in determining the observed selectivity. Iodination of ynamide having the easily modifiable electron withdrawing ester group on the aryl ring provided 4d (94%). The naphthalene and phenanthrene bearing substrate did not affect reaction efficiency affording 4e and 4f in fruitful yields. Pleasingly, the pyridyl bearing ynamide effectively participated in the iodination process, giving 4g (78%) in excellent selectivity (Scheme 3). The current method provided moderate to excellent E/Z selectivity of α-iodo/bromo-enamides, while the Iwasawa protocol exclusively delivered the corresponding E-α-Br/I-enamides.
We next investigated the synthetic potential of α-haloenamides. The iodo group of α-iodoenamide 4a was successfully utilized for the Suzuki and Sonogashira reactions to provide a non-separable mixture of tri-substituted enamides 5 (98%, E/Z = 81:19) and 6a (96%, E/Z = 63:37), respectively (Scheme 4).16b,17,18
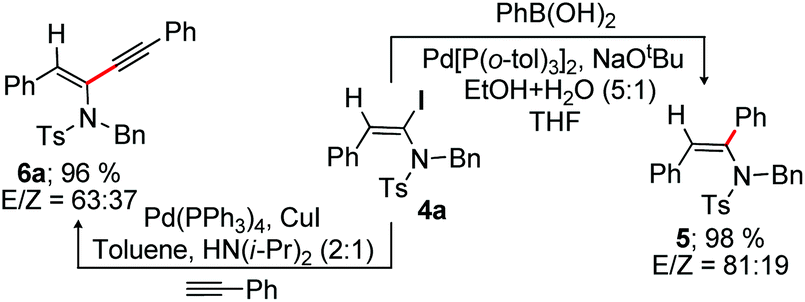 | ||
| Scheme 4 Synthetic manipulation. | ||
The regioselective alkynylation of ynamide (alkyne–ynamide cross coupling) readily constructs amidoenynes, the intermediates widely used for the fabrication of complex molecular entities. The facile reactivity of α-iodoenamide to Sonogashira reactions shown in Scheme 4 inspired us to survey one pot α-iodination and alkynylation on ynamide 1. Gratifyingly, α-iodination of 1 under the optimized conditions followed by the Sonogashira reaction with a range of alkynes readily produced synthetically valuable non-separable E/Z stereoisomers of amidoenynes 6a–d in overall good yields in a single pot (Scheme 5).18
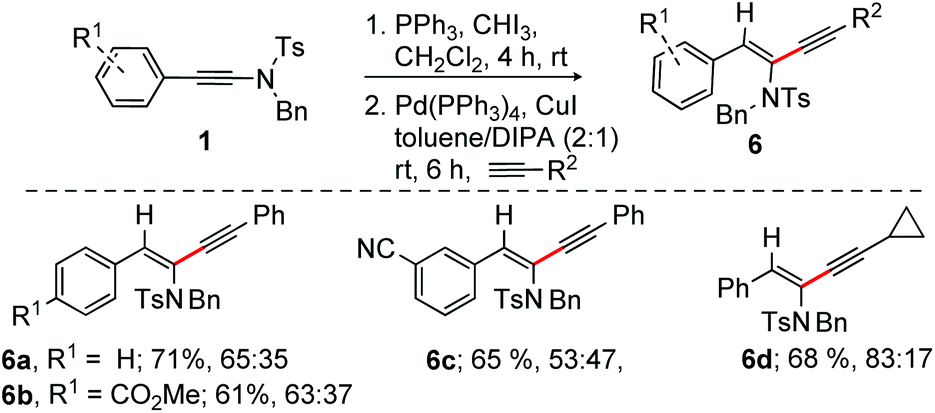 | ||
| Scheme 5 One pot sequential iodination and alkynylation of ynamides. | ||
Finally, chlorination and iodination of ynamide 1a on a gram scale were successfully tested under the optimized conditions at rt. Pleasingly, the desired products 2a (959 mg) and 4a (1.1 g) were isolated in 87% and 82% yield, respectively from 1.0 g of ynamide 1a (eqn (1), Scheme 6). Interestingly, 2-chloro-N-phenyl indole 7 was readily accessed from 2o under oxidative conditions [PhI(OCOCF3)2 at 80 °C] (eqn (2), Scheme 6).19 Deprotection of N-Boc followed by intramolecular oxidative coupling between NH and o-C–H of the alkenyl–phenyl moiety allowed the formation of 7 from 2o.
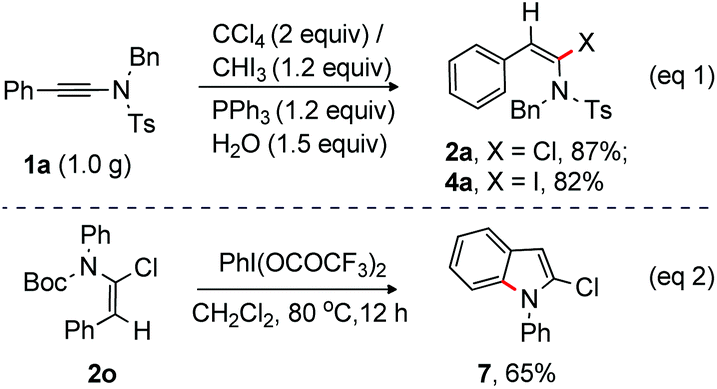 | ||
| Scheme 6 Gram scale preparation & synthesis of indole. | ||
To examine the role of H2O in the halogenation of ynamide, a control experiment reacting 1a with PPh3, CCl4 and D2O in CH2Cl2 was conducted at rt. Pleasingly, product 2a-D (80%) with a D-incorporation at the β-carbon of 1a was isolated; this suggests the occurrence of protonation in the final step of the reaction (Scheme 7).
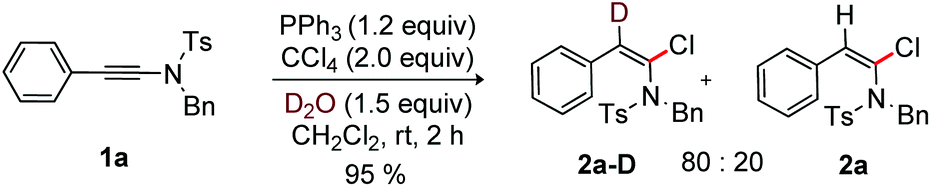 | ||
| Scheme 7 Reaction in deuterium oxide. | ||
The base mediated iodination of 3-acetoxy ynamide 8 produced Z-α-iodoacrylamides 9 at 40 °C, an elegant observation disclosed by the Skrydstrup group; this reaction proceeds through the 1,3-acetate shift (Scheme 8).8 Not surprisingly, compound 8 was efficiently converted to 9 under the optimized iodination conditions (entry 19, Table 1) at rt within 10 min (Scheme 8). Unexpectedly, the chlorination of 8 under the optimized conditions (entry 10, Table 1) exclusively delivered 3-acetoxy-α-chloro enamide 10 in excellent yield, when the reaction was independently conducted at rt or 70 °C; the formation of the corresponding α-chloroacrylamide was not detected (Scheme 8).8 Thus, two distinct products 9 and 10 can independently be accessed from 8 under different conditions.
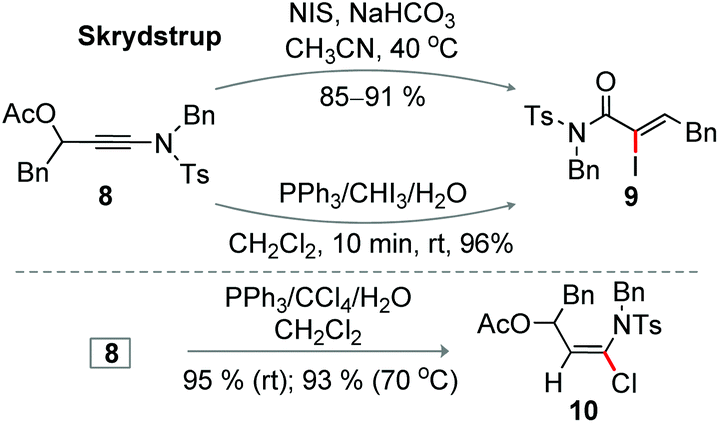 | ||
| Scheme 8 Halogenation of 3-acetoxy ynamide. | ||
To validate the probable path and the role of PPh3 in the reaction, we have performed a series of 31P NMR measurements. To begin with, the 31P NMR spectrum of a crude mixture containing PPh3/CHI3/CH2Cl2 and H2O in the absence of ynamide was recorded at regular intervals. An instant shift of PPh3 in the 31P NMR from −6.30 ppm to −8.41 ppm was observed; the respective peak at [28.1 ppm] for triphenylphosphine oxide [Ph3P(O)] was not detected. We believe that the peak at −8.41 ppm corresponds to the reactive species derived from PPh3 and CHI3, which is responsible for initiating the reaction with ynamide. In another experiment, 31P NMR of the reaction mixture containing PPh3/CHI3/CH2Cl2 and H2O in the presence of ynamide was examined at regular intervals. Interestingly, the peak at 28.1 ppm which corresponds to [Ph3P(O)] started appearing after 10 min. From this observation, we anticipate that the reaction is terminated with the liberation of Ph3P(O), which was isolated and characterized.
On the basis of the results from entry 11, Table 1 and the 31P NMR experiments, we believe that the reaction initiates with the syn-addition of phosphonium salt, generated in situ from PPh3 and CCl4/CBr4/CHI3, to the reactive ambivalent ynamide (1) in a concerted manner (I) to yield the regio- and stereoselective cationic β-phosphonium-α-haloenamide (II).21 Next, the addition of water to the cationic β-phosphonium–α-haloenamide (II) generates intermediate (III); subsequently proton quenching of (III) gives (IV). Finally, extrusion of [PPh3(O)] from IV leads to the desired (E)-α-haloenamide (Scheme 9).22 The formation of a minor Z-isomer is possible from the intermediate IV.
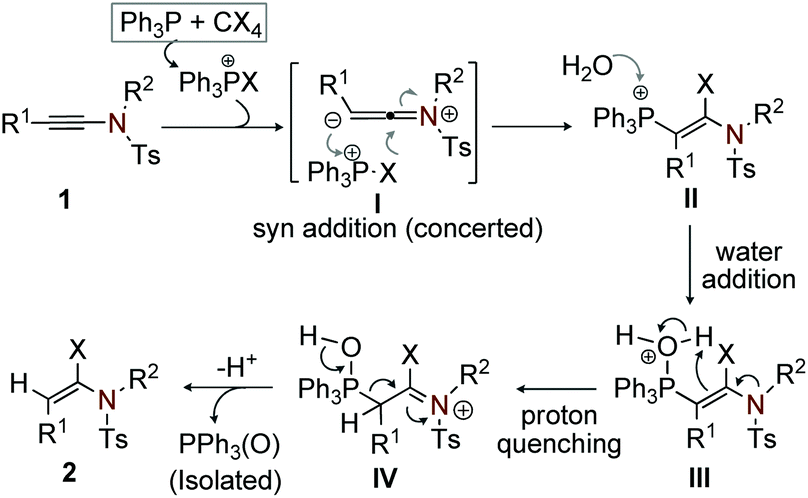 | ||
| Scheme 9 Plausible mechanistic path. | ||
Conclusions
In conclusion, we showcased a general, efficient, gram-scale synthetic method for the construction of stereodefined α-halo(Cl/Br/I)-enamides from ynamides using metal-free reagents [Ph3P and CCl4/CBr4/CHI3 in CH2Cl2 and H2O] at rt. The chlorination of ynamides at room temperature under metal-free conditions is notable. The role of H2O in the halogenation of ynamides is also elucidated through deuterium labelling experiment. The reaction displays a broad scope with the synthesis of a wide array of E-α-halo(Cl/Br/I)-enamides. Novel molecular scaffolds, such as tri-substituted enamides, amidoenynes, and 2-chloroindole, are readily accessible. 3-Acetoxy ynamides distinctly deliver diverse compounds under different conditions. Investigations to unravel the synthetic applications of the current method are underway.Acknowledgements
We thank UoH for financial support. P. B, S. N, R. K. M and R. P thank UGC, India for a fellowship. Mrs Srilakshmi and Dr Nagarjuna, UoH are thanked for X-ray crystallographic analysis.Notes and references
- (a) R. Tschesche, H. Last and H.-W. Fehlhaber, Chem. Ber., 1967, 100, 3937 CrossRef CAS PubMed; (b) K. A. Dekker, R. J. Aiello, H. Hirai, T. Inagaki, T. Sakakibara, Y. Suzuki, J. F. Thompson, Y. Yamauchi and N. Kojima, J. Antibiot., 1998, 51, 14 CrossRef CAS PubMed; (c) B. Kunze, R. Jansen, F. Sasse, G. Höfle and H. Reichenbach, J. Antibiot., 1998, 51, 1075 CrossRef CAS PubMed; (d) L. Yet, Chem. Rev., 2003, 103, 4283 CrossRef CAS PubMed; (e) P. S. Baran and R. A. Shenvi, J. Am. Chem. Soc., 2006, 128, 14028 CrossRef CAS PubMed.
- (a) G. R. Lenz, Synthesis, 1978, 489 CrossRef CAS; (b) P. W. Hickmott, Tetrahedron, 1982, 38, 1975 CrossRef CAS; (c) P. W. Hickmott, Tetrahedron, 1982, 38, 3363 CrossRef CAS; (d) J.-H. Maeng and R. L. Funk, Org. Lett., 2001, 3, 1125 CrossRef CAS PubMed; (e) J. R. Fuchs and R. L. Funk, Org. Lett., 2001, 3, 3349 CrossRef CAS PubMed; (f) G. Abbiati, F. Clerici, M. L. Gelmi, A. Gambini and T. Pilati, J. Org. Chem., 2001, 66, 6299 CrossRef CAS PubMed.
- (a) D. F. Mironova and N. A. Loginova, Ukr. Biokhim. Zh., 1986, 52, 71 CAS; (b) G. Y. Kondrat'eva, N. E. Agafonov and G. A. Stashina, Izv. Akad. Nauk SSSR, Ser. Khim., 1989, 9, 2038 Search PubMed; (c) S. Meier and E. U. Würthwein, Chem. Ber., 1990, 123, 2339 CrossRef CAS; (d) V. I. Boiko, A. A. Sinitsa and P. P. Onys'ko, Russ. J. Gen. Chem., 1999, 69, 1879 CAS; (e) S. Nayak, N. Ghosh and A. K. Sahoo, Org. Lett., 2014, 16, 2996 CrossRef CAS PubMed.
- (a) A. B. Lemay, K. S. Vulic and W. W. Ogilvie, J. Org. Chem., 2006, 71, 3615 CrossRef CAS PubMed; (b) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed; (c) D. Fujino, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2014, 136, 6255 CrossRef CAS PubMed.
- (a) H. Neumann and D. Seebach, Chem. Ber., 1978, 111, 2785 CrossRef CAS; (b) D. A. Evans, T. C. Crawford, R. C. Thomas and J. A. Walker, J. Org. Chem., 1976, 41, 3947 CrossRef CAS PubMed; (c) P. A. Lander and L. S. Hegedus, J. Am. Chem. Soc., 1994, 116, 8126 CrossRef CAS; (d) S. Miniére and J. C. Cintrat, J. Org. Chem., 2001, 66, 7385 CrossRef; (e) S. Miniére and J.-C. Cintrat, Synthesis, 2001, 705 CrossRef; (f) M. Schlosser, in Organometallics in Synthesis: A Manual, ed. M. Schlosser, Wiley, Chichester, UK, 2nd edn, 2002, pp. 1–352 Search PubMed; (g) L. Timbart and J.-C. Cintrat, Chem. – Eur. J., 2002, 8, 1637 CrossRef CAS PubMed; (h) C. Galli and Z. Rappoport, Acc. Chem. Res., 2003, 36, 580 CrossRef CAS PubMed; (i) R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292 CrossRef CAS PubMed.
- (a) J. A. Mulder, K. C. M. Kurtz, R. P. Hsung, H. A. Coverdale, M. O. Frederick, L. Shen and C. A. Zificsak, Org. Lett., 2003, 5, 1547 CrossRef CAS PubMed; (b) Y. Zhang, R. P. Hsung, M. R. Tracey, K. C. M. Kurtz and E. L. Vera, Org. Lett., 2004, 6, 1151 CrossRef CAS PubMed; (c) X. Zhang, Y. Zhang, J. Huang, R. P. Hsung, K. C. M. Kurtz, J. Oppenheimer, M. E. Petersen, I. K. Sagamanova, L. Shen and M. R. Tracey, J. Org. Chem., 2006, 71, 4170 CrossRef CAS PubMed; (d) J. Oppenheimer, W. L. Johnson, M. R. Tracey, R. P. Hsung, P.-Y. Yao, R. Liu and K. Zhao, Org. Lett., 2007, 9, 2361 CrossRef CAS PubMed.
- P. Maity, M. R. Klos and U. Kazmaier, Org. Lett., 2013, 15, 6246 CrossRef CAS PubMed.
- S. Kramer, S. D. Friis, Z. Xin, Y. Odabachian and T. Skrydstrup, Org. Lett., 2011, 13, 1750 CrossRef CAS PubMed.
- (a) G. Compain, K. Jouvin, A. Martin-Mingot, G. Evano, J. Marrot and S. Thibaudeau, Chem. Commun., 2012, 48, 5196 RSC; (b) B. Métayer, G. Compain, K. Jouvin, A. Martin-Mingot, C. Bachmann, J. Marrot, G. Evano and S. J. Thibaudeau, J. Org. Chem., 2015, 80, 3397 CrossRef PubMed.
- (a) A. H. Sato, K. Ohashi and T. Iwasawa, Tetrahedron Lett., 2013, 54, 1309 CrossRef CAS; (b) M. Ide, Y. Yauchi and T. Iwasawa, Eur. J. Org. Chem., 2014, 3262 CrossRef CAS; (c) M. Ide, Y. Yauchi, R. Shiogai and T. Iwasawa, Tetrahedron, 2014, 70, 8532 CrossRef CAS; (d) K. Ohashi, S. Mihara, A. H. Sato, M. Ide and T. Iwasawa, Tetrahedron Lett., 2014, 55, 632 CrossRef CAS.
- (a) N. Ghosh, S. Nayak and A. K. Sahoo, Chem. – Eur. J., 2013, 19, 9428 CrossRef CAS PubMed; (b) S. Nayak, N. Ghosh, B. Prabagar and A. K. Sahoo, Org. Lett., 2015, 17, 5662 CrossRef CAS PubMed; (c) S. Nayak, B. Prabagar and A. K. Sahoo, Org. Biomol. Chem. 10.1039/c5ob02262b.
- (a) G. Evano, A. Coste and K. Jouvin, Angew. Chem., 2010, 122, 2902 ( Angew. Chem., Int. Ed. , 2010 , 49 , 2840 ) CrossRef; (b) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064 CrossRef CAS PubMed; (c) X.-N. Wang, H.-S. Yeom, L.-C. Fang, S. He, Z.-X. Ma, B. L. Kedrowski and R. P. Hsung, Acc. Chem. Res., 2014, 47, 560 CrossRef CAS PubMed; (d) T. Lu and R. P. Hsung, ARKIVOC, 2014, 127 Search PubMed.
- R. Appel, Angew. Chem., Int. Ed. Engl., 1975, 14, 801 CrossRef.
- (a) O. Dmitrenko, C. Thorpe and R. D. Bach, J. Org. Chem., 2007, 72, 8298 CrossRef CAS PubMed; (b) P. J. Bunyan and J. I. G. Cadogan, J. Chem. Soc., 1963, 42 RSC; (c) S. A. Buckler, J. Am. Chem. Soc., 1962, 84, 3093 CrossRef CAS.
- See some of the PPh3 and CCl4 reactions: (a) S. G. Newman, C. S. Bryan, D. Perez and M. Lautens, Synthesis, 2011, 342 CAS; (b) E. Arstad, A. G. M. Barrett, B. T. Hopkins and J. Köbberling, Org. Lett., 2002, 4, 1975 CrossRef PubMed; (c) B. H. Lipshutz, D. W. Chung, B. Rich and R. Corral, Org. Lett., 2006, 8, 5069 CrossRef CAS PubMed.
- (a) H.-F. Klein, A. Kuhn, N. Kuhn, S. Laufer and M. Z. Ströbele, Anorg. Allg. Chem., 2012, 638, 1784 CrossRef CAS; (b) K. Sonagashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef.
- G.-P. Lu, K. R. Voigtritter, C. Cai and B. H. Lipshutz, J. Org. Chem., 2012, 77, 3700 CrossRef CAS PubMed.
- (a) G. Liu, W. Kong, J. Che and G. Zhu, Adv. Synth. Catal., 2014, 356, 3314 CrossRef CAS; (b) V. Dwivedi, M. Hari Babu, R. Kant and M. Sridhar Reddy, Chem. Commun., 2015, 51, 14996 RSC.
- Y. Du, R. Liu, G. Linn and K. Zhao, Org. Lett., 2006, 8, 5919 CrossRef CAS PubMed.
- (a) J. L. Dutton, R. Tabeshi, M. C. Jennings, A. J. Lough and P. J. Ragogna, Inorg. Chem., 2007, 46, 8594 CrossRef CAS PubMed.
- (a) A. H. Dabbagh and K. Faghihi, Tetrahedron, 2000, 56, 3611 CrossRef CAS; (b) S.-i. Kawaguchi and A. Ogawa, Org. Lett., 2010, 12, 1893 CrossRef CAS PubMed.
- See the ESI.‡.
Footnotes |
| † This manuscript is dedicated to Dr J. S. Yadav, IICT-Hyderabad, India for his outstanding contribution to Organic Chemistry on his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Copies of the 1H NMR, 13C NMR and HRMS data for all products. CCDC 1434764. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00345h |
| This journal is © the Partner Organisations 2016 |