
Copper-catalyzed tandem A3-coupling–isomerization–hydrolysis reactions of aldehydes and terminal alkynes leading to chalcones†
Yingwei
Zhao
and
Qiuling
Song
*
Institute of Next Generation Matter Transformation, College of Chemical Engineering at Huaqiao University, 668 Jimei Blvd, Xiamen, Fujian 361021, P. R. China. E-mail: qsong@hqu.edu.cn
First published on 29th December 2015
Abstract
Catalyzed by a simple copper salt, the reaction of an aryl aldehyde and a terminal alkyne in the presence of piperidine delivers the valuable chalcone products, which were rarely found in previous A3 coupling reactions. This reaction can be applied to a wide range of substrates. Mechanistic studies indicate that this reaction experiences a tandem process containing A3 coupling, copper assisted isomerization of propadienamine to allenylamine and subsequent hydrolysis.
Tandem reactions, in which multiple reactions are combined into one synthetic operation, have been extensively studied in synthetic chemistry in recent years.1 Three-component tandem reaction of an aldehyde, a terminal alkyne and an amine, often catalyzed by transition-metals and widely known as A3-coupling, generally leads to the formation of a propargylamine2 (Scheme 1, path A). Commonly, such a structure is stable under typical A3-coupling reaction conditions if there is no additional nucleophilic center3 on the substrate structure. An alternative reaction pathway is the generation of allenes via hydride shifting (1,5-H shift) of propargylamine along with the elimination of amine, which was firstly developed by Crabbe and later modified by Ma's group, et al. Formaldehyde,4 aldehydes,5 and ketones6 can be applied for the corresponding allene formations using this strategy (Scheme 1, path B). In conjunction with our current interest on copper catalyzed coupling reactions with alkynes,7 we unexpectedly discovered that these components could lead to valuable α,β-unsaturated ketones. So far, only two special instances, namely glyoxal-8 or pyridine-2-carbaldehyde-like9 substrates could achieve such products via alkyne–allene isomerization and subsequent hydrolysis, mainly due to the fact that the presence of the electron-withdrawing group in the propargylamine structure will promote deprotonation (Scheme 1, path C). Meanwhile, an expensive Au catalyst or special supported copper nanoparticles were a prerequisite to the success of the above transformations, therefore this severely hinders their application in synthesis.
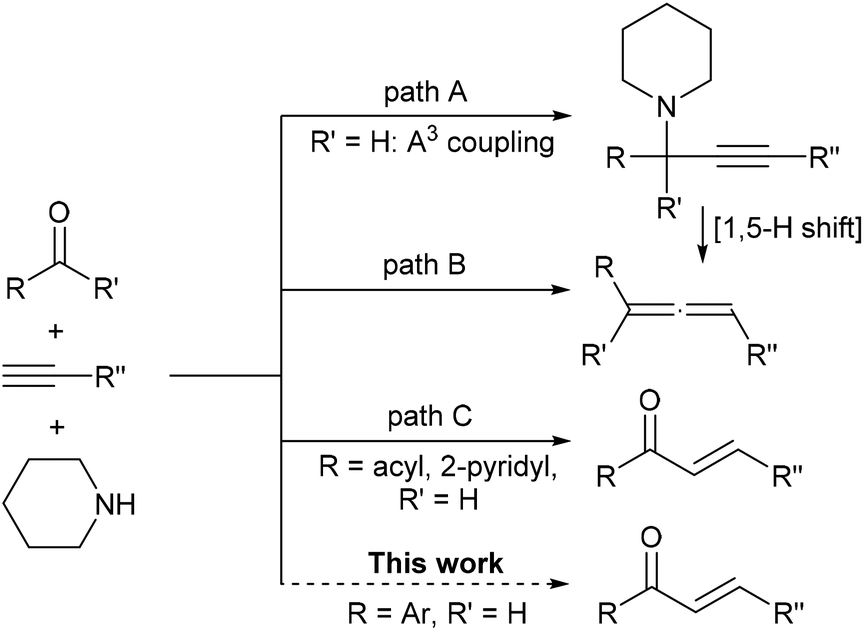 | ||
| Scheme 1 Three-component reaction of an aldehyde/ketone, a terminal alkyne and an amine. | ||
Our study showed that such a limitation as above could be conquered by using a simple and environmental benign copper salt as the catalyst in DMSO. The typical A3 starting materials with a broad scope of aryl alkynes and aldehyde could lead to the formation of chalcones. Although many strategies have been known for chalcone synthesis such as the classical Claisen–Schmidt condensation,10 catalyzed cross coupling,11a,c Meyer–Schuster rearrangements,11b carbonylation11f and hydroacylation,11h our method is a new strategy for the synthesis of such important biologically active building blocks12 from inexpensive and readily available starting materials.
We began our study on the traditional A3 coupling reaction by using benzaldehyde (1a), phenylacetylene (2a), and piperidine as the starting materials. Initially, catalyzed by Cu(OAc)2, the reaction was carried out in DMSO in the presence of pyridine as a base at 100 °C for 12 h. Unsurprisingly, the A3 product propargylamine (3aa′) was obtained in 27% GC yield as the major product. In addition, the unexpected product chalcone (3aa) was formed in 11% yield (Table 1, entry 1). We then decided to optimize the reaction conditions to make 3aa as the main product. To our delight, raising the reaction temperature to 130 °C efficiently reversed the product distribution (Table 1, entry 2). The solvent obviously played an important role in the selectivity of this reaction and DMSO was the only one among all solvents we screened which could lead to the formation of chalcone as the main product (Table 1, entries 3–7). Further screening on bases showed that pyridine was the optimal one (Table 1, entries 8 and 9). Interestingly, under pyridine-free conditions, only a slight decrease of chalcone formation was observed (Table 1, entry 10), which suggested that pyridine might not act as a base but a ligand in this transformation. The reactions performed under solvent free conditions did not lead to satisfactory results, showing the important role that DMSO played (Table 1, entries 11 and 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Base | Solvent |
3aa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
3aa′b (%) |
| a Reaction conditions: benzaldehyde (1a) (52 μL, 0.5 mmol), phenylacetylene (2a) (75 μL, 0.75 mmol), piperidine (74 μL, 0.75 mmol), catalyst (0.1 mmol), base (1 mmol), solvent (1.5 mL), N2 atmosphere. b Yields were based on GC analyses using n-dodecane as the internal standard. c The reaction was carried out at 100 °C. d Isolated yield. | |||||
| 1c | Cu(OAc)2 | Pyridine | DMSO | 11 | 27 |
| 2 | Cu(OAc)2 | Pyridine | DMSO | 50 | 5 |
| 3 | Cu(OAc)2 | Pyridine | Toluene | Trace | 72 |
| 4 | Cu(OAc)2 | Pyridine | o-Xylene | 4 | 78 |
| 5 | Cu(OAc)2 | Pyridine | t-AmOH | 0 | 88 |
| 6 | Cu(OAc)2 | Pyridine | Dioxane | Trace | 74 |
| 7 | Cu(OAc)2 | Pyridine | NMP | 16 | 35 |
| 8 | Cu(OAc)2 | Et3N | DMSO | 35 | 9 |
| 9 | Cu(OAc)2 | DABCO | DMSO | 38 | 1 |
| 10 | Cu(OAc)2 | — | DMSO | 45 | 7 |
| 11 | Cu(OAc)2 | Pyridine | — | 25 | 32 |
| 12 | Cu(OAc)2 | — | — | 27 | 5 |
| 13 | — | Pyridine | DMSO | 0 | 0 |
| 14 | CuBr | Pyridine | DMSO | 42 | 8 |
| 15 | CuBr2 | Pyridine | DMSO | 43 | 17 |
| 16 | CuCl | Pyridine | DMSO | 55 | 10 |
| 17 | CuCl2 | Pyridine | DMSO | 39 | 6 |
| 18 | CuI | Pyridine | DMSO | 33 | 7 |
| 19 | Cu(TFA)2 | Pyridine | DMSO | 28 | 21 |
| 20 | CuSO4 | Pyridine | DMSO | 76 (70)d | 1 |
| 21 | Cu(OTf)2 | Pyridine | DMSO | 46 | 2 |
| 22 | AuCl3 | Pyridine | DMSO | 39 | 4 |
| 23 | AgOTf | Pyridine | DMSO | 32 | 4 |
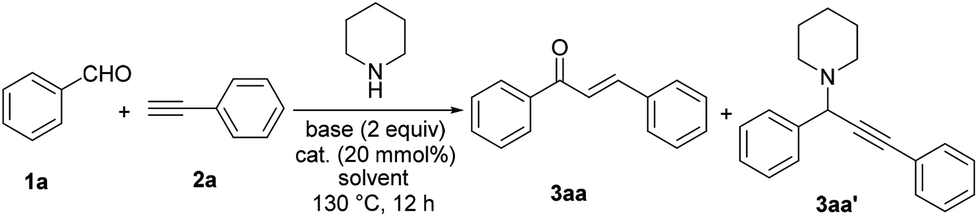
The control experiment showed that no reaction occurred in the absence of a copper catalyst (Table 1, entry 13). As a result of catalyst screening (Table 1, entries 14–23), CuSO4 exhibited the best reactivity and selectivity for chalcone formation (Table 1, entry 20). Other copper salts, either Cu(I) or Cu(II) gave low to medium yields. Notably, Au and Ag catalysts which were widely used in traditional A3 couplings only gave inferior yields of chalcones under our conditions. We also used other secondary amines such as morpholine and pyrrolidine instead of piperidine, however, they were ineffective for this reaction (see section 3.1 in the ESI†).
Next, we explored the scope of this tandem reaction by using various aryl aldehydes 1 and terminal alkynes 2 under the optimized conditions and the results are summarized in Scheme 2. A wide range of chalcones could be readily obtained by this protocol. We first examined the reaction of phenylacetylene (2a) with a series of aryl aldehydes 1. It could be seen that a wide range of substituents such as halo (Cl and Br), trifluoromethyl, cyano, acetyl and even the strong electron-withdrawing nitro group were all tolerated, affording the corresponding chalcones (3ba–3ja) in moderate to high yields. 2-Naphthaldehyde (1k) and the heteroaromatic aldehyde (1l) could also be applied for this reaction. We then explored the scope of terminal alkynes 2. Both electron-rich (2b) and electron-deficient (2c–2g) substituted phenylacetylenes exhibited good reactivity and the corresponding desired chalcones were obtained in moderate yields. All the reactions above had a clear preference for the E geometrical isomer demonstrated by a 15.5 Hz olefinic proton coupling constant. In a very few cases trace amounts of the Z products (<1:20) were detected. The exception is the reaction of 2-naphthaldehyde (1k), in which the corresponding chalcone (3ka) was obtained as a mixture of isomers with an E/Z ratio of 8:1. Unfortunately, this reaction could not be extended to strong electron-rich aldehydes, aliphatic aldehydes and aliphatic alkynes, wherein either A3 coupling products were afforded or only the starting material remained (for Me2N- or AcHN-substituted benzaldehyde).
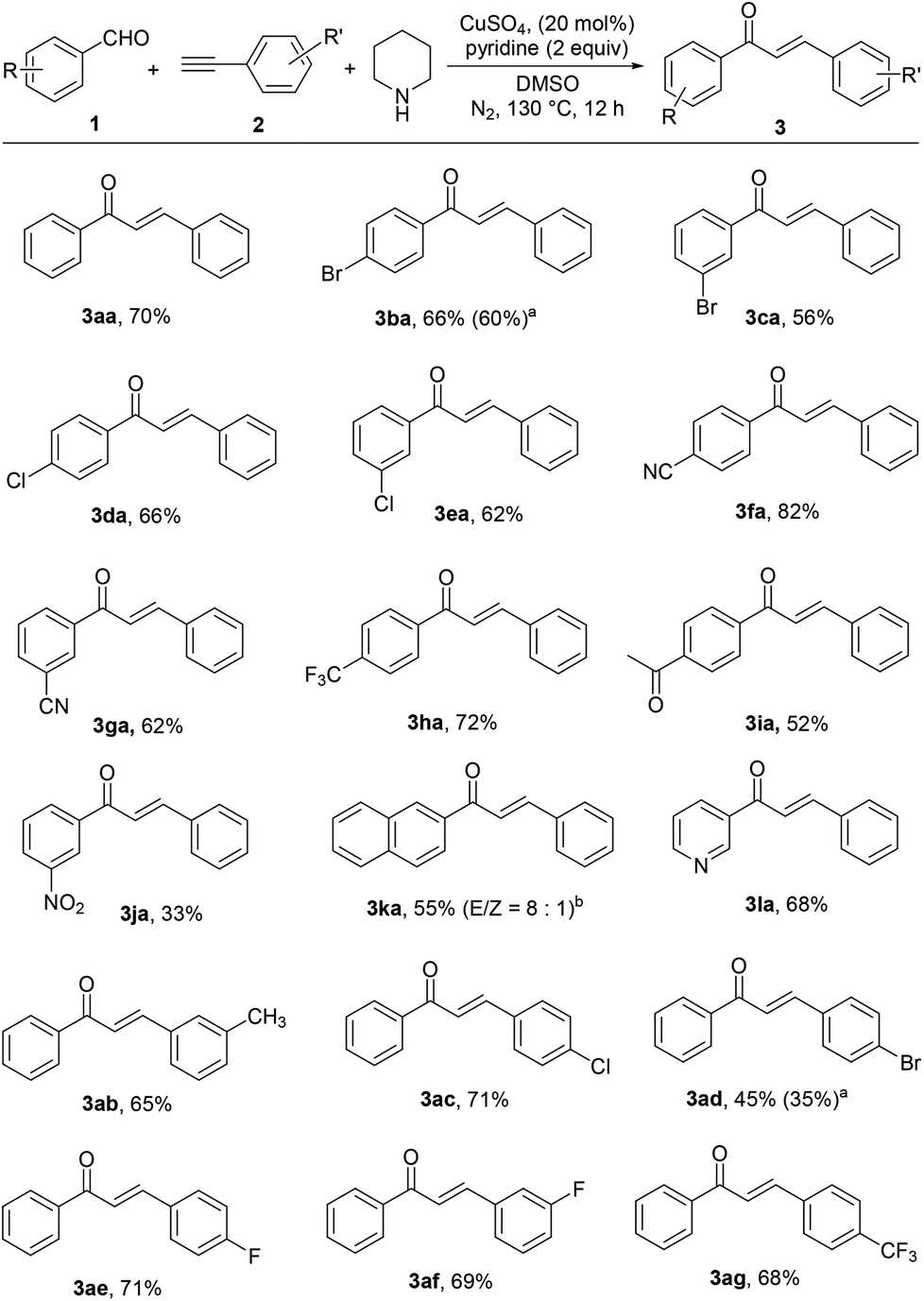 | ||
| Scheme 2 Substrate scope. aIn the absence of pyridine; bbased on GC. | ||
To gain insight into the reaction mechanism, we initially carried out some control experiments. Firstly, when the reaction was performed in the absence of piperidine, no chalcone product was formed (Scheme 3, eqn (1)). This result excludes the possible route containing the alkynylation of 1a leading to the formation of propargylic alcohol (1,3-diphenylprop-2-yn-1-ol) and the subsequent isomerization via 1,3-H shift.13 Next, the reaction of phenylacetylene (2a) with water under standard conditions afforded only a trace of acetophenone, illustrating that our reaction was not proceeded through the well-known Claisen–Schmidt condensation10 (Scheme 3, eqn (2)). These above experiments clearly supported that the A3 coupling was the first step. Therefore propargylamine 3aa′ was prepared and employed with water under different conditions. No reaction occurred when it was just heated at 130 °C in DMSO. Yet 44% of the desired product 3aa was obtained in the presence of CuSO4, which suggested that CuSO4 could promote chalcone formation in the absence of a base (Scheme 3, eqn (3) and (4)). Subsequently, our labeling experiment was conducted with D2O and it was found that two deuterium atoms were incorporated into the final product 3aa with 44% and 55% incorporation (Scheme 3, eqn (5)). In the 18O labeling experiment, H218O was added to the reaction system under standard conditions, resulting in the 18O-labeled product 18O-3aa (Scheme 3, eqn (6)). Finally, the A3 product with the propargylic proton alpha to the amine being replaced by deuterium (D-3aa′) was allowed to react with water. The low deuterated ratios in the product chalcone (D-3aa) indicated that this reaction did not proceed via 1,2-hydride transfer.
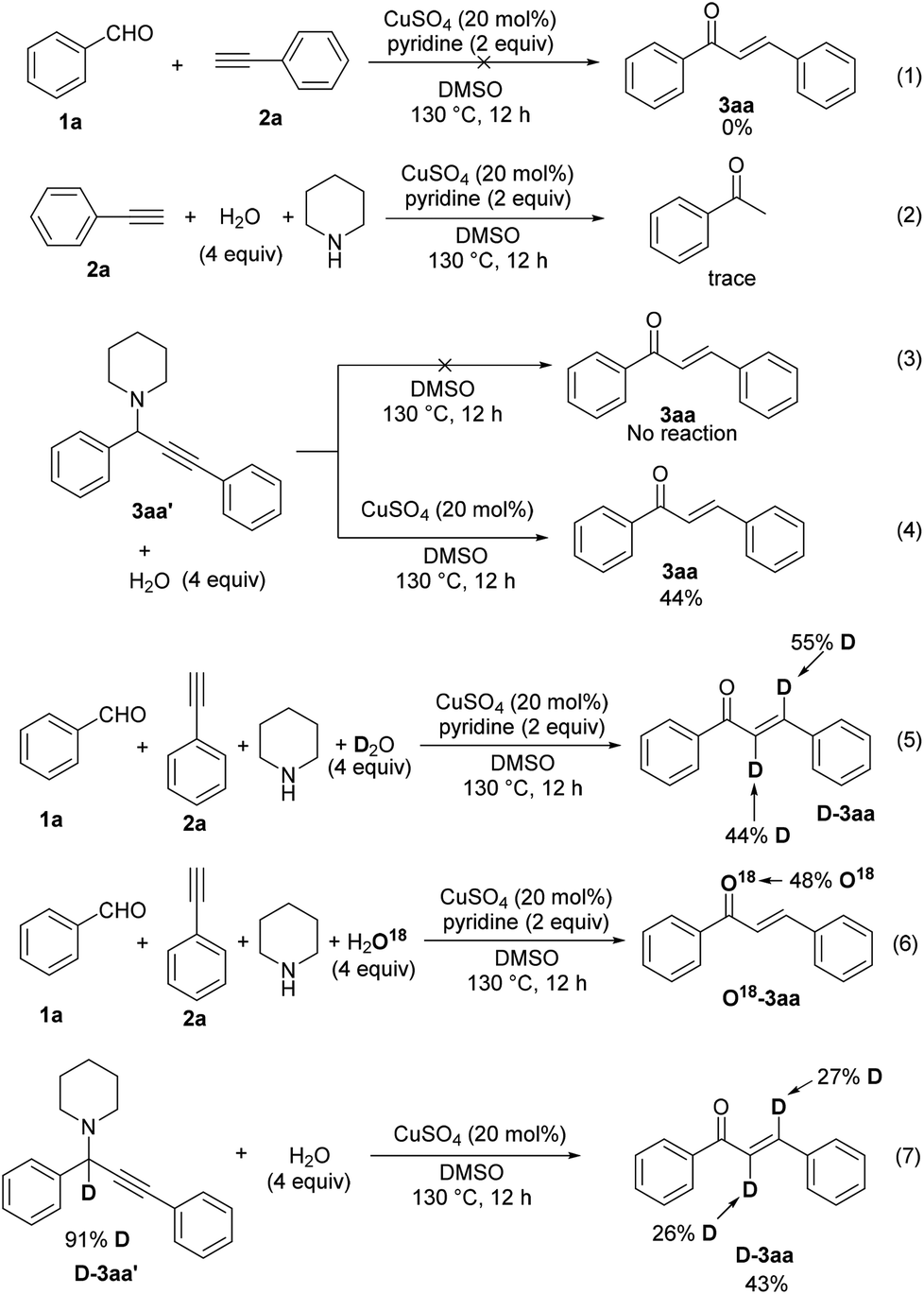 | ||
| Scheme 3 Control experiments. | ||
On the basis of all the above results, the reaction mechanism is postulated in Scheme 4. Initially, copper-catalyzed A3 coupling results in the formation of propargylamine 3aa′. Coordination of copper with a triple bond facilitates deprotonation by a base, which readily proceeds only in DMSO.14 The existence of electron-withdrawing substituents is generally beneficial for enhancing the acidity of the propargylic proton. Subsequent protonation affords the allenylamine intermediate 3aa′′.15 Hydrolysis of the enamine substructure then leads to the final product 3aa, and the thermodynamically more stable E geometrical isomer is formed preferentially.
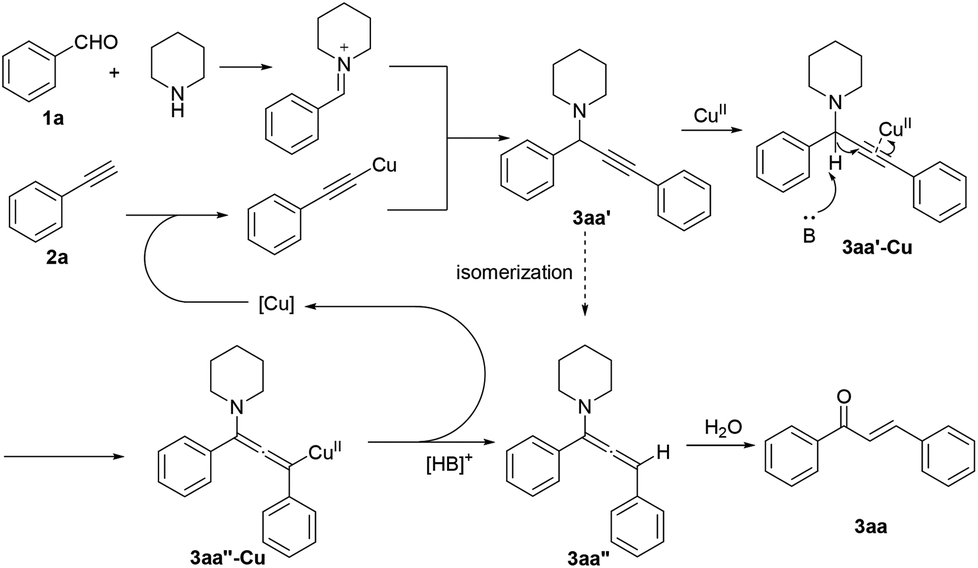 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a novel copper-catalyzed A3-coupling–isomerization–hydrolysis tandem reaction to afford valuable chalcones from aryl aldehydes, terminal alkynes and piperidine, instead of stopping at the A3-coupling step. A mechanistic study showed that the copper catalyst might play dual roles in this transformation: facilitate the A3 coupling as well as promote the isomerization of the propargylamine intermediate via coordination with a triple bond. Further research on understanding of this new reaction is ongoing in our laboratory.Financial support from the National Science Foundation of China (21202049), the Recruitment Program of Global Experts (1000 Talents Plan) and the Fujian Hundred Talents Plan and the Program of Innovative Research Team of Huaqiao University (Z14X0047) are gratefully acknowledged.
Notes and references
- (a) P. J. Parsons, C. S. Penkett and A. J. Shell, Chem. Rev., 1996, 96, 195–206 CrossRef CAS PubMed; (b) J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed.
- (a) V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790–3807 RSC; (b) C.-J. Li and C. Wei, Chem. Commun., 2002, 268–269 RSC; (c) C. Wei and C.-J. Li, J. Am. Chem. Soc., 2003, 125, 9584–9585 CrossRef CAS PubMed; (d) C. Zhang and D. Seidel, J. Am. Chem. Soc., 2010, 132, 1798–1799 CrossRef CAS PubMed; (e) C. Wei, Z. Li and C.-J. Li, Synlett, 2004, 1472–1483 CAS; (f) A. B. Dyatkin and R. A. Rivero, Tetrahedron Lett., 1998, 39, 3647–3650 CrossRef CAS; (g) C.-J. Li, Acc. Chem. Res., 2010, 43, 581–590 CrossRef CAS PubMed.
- (a) N. T. Patil and V. S. Raut, J. Org. Chem., 2010, 75, 6961–6964 CrossRef CAS PubMed; (b) N. Chernyak and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2743–2746 CrossRef CAS PubMed; (c) B. Yan and Y. Liu, Org. Lett., 2007, 9, 4323–4326 CrossRef CAS PubMed; (d) H. Ohno, Y. Ohta, S. Oishi and N. Fujii, Angew. Chem., Int. Ed., 2007, 46, 2295–2298 CrossRef CAS PubMed; (e) Y. Ohta, H. Chiba, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2009, 74, 7052–7058 CrossRef CAS PubMed; (f) Q. Zhang, M. Cheng, X. Hu, B.-G. Li and J.-X. Ji, J. Am. Chem. Soc., 2010, 132, 7256–7257 CrossRef CAS PubMed; (g) N. Sakai, N. Uchida and T. Konakahara, Tetrahedron Lett., 2008, 49, 3437–3440 CrossRef CAS.
- (a) S. Searles, Y. Li, B. Nassim, M.-T. R. Lopes, P. T. Tran and P. Crabbe, J. Chem. Soc., Perkin Trans. 1, 1984, 747–751 RSC; (b) P. Crabbe, H. Fillion, D. Andre and J.-L. Luche, J. Chem. Soc., Chem. Commun., 1979, 859–860 RSC.
- (a) J. Kuang and S. Ma, J. Am. Chem. Soc., 2010, 132, 1786–1787 CrossRef CAS PubMed; (b) X. Huang, T. Cao, Y. Han, X. Jiang, W. Lin, J. Zhang and S. Ma, Chem. Commun., 2015, 51, 6956–6959 RSC; (c) J. Zhang, J. Ye and S. Ma, Org. Biomol. Chem., 2015, 13, 4080–4089 RSC.
- X. Tang, C. Zhu, T. Cao, J. Kuang, W. Lin, S. Ni, J. Zhang and S. Ma, Nat. Commun., 2013, 4, 2450, DOI:10.1038/ncomms3450.
- M. Zhou, M. Chen, Y. Zhou, K. Yang, J. Su, J. Du and Q. Song, Org. Lett., 2015, 17, 1786–1789 CrossRef CAS PubMed.
- S. Shi, T. Wang, V. Weingand, M. Rudolph and A. S. Hashmi, Angew. Chem., Int. Ed., 2014, 53, 1148–1151 CrossRef CAS PubMed.
- M. J. Albaladejo, F. Alonso and M. Yus, Chem. – Eur. J., 2013, 19, 5242–5245 CrossRef CAS PubMed.
- (a) S. Sebti, A. Solhy, R. Tahir and A. Smahi, Appl. Catal., A, 2002, 235, 273–281 CrossRef CAS; (b) K. Mogilaiah and N. Vasudeva Reddy, Synth. Commun., 2003, 33, 73–78 CrossRef CAS; (c) B. J. G. A. G. Doshi, Curr. Sci., 1986, 55, 502–503 Search PubMed; (d) F. Toda, K. Tanaka and K. Hamai, J. Chem. Soc., Perkin Trans. 1, 1990, 3207–3209 RSC; (e) J.-T. Li, W.-Z. Yang, S.-X. Wang, S.-H. Li and T.-S. Li, Ultrason. Sonochem., 2002, 9, 237–239 CrossRef CAS PubMed; (f) S. Sebti, A. Saber, A. Rhihil, R. Nazih and R. Tahir, Appl. Catal., A, 2001, 206, 217–220 CrossRef CAS.
- For other methods for chalcone synthesis, see: (a) J. Zhou, G. Wu, M. Zhang, X. Jie and W. Su, Chem. – Eur. J., 2012, 18, 8032–8036 CrossRef CAS PubMed; (b) S. Swaminathan and K. V. Narayanan, Chem. Rev., 1971, 71, 429–438 CrossRef CAS; (c) J. Wang, C. Liu, J. Yuan and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 2256–2259 CrossRef CAS PubMed; (d) F. Fang, Y. Li and S.-K. Tian, Eur. J. Org. Chem., 2011, 1084–1091 CrossRef CAS; (e) X.-F. Wu, H. Neumann and M. Beller, Chem. – Asian J., 2012, 7, 282–285 CrossRef CAS PubMed; (f) J. Schranck, X.-F. Wu, H. Neumann and M. Beller, Chem. – Eur. J., 2012, 18, 4827–4831 CrossRef CAS PubMed; (g) S. Kim, S. W. Bae, J. S. Lee and J. Park, Tetrahedron, 2009, 65, 1461–1466 CrossRef CAS; (h) M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed; (i) J. Safaei-Ghomi, H. Shahbazi-Alavi, A. Ziarati, R. Teymuri and M. R. Saberi, Chin. Chem. Lett., 2014, 25, 401–405 CrossRef CAS; (j) M. von Delius, C. M. Le and V. M. Dong, J. Am. Chem. Soc., 2012, 134, 15022–15032 CrossRef CAS PubMed; (k) M. Castaing, S. L. Wason, B. Estepa, J. F. Hooper and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 13280–13283 CrossRef CAS PubMed.
- (a) N. K. Sahu, S. S. Balbhadra, J. Choudhary and D. V. Kohli, Curr. Med. Chem., 2012, 19, 209–225 CrossRef CAS PubMed; (b) A. Kamal, G. Ramakrishna, P. Raju, A. Viswanath, M. Janaki Ramaiah, G. Balakishan and M. Pal-Bhadra, Bioorg. Med. Chem. Lett., 2010, 20, 4865–4869 CrossRef CAS PubMed; (c) R. Romagnoli, P. G. Baraldi, M. D. Carrion, O. Cruz-Lopez, C. L. Cara, J. Balzarini, E. Hamel, A. Canella, E. Fabbri, R. Gambari, G. Basso and G. Viola, Bioorg. Med. Chem. Lett., 2009, 19, 2022–2028 CrossRef CAS PubMed; (d) R. Li, G. L. Kenyon, F. E. Cohen, X. Chen, B. Gong, J. N. Dominguez, E. Davidson, G. Kurzban, R. E. Miller, E. O. Nuzum, P. J. Rosenthal and J. H. McKerrow, J. Med. Chem., 1995, 38, 5031–5037 CrossRef CAS PubMed.
- (a) T. Baba, H. Kizuka, H. Handa and Y. Ono, Appl. Catal., A, 2000, 194–195, 203–211 CrossRef CAS; (b) T. Ishikawa, T. Mizuta, K. Hagiwara, T. Aikawa, T. Kudo and S. Saito, J. Org. Chem., 2003, 68, 3702–3705 CrossRef CAS PubMed; (c) M. Egi, M. Umemura, T. Kawai and S. Akai, Angew. Chem., Int. Ed., 2011, 50, 12197–12200 CrossRef CAS PubMed.
- Y. Fang, S. MacMillar, J. Eriksson, M. Kołodziejska-Huben, A. Dybała-Defratyka, P. Paneth, O. Matsson and K. C. Westaway, J. Org. Chem., 2006, 71, 4742–4747 CrossRef CAS PubMed.
- W. Fan and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 14542–14545 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and full spectroscopic data for all products. See DOI: 10.1039/c5qo00282f |
| This journal is © the Partner Organisations 2016 |