
Cu-catalyzed hydrofluoroacetylation of alkynes or alkynyl carboxylic acids leading highly stereoselectively to fluoroacetylated alkenes†
Miaolin
Ke
a,
Qiang
Feng
b,
Kai
Yang
b and
Qiuling
Song
*bc
aCollege of Materials Science & Engineering, Huaqiao University, China
bInstitute of Next Generation Matter Transformation, College of Chemical Engineering, Huaqiao Univeristy, 668 Jimei Blvd, Xiamen, Fujian, 361021, P. R. China
cBeijing National Laboratory for Molecular Sciences, Beijing, 100190, P. R. China. E-mail: qsong@hqu.edu.cn; Fax: +86-592-6162990
First published on 7th December 2015
Abstract
A copper-catalyzed hydrofluoroacetylation of arylacetylenes or alkynyl carboxylic acids with bromofluoroacetates was developed. The reaction shows excellent stereoselectivity to afford fluoroacetylated alkenes in good to excellent yields with a broad substrate scope. A preliminary mechanism study suggested that a radical pathway was involved in the reaction and (phenylethynyl)copper might be the key intermediate for the reaction. B2pin2 also plays an indispensable role in this novel hydrofluoroacetylation reaction.
The incorporation of fluorinated functional groups into organic molecules has attracted great attention and become an appealing topic in synthetic organic chemistry, owing to their prevailing presence in many bioactive compounds1 and their unique properties in enhancing the lipophilicity, metabolic stability and bioavailability of the parent molecules.2 Although significant advances have been made in the fluoroalkylation of aromatic compounds using functionalized fluoroalkyl reagents,3 efficient and general methods for the synthesis of alkenes containing difluorinated functional groups with high stereoselectivity are relatively rare. There are several one-step ways to obtain fluoroalkylated alkene compounds: (1) transition-metal-catalyzed cross coupling between fluoroalkyl metal species and alkenyl halides (Scheme 1a);4 (2) transition-metal-catalyzed cross coupling between alkenyl metal species and fluoroalkyl halides (Scheme 1b);5 (3) transition-metal-catalyzed Heck-type reactions between fluoroalkyl halides and alkenes6 or radical addition of halofluoroalkanes to alkenes7 (Scheme 1c); (4) halo or hydrofluoroalkylation of alkynes with fluoroalkyl halides8 (Scheme 1d). However, there are several drawbacks of the first two processes: stoichiometric amounts of transition metals are always required and the substrate alkenyl metals (such as alkenyl Zr) and alkenyl halides are not readily available. The Heck-type process provides a straightforward alternative. Recently, Zhang and co-workers reported an elegant Pd catalyzed Heck-type fluoroalkylation of alkenes using perfluoroalkyl bromides,6a however, aliphatic alkenes and internal linear alkenes failed to provide difluoroacetylated alkenes in this strategy. In terms of halo- or hydrofluoroalkylation of alkynes, both transition-metal-catalyzed processes and transition-metal-free protocols have been developed. For instance, Besset, Poisson et al. reported a Cu-mediated difunctionalization of alkynes with BrCF2COOEt leading to halofluoroalkylation of the alkynes;8a and in 1999 Chen et al. reported a transition-metal free hydrodifluoroacetylation of alkynes with Na2S2O3 as a radical initiator.8b In the latter report, the authors claimed that the stereoselectivity of their reaction provided an E-type product,8b unfortunately Réglier et al. later on proved that the stereochemistry in the above paper was wrong and the major products turned out to be Z isomers!8c
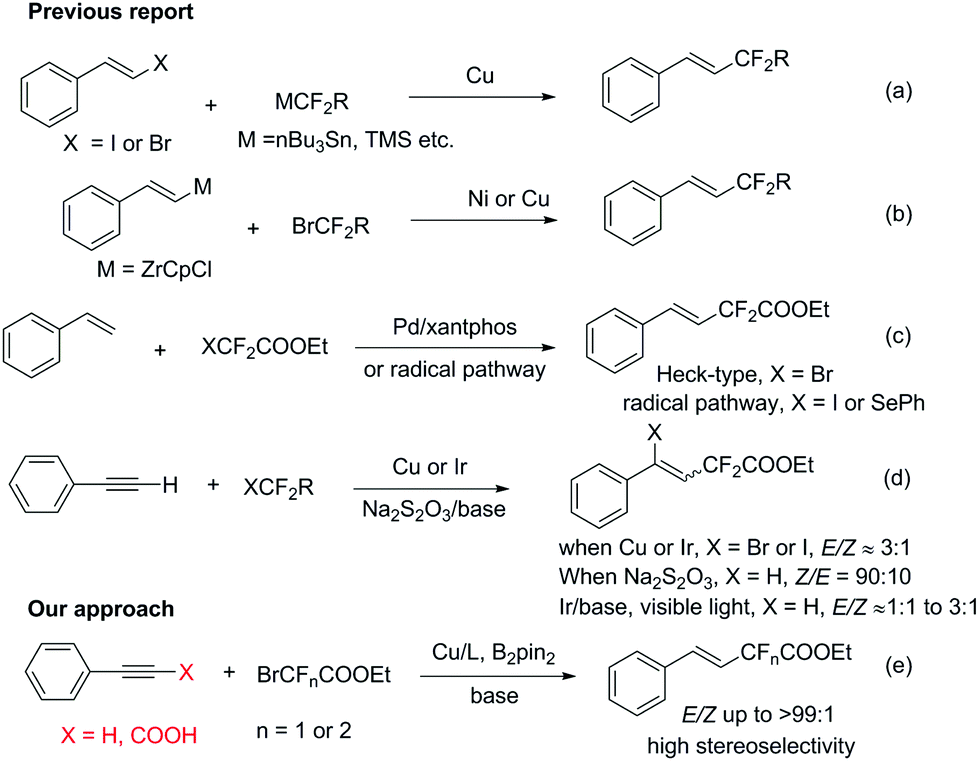 | ||
| Scheme 1 Synthetic methods for obtaining difluoroalkylated alkenes. | ||
To date, the hydrofluoroalkylation of alkynes with high E-stereoselectivity is relatively rare and remains appealing and challenging in modern synthetic chemistry, due to the prevalence and easy accessibility of alkynes and fluoroalkyl halides. In addition, copper salts are much cheaper and more environmentally benign than other precious nobel metals. Herein, we would like to report a highly stereoselective hydrodifluoroacetylation or hydromonofluoroacetylation of alkynes under a Cu/diboron system leading to (E)-fluoroacetylated alkenes in good to excellent yields, from low cost and readily available bromodi- or bromomono-fluoroacetates, with a broad substrate scope under mild reaction conditions. Remarkably, alkynyl carboxylic acids were competent substrates for this transformation as well and showed even better stereoselectivities than the corresponding alkynes with good to excellent yields.
Our investigation commenced with phenylacetylene (1) and ethyl bromodifluoroacetate (2) as the model substrates with 1 equivalent of B2pin2 in an attempt to obtain compound 4. Surprisingly, with copper salts, no compound 4 was ever detected, instead, the difluoroacetylated alkene 3 was obtained in a decent yield. This result inspired us, since the available literature on transition-metal-catalyzed efficient and highly stereoselective hydrodifluoroacetylations of alkynes to provide difluorinated alkenes is scant to date. After extensive screening of copper catalysts, ligands, bases and solvents (see Tables S1–S4 in the ESI† for details), it turned out that CuBr2, DTBDPy (4,4′-di-tert-butyl-2,2′-dipyridyl) and KOAc with 1 equivalent of B2pin2 in 2 mL of dioxane is the optimal combination (Table 1, entry 5). Potassium carbonate could be an alternative to KOAc since it gave a similar result as the latter for the reaction (entry 4 vs. entry 5 in Table 1). Control experiments suggested that CuBr2, DTBDPy and KOAc are all prerequisites for the success of the reaction (Table 1, entries 6, 7 and 9). Notably, additive B2pin2 is also indispensable for the reaction and no reaction occurred in its absence (Table 1, entry 8). The reaction is anaerobic and could not proceed under air or molecular oxygen (Table 1, entry 12 and Table S5,† entries 12 and 13). The temperature had an obvious effect on the reaction, when the temperature was reduced to 70 °C, the corresponding yield of product 3 was reduced to 83% vs. 99% by GC (Table 1, entry 13). Increasing the temperature to 90 °C did not increase the yield of the product, surprisingly, a much lower yield of product was obtained (77% in Table S5 in the ESI†). Further fine tuning of the amount of catalyst and ligand as well as ethyl bromodifluoroacetate (2) eventually gave the optimized conditions listed in Table 1, entry 5.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Ligand | Base (2 equiv.) | Solvent | Additive | Yieldb,c (%) |
| a Reaction conditions: phenylacetylene (1) (0.25 mmol), ethyl bromodifluoroacetate (2) (1 mmol), bis(pinacolato)diboron (additive) (0.25 mmol), catalyst (10 mol%), ligand (10 mol%), base (0.5 mmol), and solvent (2 mL) under a N2 atmosphere at 80 °C for 16 h. b GC yield using n-dodecane as an internal standard. c The isolated yield is listed in parenthesis. d Using CuBr2 (5 mol%). e Under air. f At 70 °C. g Using 2 equiv. of BrCF2COOEt. h Using 3 equiv. of BrCF2COOEt. DTBDPy = 4,4′-di-tert-butyl-2,2′-dipyridyl. | ||||||
| 1 | CuBr | 1,10-Phen | K2CO3 | Dioxane | B2pin2 | 67 |
| 2 | CuBr2 | 1,10-Phen | K2CO3 | Dioxane | B2pin2 | 83 |
| 3 | CuCl | 1,10-Phen | K2CO3 | Dioxane | B2pin2 | 33 |
| 4 | CuBr2 | DTBDPy | K2CO3 | Dioxane | B2pin2 | 98 |
| 5 | CuBr2 | DTBDPy | KOAc | Dioxane | B2pin2 | >99 (78) |
| 6 | — | DTBDPy | KOAc | Dioxane | B2pin2 | 0 |
| 7 | CuBr2 | — | KOAc | Dioxane | B2pin2 | 0 |
| 8 | CuBr2 | DTBDPy | KOAc | Dioxane | — | 0 |
| 9 | CuBr2 | DTBDPy | — | Dioxane | B2pin2 | 0 |
| 10 | CuBr2 | DTBDPy | KOAc | CH3CN | B2pin2 | 76 |
| 11d | CuBr2 | DTBDPy | KOAc | Dioxane | B2pin2 | 51 |
| 12e | CuBr2 | DTBDPy | KOAc | Dioxane | B2pin2 | Trace |
| 13f | CuBr2 | DTBDPy | KOAc | Dioxane | B2pin2 | 83 |
| 14g | CuBr2 | DTBDPy | KOAc | Dioxane | B2pin2 | 70 |
| 15h | CuBr2 | DTBDPy | KOAc | Dioxane | B2pin2 | 75 |
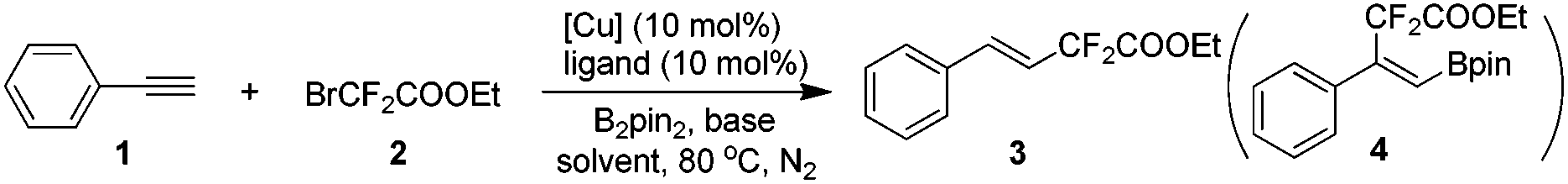
The scope of the Cu-catalyzed hydrodifluoroacetylation was surveyed using the optimized reaction conditions. A variety of alkynes, including both aromatic and aliphatic ones, could be hydrodifluoroacetylated smoothly using this transformation (Scheme 2). For the aromatic alkynes, a variety of substituents on the aromatic ring were compatible under the standard conditions, indicating that the reaction has a broad substrate scope. In addition to the normal electron-neutral (5–9) and electron-rich (10–11) substituents, halo substituents (12–16) were well-tolerated in this reaction leading to products which could be further structurally elaborated, with good stereoselectivity. Meanwhile, electron-deficient groups (17–19) were compatible under the standard conditions as well, albeit with relatively lower yields and lower stereoselectivities. Polyphenylalkynes also performed well under the standard conditions and the corresponding hydrodifluoroacetylated products (20 and 21) were obtained in reasonable yield. Noteworthily, heteroaromatic alkynes, such as 2-thiophene and 2-pyridine alkynes, were proven to be good candidates for this transformation with excellent stereoselectivities (22 and 23). Gratifyingly, both aliphatic alkynes and non-terminal alkynes were good substrates in this transformation and the desired products were obtained in good yields, albeit the stereoselectivities were relatively lower (24–27).
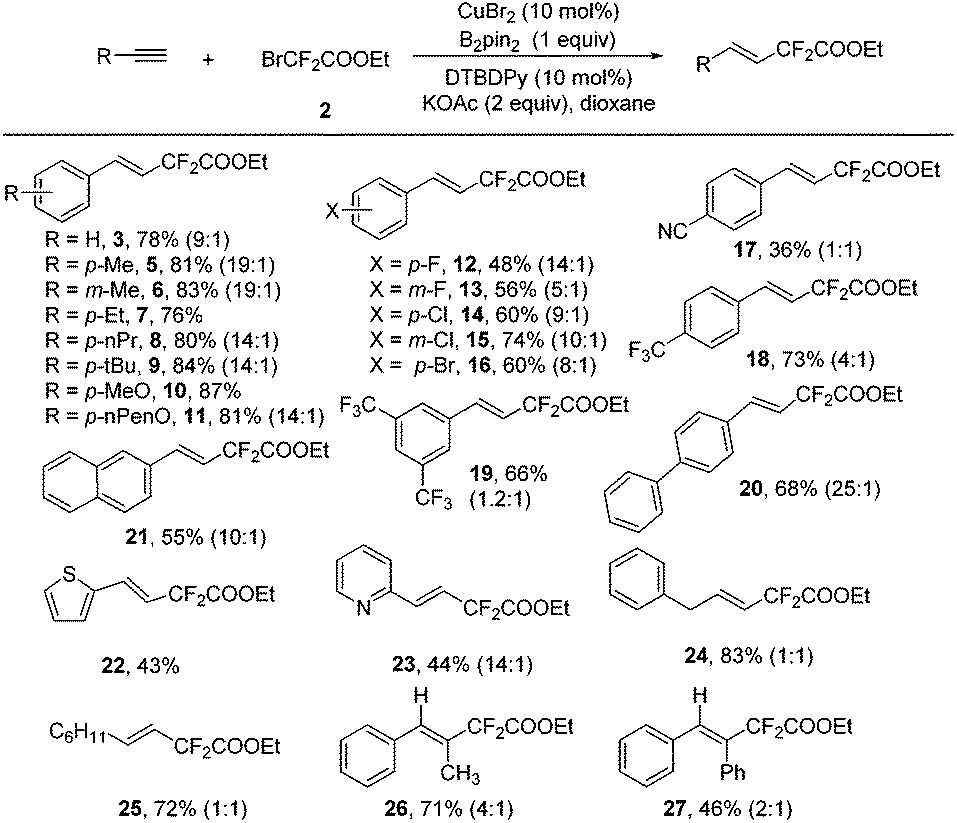 | ||
| Scheme 2 Substrate scope of the alkynes for the hydrodifluoroacetylation. Reaction conditions: alkyne (0.25 mmol), BrCF2COOEt (2) (1.0 mmol), CuBr2 (10 mol%), B2pin2 (0.5 mmol), DTBDPy (10 mol%), KOAc (2 equiv.), dioxane (2 mL), 80 °C, 16 h, under N2 in a sealed tube. Isolated yield based on the alkyne. The ratio in parenthesis is the E/Z ratio. | ||
As a practical alternative to terminal alkynes, alkynyl carboxylic acids have attracted great attention recently in the synthetic organic community and sometimes they showed superior reactivity compared to their terminal alkyne counterparts.9 Therefore, alkynyl carboxylic acids were employed in the above transformation to determine the feasibility of this type of compound. Delightfully, hydrodifluoroacetylated 3 was obtained in 62% yield upon isolation. Further screening demonstrated that a Cu(TFA)2/1,10-phenathroline/Na2CO3 system showed superior activity (see Table S6 in the ESI† for details), and that using 70 °C provided the same result as 80 °C. Once again, the copper, ligand, base and additive B2pin2 were all essential to the reaction since no product was generated in the absence of any of them (Table S6, ESI†).
Various alkynyl carboxylic acids were investigated using bromodifluoroacetate under the above optimal conditions (Scheme 3). A series of difluoroacetylated alkenes were obtained in good to excellent yields. Noteworthily, in most cases, the alkynyl carboxylic acids showed better a stereoselectivity in this transformation than their terminal alkyne counterparts (3, 5–6, 9–10, 12–14, 16, 18, 20 and 22 in Schemes 2 and 3). Most remarkably, for the aliphatic products, the alkynyl carboxylic acid gave compound 30 in exclusively the trans isomer form in 83% yield, yet for the terminal alkyne case, desired products 24 and 25 were obtained in 83% and 72% yield with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E:Z ratio. This clearly underscores the advantages of using alkynyl carboxylic acids in this transformation; this might be a complementary method to the recent elegant publication from Zhang and co-workers, since they reported a Pd-catalyzed Heck-type fluoroalkylation of alkenes with perfluoroalkyl bromides leading to fluoroalkylated alkenes,6a however, aliphatic alkenes and internal linear alkenes failed to provide difluoroacetylated alkenes in their strategy. The exact reasons for the differences between the terminal alkynes and alkynyl carboxylic acids are unknown, most likely the carboxylic acid group and the different ligands play critical roles in determining the stereoselectivity.
:1 E:Z ratio. This clearly underscores the advantages of using alkynyl carboxylic acids in this transformation; this might be a complementary method to the recent elegant publication from Zhang and co-workers, since they reported a Pd-catalyzed Heck-type fluoroalkylation of alkenes with perfluoroalkyl bromides leading to fluoroalkylated alkenes,6a however, aliphatic alkenes and internal linear alkenes failed to provide difluoroacetylated alkenes in their strategy. The exact reasons for the differences between the terminal alkynes and alkynyl carboxylic acids are unknown, most likely the carboxylic acid group and the different ligands play critical roles in determining the stereoselectivity.
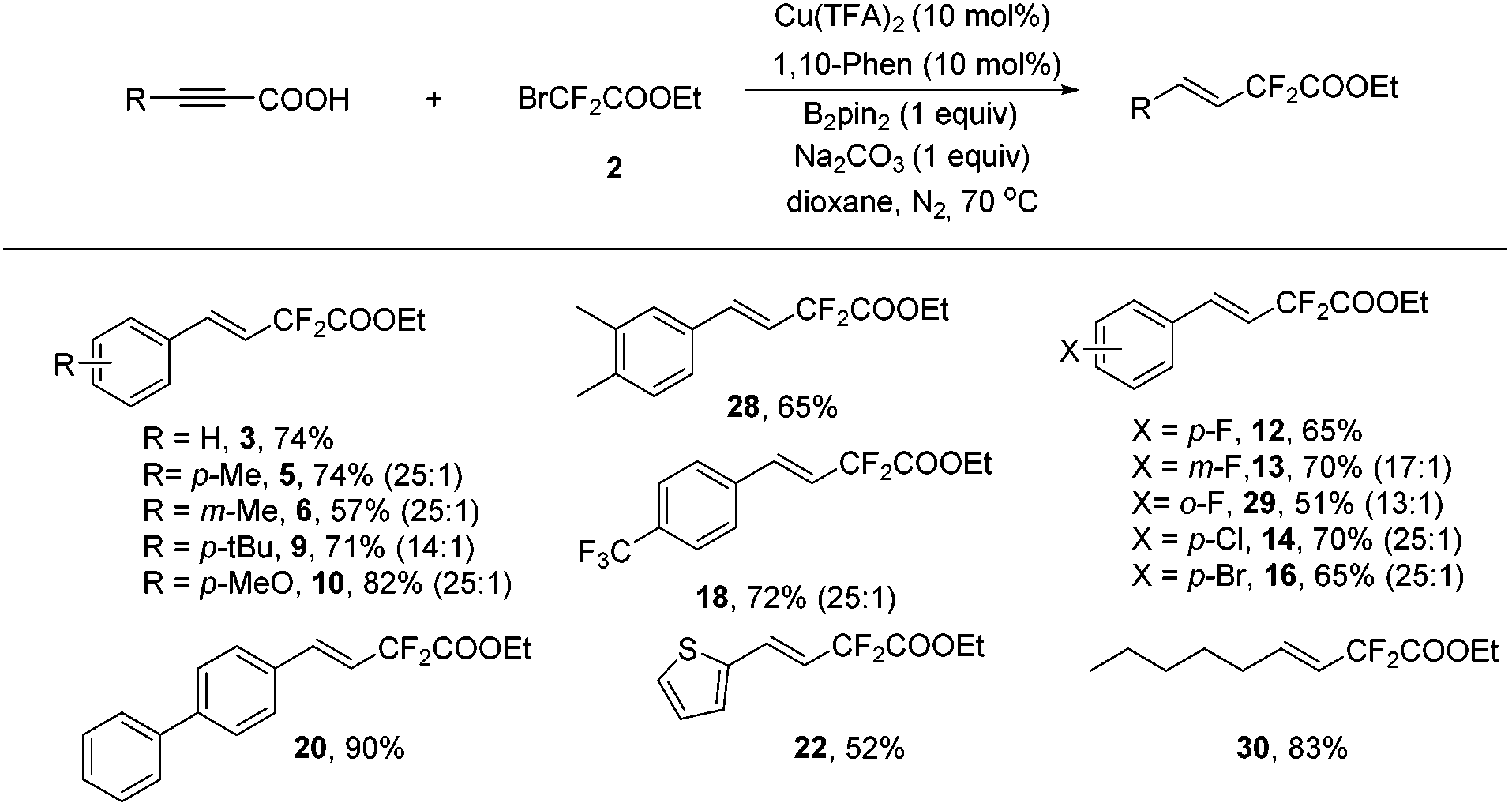 | ||
| Scheme 3 Substrate scope of the alkynyl carboxylic acids for the hydrodifluoroacetylation. Reaction conditions: alkynyl carboxylic acid (0.25 mmol), BrCF2COOEt (2) (0.75 mmol), Cu(TFA)2 (10 mol%), B2pin2 (0.5 mmol), 1,10-phen (10 mol%), Na2CO3 (1 equiv.), dioxane (1 mL), 70 °C, 24 h, under N2 in a sealed tube. Isolated yield based on the alkynyl carboxylic acid. The ratio in parenthesis is the E/Z ratio. | ||
The reaction was also extended to phenylalanine derived bromodifluoroamide 31, which was prepared from bromodifluoroacetate 2 with a phenylalanine ester.10 To our delight, when two terminal alkynes were employed with the difluoro reagent under the standard conditions, the corresponding desired products 32 and 33 were obtained in 66% and 55% yields with excellent stereoselectivity (Scheme 4), thus providing a valuable strategy to access complex difluorinated molecules.
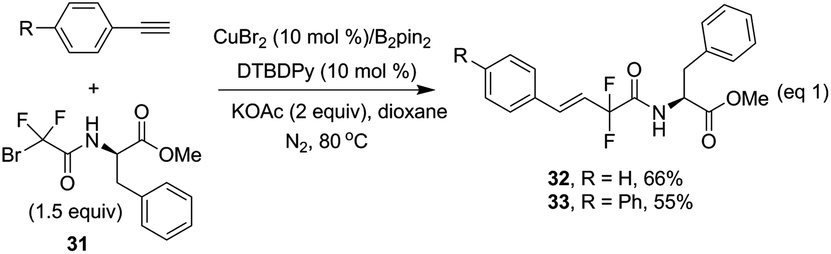 | ||
| Scheme 4 Hydrodifluoroamidation of alkynes with new difluoro reagent 31. | ||
When commercially available ethyl bromomonofluoroacetate was employed under the standard conditions, without further optimization of the conditions, remarkably, the corresponding hydromonofluoroacetylated alkenes 34–37 were obtained in reasonable yields in exclusively the E isomer form (Scheme 5). Although one might expect double bond migration to provide conjugation with the ester group, it turned out that the corresponding hydromonofluoroacetylated alkenes 34–37 were the exclusive structures.
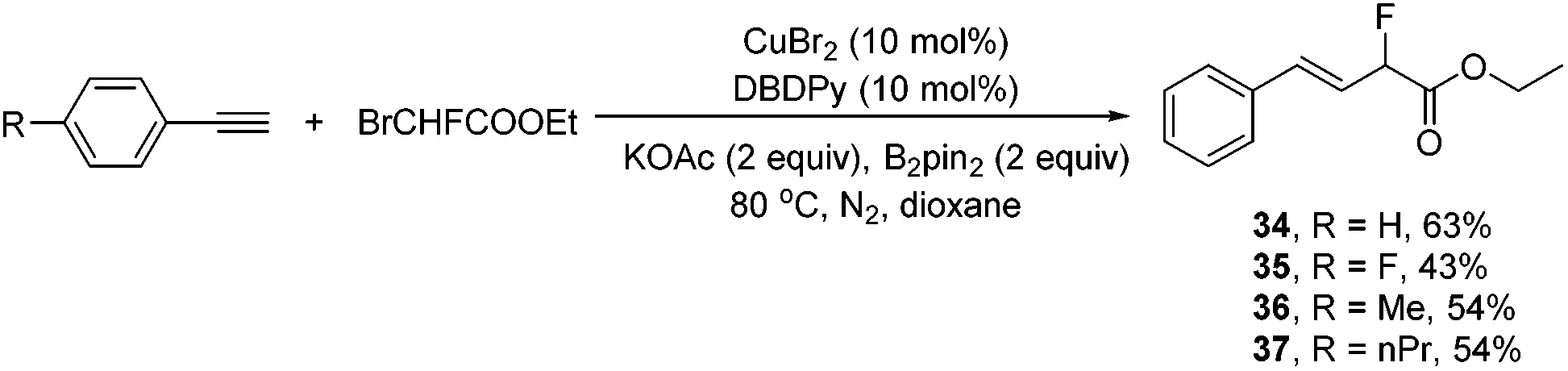 | ||
| Scheme 5 Hydromonofluoroacetylation of alkynes with ethyl bromomonofluoroacetate. | ||
When the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was added, the desired product 3 was not obtained, instead, TEMPO–CF2COOEt was generated in ca. 30% yield, implying that a radical pathway was involved in this reaction (ESI†). In order to determine the origin of ˙CF2COOEt, the following control experiments were performed: without the copper salt and B2pin2, difluoro compound 2 was directly treated with base and TEMPO, and no TEMPO–CF2COOEt was detected (eqn (2)); difluoro compound 2 was exposed to the copper salt and B2pin2 under base-free conditions, and remarkably the addition of TEMPO lead to ca. 20% of TEMPO–CF2COOEt (eqn (3)). These two results revealed that the formation of radical species ˙CF2COOEt was not related to the base as in a precedent report,11 instead, CuBr2 and B2pin2 play key roles in its generation.
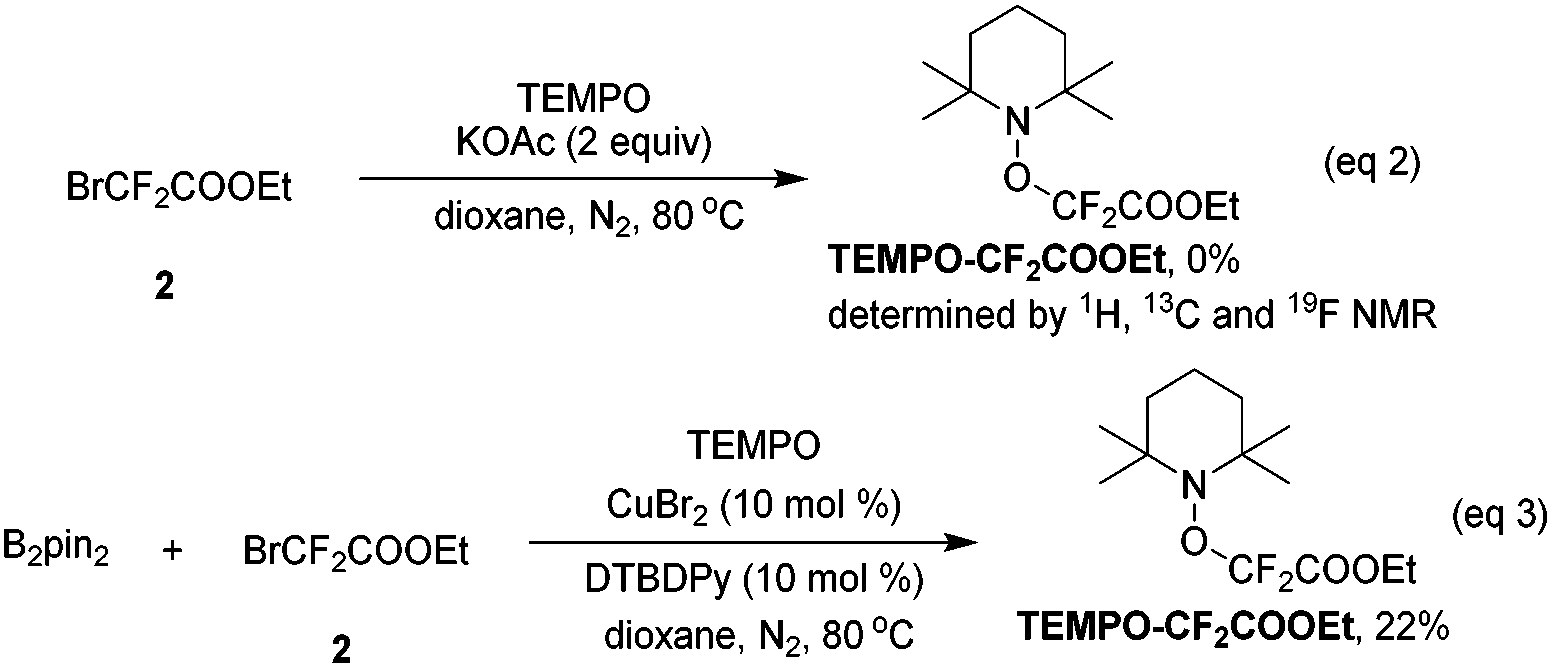
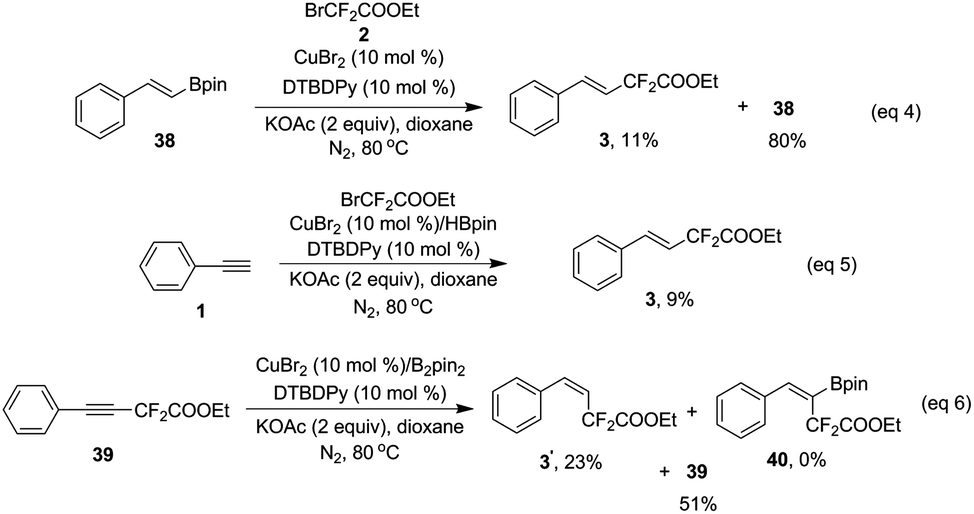 :1 vs. 9:1 for CuBr2) (eqn (7) and (8)). This result suggests that (phenylethynyl)copper might be a key intermediate in this transformation.
:1 vs. 9:1 for CuBr2) (eqn (7) and (8)). This result suggests that (phenylethynyl)copper might be a key intermediate in this transformation.
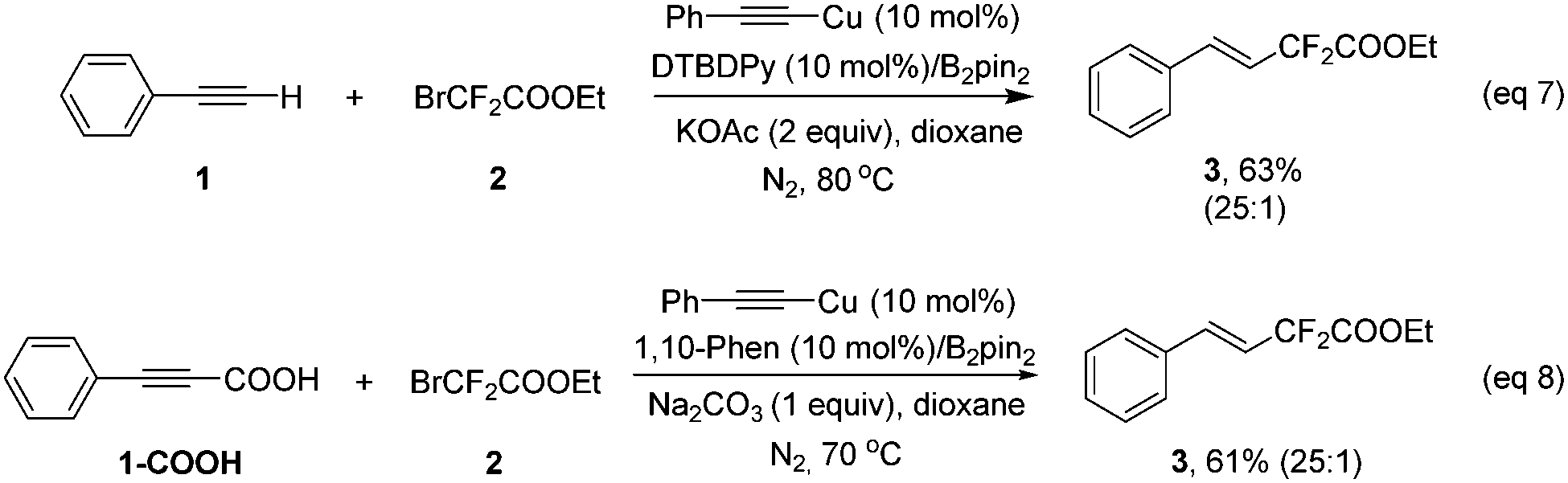
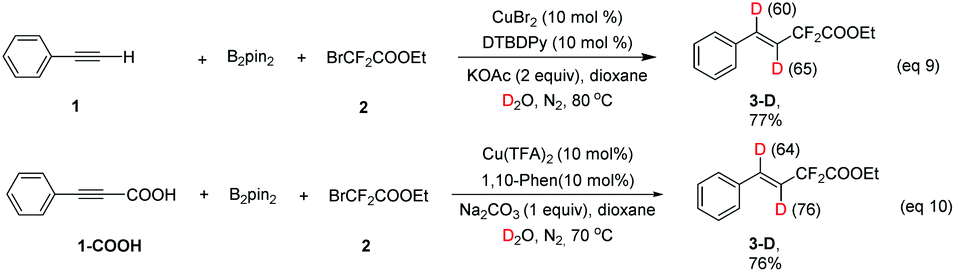
|
|
||||
|---|---|---|---|---|
| Time | 3′ | B2pin2 | 3 | 38 |
| 2 h | 5% | 37% | 43% | <1% |
| 4 h | 7% | 30% | 39% | 0 |
| 6 h | 6% | 3% | 65% | 0 |
| 10 h | 6% | 0 | 74% | 0 |
| 14 h | 8% | 0 | 75% | 0 |
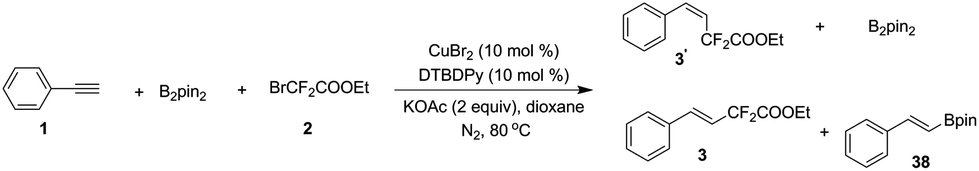
Recently, two elegant publications reported Ni-catalyzed couplings with diboron as a reductant.16 Although the role of B2pin2 in this transformation is still obscure at this point, based on our observations and precedent reports, we proposed that diboron might be a reductant in our reactions as well and a preliminary mechanism is outlined in Scheme 6: borylation of the Cu–X complex (A) leads to Cu–Bpin B, which undergoes one electron transfer with the bromodifluoro reagent to generate complex C (consistent with what we observed in eqn (3)), which further converts into complex D. Elimination of X–Bpin from complex D leads to R–Cu–Ln species E, which adds to the in situ formed (phenylethynyl)copper G from phenylacetylene or phenylpropiolic acid to afford the dicopper alkene F. The dicopper species F subsequently accepts protons from water in the reaction system,17 and eventually, desired product 3 is released with copper catalyst regeneration.
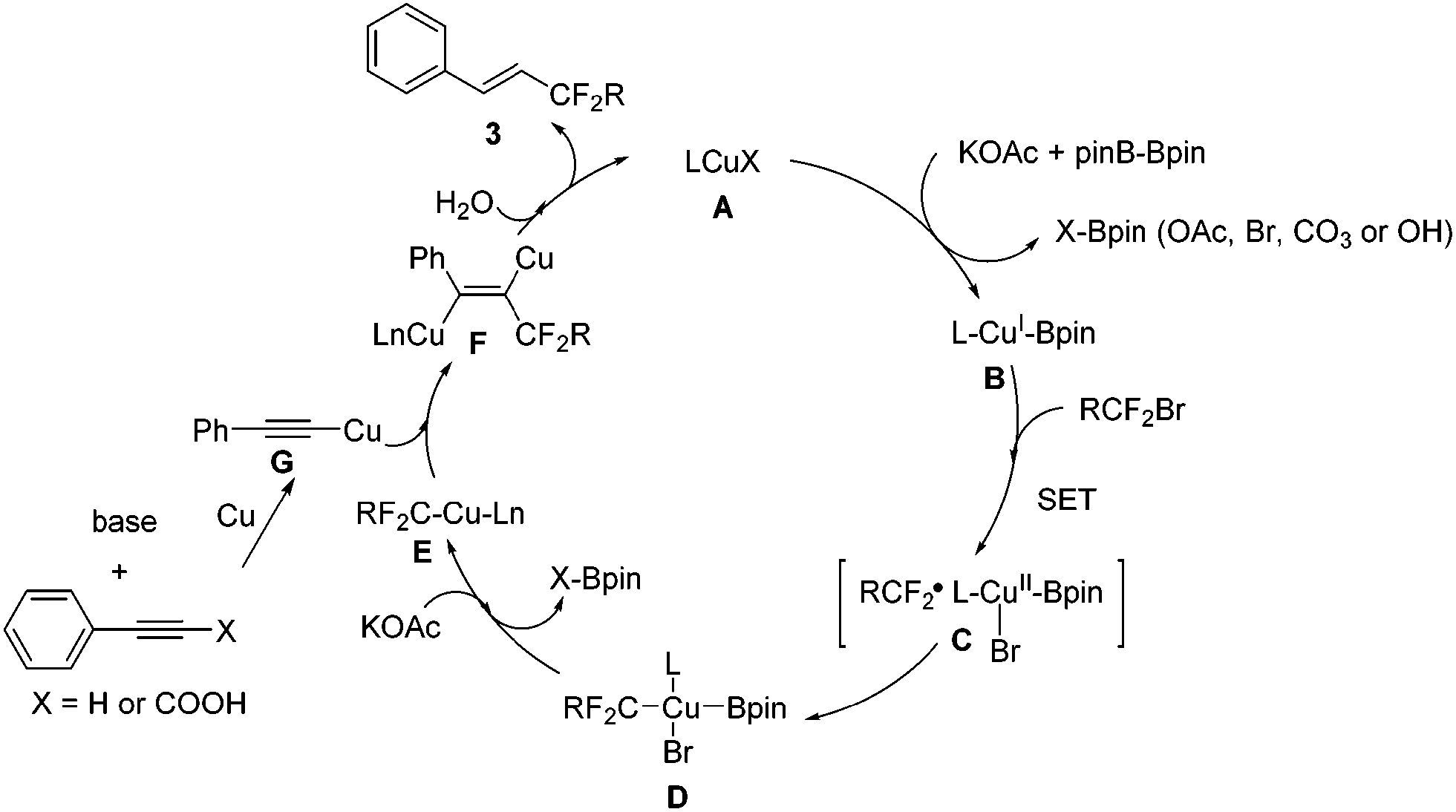 | ||
| Scheme 6 Proposed mechanism. | ||
In conclusion, a highly stereoselective copper-catalyzed hydrofluoroacetylation of arylacetylenes or alkynyl carboxylic acids with bromofluoroacetates leading to fluoroacetylated alkenes was depicted with broad substrate scope. This reaction provides an alternative to current approaches for the formation of di- or mono-fluoroacetylated alkenes. A mechanism study suggested that a radical pathway was involved in the reaction and that (phenylethynyl)copper might be the key intermediate for the reaction. The mechanism was quite different from a hydroboration reaction even though a Cu salt and B2pin2 were both existing in the reaction mixture. Further investigations of the reactivity, synthetic utility and mechanism of this catalytic system are under way in our laboratory.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21202049), the Recruitment Program of Global Experts (1000 Talents Plan), Fujian Hundred Talents Program, the Huaqiao University Research Innovation Fund for Graduate Student and the Program of Innovative Research Team of Huaqiao University is gratefully acknowledged. We also thank the Instrumental Analysis Center of Huaqiao University for analysis support.Notes and references
- (a) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis Reactivity, Applications, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) M. J. Tozer and T. F. Herpin, Tetrahedron, 1996, 52, 8619 CrossRef CAS; (c) Special issue on “Fluorine in the Life Sciences”, ChemBioChem, 2004, 5, 557;; (d) S. Pruser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- (a) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) J.-P. Begue and D. Bonnet-Delpon, Bioorganic and Medicinal Chemistry of Fluorine, Wiley, Hoboken, 2008 Search PubMed.
- (a) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (c) F. L. Qing, J. Org. Chem., 2012, 32, 815 CAS; (d) T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048 CrossRef CAS PubMed; (e) Q. Q. Min, Z. Yin, Z. Feng, W. H. Guo and X. Zhang, J. Am. Chem. Soc., 2014, 136, 1230 CrossRef CAS PubMed.
- (a) L. Chu and F. L. Qing, Org. Lett., 2010, 12, 5060 CrossRef CAS PubMed; (b) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524 CrossRef CAS PubMed; (c) G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., Int. Ed., 2012, 51, 12090 CrossRef CAS PubMed; (d) T. Taguchi, O. Kitagawa, T. Morikawa, T. Nishiwaki, H. Uehara, H. Endo and Y. Kobayashi, Tetrahedron Lett., 1986, 27, 6103 CrossRef CAS; (e) T. Yokomatsu, K. Suemune, T. Murano and S. Shibuya, J. Org. Chem., 1996, 61, 7207 CrossRef CAS PubMed.
- (a) M. K. Schwaebe, J. R. McCarthy and J. P. Whitten, Tetrahedron Lett., 2000, 41, 791 CrossRef CAS.
- (a) Z. Feng, Q. Q. Min, H. Y. Zhao, J. W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270 CrossRef CAS PubMed; (b) C. Yu, N. Iqbal, S. Park and E. Cho, Chem. Commun., 2014, 50, 12884 RSC.
- (a) S. Murakami, H. Ishii, T. Tajima and T. Fuchigami, Tetrahedron, 2006, 62, 3761 CrossRef CAS; (b) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875 CrossRef CAS PubMed; (c) H. Jiang, C. Huang, J. Guo, C. Zeng, Y. Zhang and S. Yu, Chem. – Eur. J., 2012, 18, 15158 CrossRef CAS PubMed; (d) S. Murakami, H. Ishii and T. Fuchigami, J. Fluorine Chem., 2004, 125, 609 CrossRef CAS.
- (a) M.-C. Belhomme, D. Dru, H.-Y. Xiong, D. Cahard, T. Besset, T. Poisson and X. Pannecoucke, Synthesis, 2014, 46, 1859–1870 CrossRef; (b) Z.-Y. Long and Q.-Y. Chen, J. Org. Chem., 1999, 64, 4775–4782 CrossRef CAS PubMed; (c) W. Ghattas, C. R. Hess, G. Iacazio, R. Hardré, J. P. Klinman and M. Réglier, J. Org. Chem., 2006, 71, 8618–8621 CrossRef CAS PubMed; (d) N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539 CrossRef CAS PubMed; (e) T. Basset, T. Poisson and X. Pannecoucke, Eur. J. Org. Chem., 2015, 2765 CrossRef; (f) M. C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2015, 21, 12836 CrossRef CAS PubMed.
- (a) J. Hu, N. Zhao, B. Yang, G. Wang, L. N. Guo, Y. M. Liang and S. D. Yang, Chem. – Eur. J., 2011, 17, 5516 CrossRef CAS PubMed; (b) S. Ranjit, Z. Duan, P. Zhang and X. Liu, Org. Lett., 2010, 12, 4134 CrossRef CAS PubMed; (c) J. Park, E. Park, A. Kim, S. A. Park;, Y. Lee, K. W. Chi, Y. H. Jung and I. S. Kim, J. Org. Chem., 2011, 76, 2214 CrossRef CAS PubMed; (d) D. L. Priebbenow, P. Becker and C. Bolm, Org. Lett., 2013, 15, 6155 CrossRef CAS PubMed; (e) W. W. Zhang, X. G. Zhang and J. H. Li, J. Org. Chem., 2010, 75, 5259 CrossRef CAS PubMed; (f) G. Hu, Y. Gao and Y. Zhao, Org. Lett., 2014, 16, 4464 CrossRef CAS PubMed; (g) D. Zhao, C. Gao, X. Su, Y. He, J. You and Y. Xue, Chem. Commun., 2010, 46, 9049 RSC; (h) M. Zhou, M. Chen, Y. Zhou, K. Yang, J. Su, J. Du and Q. Song, Org. Lett., 2015, 17, 1786–1789 CrossRef CAS PubMed.
- Z. Feng, Q. Q. Min, Y. L. Xiao, B. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1669 CrossRef CAS PubMed.
- (a) H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang and S. Yu, Angew. Chem., 2013, 125, 13531 CrossRef; (b) X. Sun and S. Yu, Org. Lett., 2014, 16, 2938 CrossRef CAS PubMed; (c) T. Xu, C. W. Cheung and X. Hu, Angew. Chem., Int. Ed., 2014, 53, 4910 CrossRef CAS PubMed; (d) W. Fu, M. Zhu, G. Zou, C. Xu, Z. Wang and B. Ji, J. Org. Chem., 2015, 4776 Search PubMed.
- (a) J. E. Lee, J. Kwon and J. Yun, Chem. Commun., 2008, 733 RSC; (b) J. Zhao, Z. Niu, H. Fu and Y. Li, Chem. Commun., 2014, 50, 2058 RSC.
- For recent reviews on the hydroboration of alkynes, see: (a) I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320 CrossRef CAS PubMed; (b) J. Yun, Asian J. Org. Chem., 2013, 2, 1016 CrossRef CAS; (c) G. J. Irvine, M. J. Gerald Lesley, T. B. Marder, N. C. Norman and C. R. Rice, Chem. Rev., 1998, 98, 2685 CrossRef CAS PubMed.
- T. Besset, T. Poisson and X. Pannecoucke, Eur. J. Org. Chem., 2014, 7220 CrossRef CAS.
- K. Jouvin, J. Heimburger and G. Evano, Chem. Sci., 2012, 3, 756 RSC.
- (a) H. Xu, C. Zhao, Q. Qian, W. Deng and H. Gong, Chem. Sci., 2013, 4, 4022 RSC; (b) G. Zhang, Y. Xie, Z. Wang, Y. Liu and H. Huang, Chem. Commun., 2015, 51, 1850 RSC.
- Q. Feng, K. Yang and Q. Song, Chem. Commun., 2015, 51, 15394 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00324e |
| This journal is © the Partner Organisations 2016 |