
Rhodium-catalyzed oxidative coupling of N-acyl anilines with alkynes using an acylamino moiety as the traceless directing group†
Kaijun
Geng‡
,
Zhoulong
Fan‡
and
Ao
Zhang
*
CAS Key Laboratory of Receptor Research, and Synthetic Organic & Medicinal Chemistry Laboratory (SOMCL), Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences, Shanghai 201203, China. E-mail: aozhang@simm.ac.cn
First published on 14th January 2016
Abstract
A rhodium-catalyzed oxidative annulation of N-acyl anilines with alkynes was developed by using the acylamino group as a traceless directing group for the first time. Various N-acyl anilines and para- or meta-substituted diphenylacetylenes were well tolerated, and a series of 1,2,3,4-tetrasubstituted naphthalenes were readily synthesized in good to excellent yields. Meanwhile, this method also provides a new strategy for the N-dearylation of N-phenylamides.
Polycyclic aromatic compounds are widely used as organic semiconductors and luminescent materials in materials science due to their unique electron and photochemical properties.1 Traditional synthetic methods for this class of compounds generally suffer from harsh reaction conditions and low yields. In the past few decades, a significant breakthrough has been achieved in directing group-assisted transition-metal catalyzed oxidative coupling of aromatic substrates with internal alkynes, providing an alternative option to the synthesis of such π-conjugated molecules.2 Among which, the recently developed traceless directing group (TDG) strategy is particularly appealing, due to the easy pre-attaching and traceless cutting-off.3 For example, carboxylic acid,4 boronic acid,5 sodium sulfonate6 and aldehyde7 have been successfully used as the TDGs in transition metal-catalyzed oxidative couplings for the synthesis of fused aromatic or heteroaromatic compounds.
The acylamino group, as a directing group, has been widely used in diversified ortho C–H functionalizations.8 For example, Fagnou, Tanaka and Lu's laboratories have reported that multisubstituted indoles could be synthesized via Rh- or Pd-catalyzed intermolecular C–H activation/annulation of alkynes and acetanilides (Scheme 1a).9 Moreover, the Wu group disclosed an ortho- and meta-position dual C–H activation strategy to synthesize highly substituted naphthalenes with similar substrates under a Pd(OAc)2/K2S2O8 catalytic system (Scheme 1b).10 In these examples, the acylamino group served as the directing group and was retained as a part of the products.
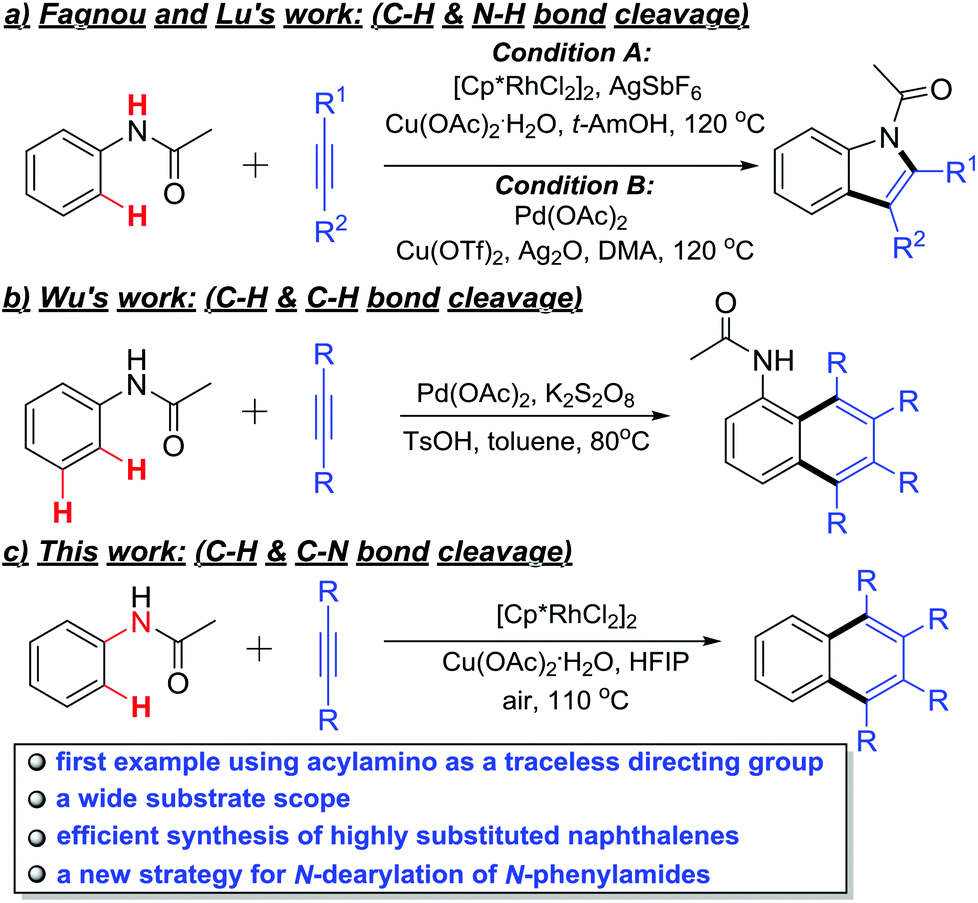 | ||
| Scheme 1 Metal-catalyzed oxidative coupling of acetanilines with alkynes. | ||
In view of the advantages of the TDGs, we decided to explore the possibility of using an acylamino group as a TDG in the rhodium-catalyzed ortho- and ipso-selective oxidative annulation of N-acyl anilines with alkynes that would lead to easy synthesis of 1,2,3,4-tetrasubstituted naphthalenes (Scheme 1c). This method not only provides a new example of TDGs in the C–H activation toolbox, but also offers a new strategy to remove the N-phenyl protecting group of N-phenylamides.
Our investigation on the rhodium-catalyzed C–H activation/annulation began with the reaction of N-phenylpivalamide (1a) with diphenylacetylene (2a) in the presence of 2.5 mol% [Cp*RhCl2]2. The results of screening of oxidants and solvents are shown in Table 1. Using Cu(OAc)2·H2O as the oxidant and methanol as the solvent gave only a trace of 1,2,3,4-tetraphenylnaphthalene (3a) (entry 1). After several attempts, it was found that 3a was obtained in 83% yield in the presence of hexafluoroisopropanol (HFIP) (entry 4), and nearly no reaction took place using other solvents, including DMF, toluene and i-PrOH. Among the various oxidants tested, no better one was found than Cu(OAc)2·H2O (entries 6–9). Further optimization of the loading amounts of Cu(OAc)2·H2O showed that 1 equivalent of Cu(OAc)2·H2O is optimal (entries 10–12). Meanwhile, an excellent isolated yield of 3a (90%, entry 13) was achieved when increasing the rhodium catalyst from 2.5 to 5 mol%. Based on these screening results, the best reaction conditions were reached as follows: N-acyl anilines 1 (1 equiv.), internal alkynes 2 (2 equiv.), [Cp*RhCl2]2 (5 mol%) and Cu(OAc)2·H2O (1 equiv.) in HFIP at 110 °C in a sealed tube.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
|||
|---|---|---|---|
| Entry | Oxidant (× equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), [Cp*RhCl2]2 (2.5 mol%), oxidant (×equiv.), solvent (1 ml) in a sealed tube at 110 °C for 5 h. b Yield was determined by 1H NMR analysis using 1,2-dibromomethane as an internal standard; the dashed line indicates that the product is not detected. c 5 mol% [Cp*RhCl2]2. d Isolated yield when using 0.5 mmol 1a. HFIP = hexafluoroisopropanol. | |||
| 1 | Cu(OAc)2·H2O (1.0) | MeOH | Trace |
| 2 | Cu(OAc)2·H2O (1.0) | DMF | — |
| 3 | Cu(OAc)2·H2O (1.0) | Toluene | — |
| 4 | Cu(OAc)2·H2O (1.0) | HFIP | 83 |
| 5 | Cu(OAc)2·H2O (1.0) | i-PrOH | — |
| 6 | AgOAc (2.0) | HFIP | Trace |
| 7 | Ag2CO3 (1.0) | HFIP | Trace |
| 8 | PhI(OAc)2 (1.0) | HFIP | Trace |
| 9 | K2S2O8 (1.0) | HFIP | Trace |
| 10 | Cu(OAc)2·H2O (1.5) | HFIP | 78 |
| 11 | Cu(OAc)2·H2O (0.5) | HFIP | 65 |
| 12 | Cu(OAc)2·H2O (0.1) | HFIP | 52 |
| 13c | Cu(OAc)2·H2O (1.0) | HFIP | 94(90)d |
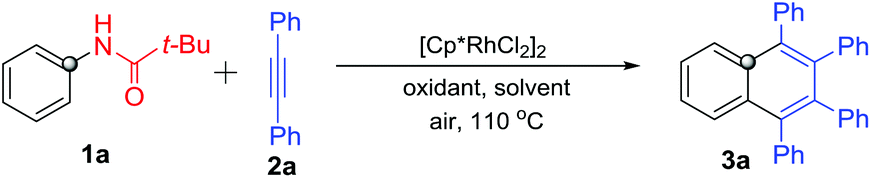
With the optimized reaction conditions in hand, we firstly investigated various acylamino groups as the traceless directing groups and the results are summarized in Scheme 2. The cyclic alkyl acylanilines (1c–1d) gave higher yields than the acyclic alkyl acyl and aromatic acyl substrates (1b, 1e and 1f). We speculated that the steric hindrance on the aliphatic alkyl portion likely makes the TDG more prone to cleavage during the C–H activation process. N-Phenyl lactams 1g–1h proceeded smoothly in the reaction but afforded product 3a in much lower yields. For the five-membered (1i) or six-membered (1j) heterocycle-bearing TDGs, such as pyrazolones11 and pyridazinones,12 the corresponding product 3a was obtained in 15% and 70% yields, respectively, suggesting that the pyridazinonyl-TDG has a higher inductive effect. Interestingly, 1-acetyl-2-phenylhydrazine (1k) took part in the reaction as well, though in a lower yield (17%). In addition, N-methyl-acetanilide 1l was applied in this reaction, but no product was obtained. We propose that the steric congestion of the N-methyl on the TDG might deteriorate the reaction.13N-Methylaniline 1m lacking the carbonyl group as the DG also failed in this reaction.
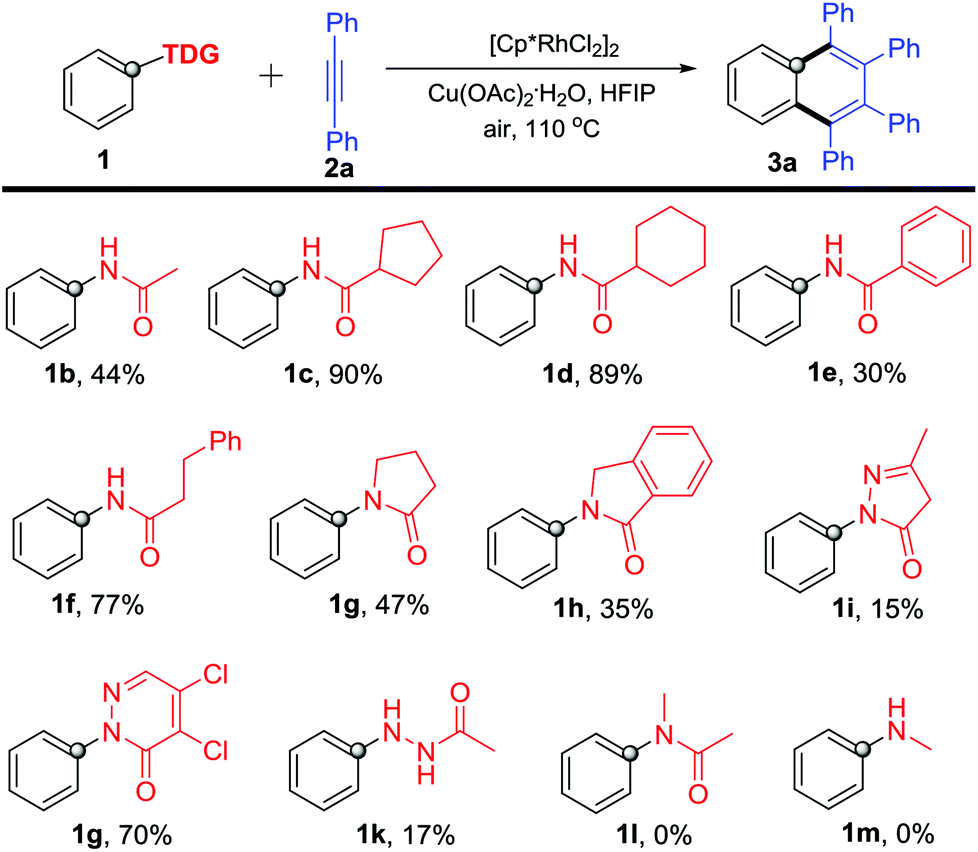 | ||
| Scheme 2 Reaction of various N-acyl anilines with diphenylacetylene (2a). | ||
Next, various substituted N-phenylpivalamides were used to explore the reaction scope and limitation. As shown in Scheme 3, all these substrates reacted with diphenylacetylene (2a) and gave the corresponding highly substituted naphthalenes in good to excellent yields (Scheme 3). N-Phenylpivalamides bearing electron-donating or -withdrawing substituents on the para-position offered the corresponding products 3b–3g in 80–91% yields. Some meta-substituents (Cl–, Me–, OMe– and CF3–) were also well tolerated and afforded products 3b–3e in 81–89% yields, which are shown in the parentheses in Scheme 3. Subsequently, ortho-methyl or -chloro substituted N-phenylpivalamides were explored, products 3h–3i were obtained in 56% and 73% yields, respectively. Moreover, bicyclo-N-phenylpivalamides were found to participate in the reaction as well and afforded the annulated products 3j–l in 65–77% yields.
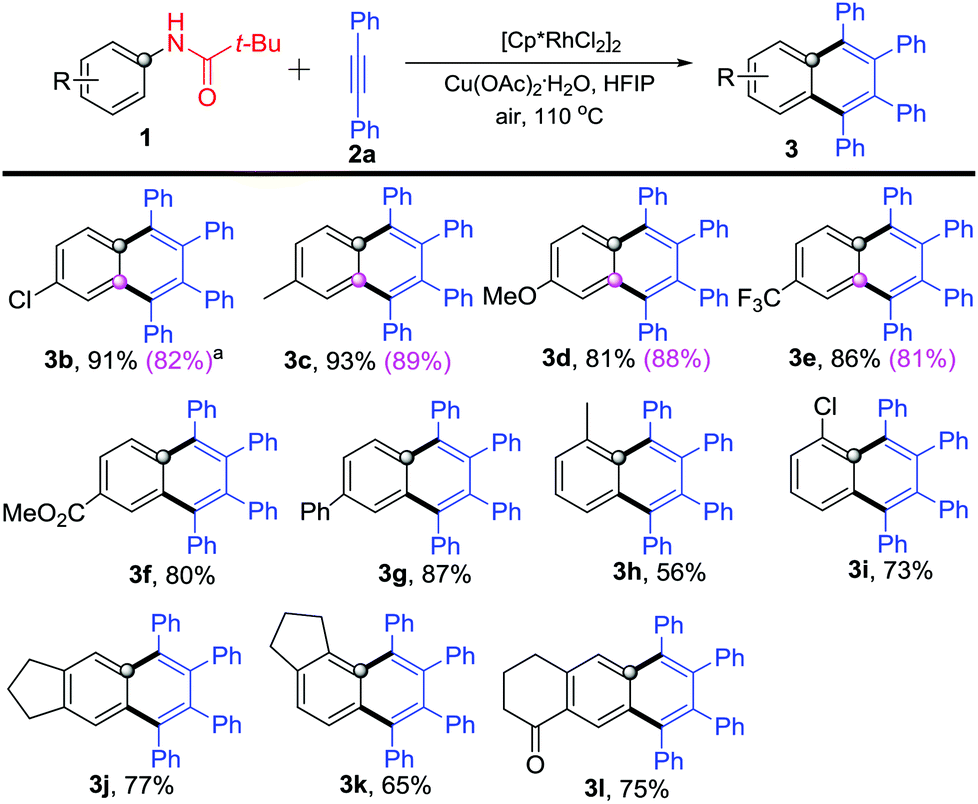 | ||
| Scheme 3 Reaction of substituted N-phenylpivalamides 1 with diphenylacetylene (2a). aYields are listed in the parentheses when employing meta-substituted N-phenylpivalamides. | ||
Further, different internal alkynes were used to react with N-phenylpivalamide 1a under the optimized reaction conditions (Scheme 4). The para-methyl, -methoxyl, and -tert-butyl substituted phenylacetylenes participated in the reaction very well and gave 1,2,3,4-tetrasubstituted naphthalenes 3m–3o in moderate to good yields. It is worthy of note that similar to the para-substituted congeners, reactions of 1a with meta-substituted diphenylacetylenes occurred smoothly to afford products 3p–3r in 63–70% yields. In addition, 1-phenyl-1-propyne and 1-phenyl-1-hexyne also reacted with 1a to give symmetric 1,2,3,4-tetrasubstituted naphthalenes 3s and 3t, along with unsymmetric isomers 3s′ and 3t′. The structures were deduced by NOE experiments (see the ESI†). Although the reaction generally occurred smoothly, it still suffered from inevitable substrate limitation. For example, the alkyl–alkyl disubstituted alkynes and terminal alkynes failed in the reaction, and the unsymmetric aryl-aryl disubstituted alkynes gave inseparable complex mixtures.
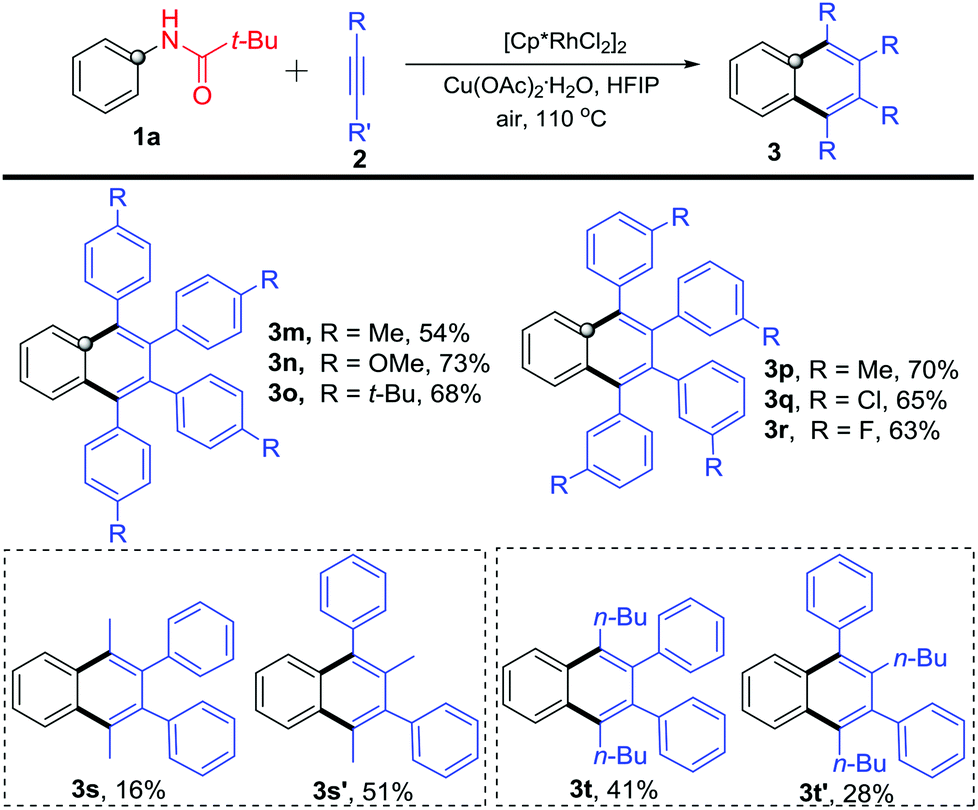 | ||
| Scheme 4 Reactions of N-phenylpivalamide (1a) with substituted diphenylacetylenes 2. | ||
In addition to the TDG-assisted rhodium-catalyzed oxidative annulation of N-acyl anilines, the current protocol also provides a new strategy for the N-dearylation of amides.14 To validate the practicality, N-acyl anilines 1m and 1n were reacted with diphenylacetylene (2a) under the optimized reaction conditions and the corresponding dephenylated products 4 and 5 were obtained in 65% and 62% yields, respectively (Scheme 5). This approach would be valuable in the peptide synthesis.
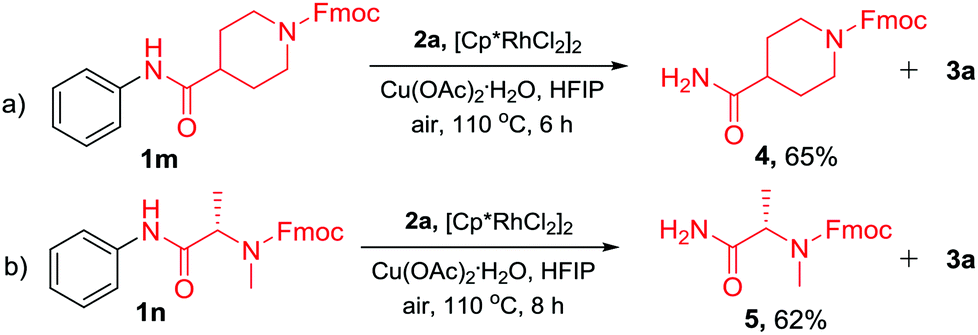 | ||
| Scheme 5 N-Dearylation of N-phenylamides. | ||
To gain more insights on the reaction pathway, additional experiments were performed. In order to demonstrate the sequence of C–N cleavage and C–H activation, we re-conducted the reaction with 1m as the substrate but without 2a under the standard conditions. The C–N cleavage product 4 was not detected, even when increasing the amount of the Rh-catalyst or adding HOAc as the proton source (Scheme 6a). Therefore, it is unlikely that the C–N cleavage occurred prior to the C–H activation in the protocol.
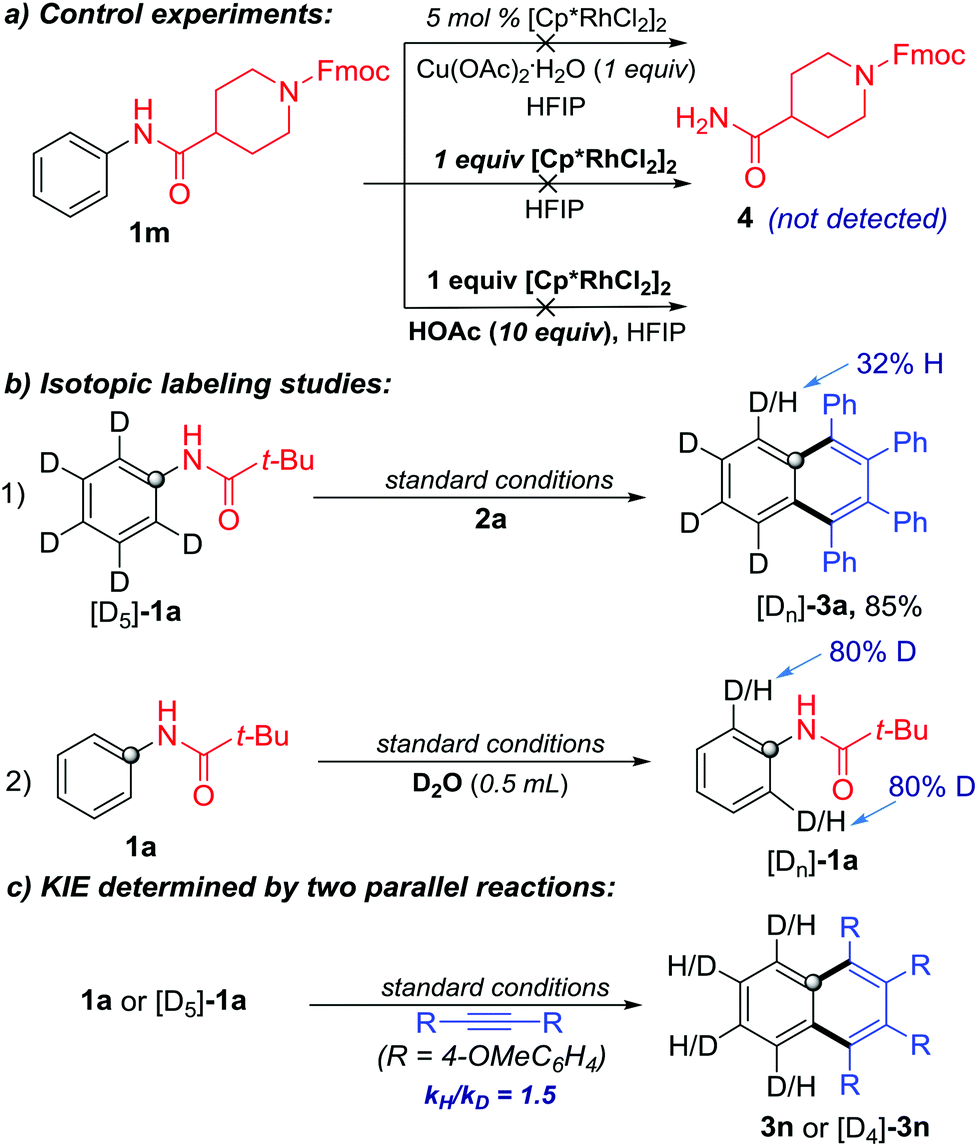 | ||
| Scheme 6 Preliminary mechanistic studies. | ||
To probe the C–H activation process, we conducted C–H functionalization with isotopically labeled substrates. First, [D5]-1a was used to react with 2a under the standard conditions. The ortho-D/H partially exchanged product [Dn]-3a was obtained in 85% yield. Meanwhile, ortho-D/H exchange was observed when treating 1a with D2O under the same catalytic conditions but without 2a (Scheme 6b). These results confirmed that the C–H bond metalation/activation in the ortho-position of the N-phenylpivalamide was the first step and this process was reversible. Subsequently, the kinetic isotopic effect (KIE) of ortho-C–H cleavage was determined to be 1.5 by two independent reactions of substrates 1a and [D5]-1a, indicating that the C–H cleavage was probably involved in the rate-limiting step (Scheme 6c).
A plausible mechanism is presented in Scheme 7 (based on the model reaction of 1a and 2a).9a,d Firstly, the combination of the catalyst precursor [Cp*RhCl2]2 and Cu(OAc)·H2O would give the cationic rhodium(III) complex A, which is then converted to a rhodacycle Bvia a C–H bond cleavage process. Insertion of alkyne 2a to complex B affords the rhodium complex C. Complex C is then converted to a five-membered rhodium complex D, accompanied by the C–N bond cleavage. The intermediate D then undergoes the second insertion of alkyne 2a to give the 7-membered metallacycle E, which then proceeds via a reductive elimination to form the desired product 3a and rhodium(I) complex F. The active cationic rhodium(III) complex A was regenerated under the oxidation of Cu(OAc)2·H2O and air and used for further catalysis.
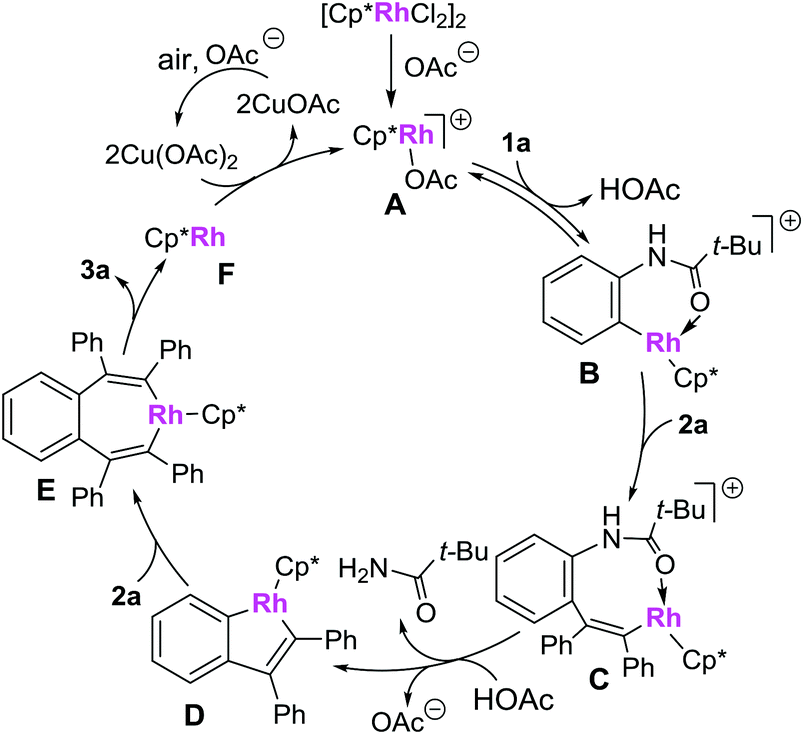 | ||
| Scheme 7 Proposed mechanism. | ||
In conclusion, we have reported the first example of using the acylamino group as a traceless directing group to initiate a rhodium-catalyzed oxidative annulation of N-acyl anilines with internal alkynes. In this approach, various N-acyl anilines and para- or meta-substituted diphenylacetylenes were well tolerated, and a series of 1,2,3,4-tetrasubstituted naphthalenes were readily synthesized in good to excellent yields. Meanwhile, this method also represents a new strategy of removal of the N-phenyl protective group of N-phenylamides.
This work was supported by grants from Chinese NSF (81430080, 81125021), and the Major State Basic Research Development Program (2015CB910603). Seeding Grants from SIMM (CASIMM0120154002/2002) are also appreciated.
Notes and references
- For recent reviews, see: (a) J. Yang, D. Yan and T. S. Jones, Chem. Rev., 2015, 115, 5570 CrossRef CAS PubMed; (b) L. Zhang, Y. Cao, N. S. Colella, Y. Liang, J. L. Bredas, K. N. Houk and A. L. Briseno, Acc. Chem. Res., 2015, 48, 500 CrossRef CAS PubMed; (c) K. Takimiya, I. Osaka, T. Mori and M. Nakano, Acc. Chem. Res., 2014, 47, 1493 CrossRef CAS PubMed; (d) W. Jiang, Y. Li and Z. Wang, Acc. Chem. Res., 2014, 47, 3135 CrossRef CAS PubMed; (e) M. Watanabe, K.-Y. Chen, Y. J. Chang and T. J. Chow, Acc. Chem. Res., 2013, 46, 1606 CrossRef CAS PubMed; (f) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed.
- For recent reviews, see: (a) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2015, 6, 498 CrossRef; (b) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007 CrossRef CAS PubMed; (c) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155 RSC; (d) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (e) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef CAS; (f) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (g) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (h) T. Satoh and M. Miura, Chemistry, 2010, 16, 11212 CrossRef CAS PubMed.
- (a) Y. Zhang, H. Zhao, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 3817 CrossRef CAS PubMed; (b) X. Qin, D. Sun, Q. You, Y. Cheng, J. Lan and J. You, Org. Lett., 2015, 17, 1762 CrossRef CAS PubMed; (c) K. Pati, G. dos Passos Gomes, T. Harris, A. Hughes, H. Phan, T. Banerjee, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 1165 CrossRef CAS PubMed; (d) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (e) U. Sharma, Y. Park and S. Chang, J. Org. Chem., 2014, 79, 9899 CrossRef CAS PubMed; (f) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109 CrossRef CAS PubMed; (g) D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426 CrossRef CAS PubMed; (h) S. M. Preshlock, D. L. Plattner, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr. and M. R. I. Smith, Angew. Chem., Int. Ed., 2013, 52, 12915 CrossRef CAS PubMed; (i) X. Huang, J. Huang, C. Du, X. Zhang, F. Song and J. You, Angew. Chem., Int. Ed., 2013, 52, 12970 CrossRef CAS PubMed; (j) A. V. Gulevich, F. S. Melkonyan, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2012, 134, 5528 CrossRef CAS PubMed; (k) C. Wang and H. Ge, Chemistry, 2011, 17, 14371 CrossRef CAS PubMed; (l) R. Samanta and A. P. Antonchick, Angew. Chem., Int. Ed., 2011, 50, 5217 CrossRef CAS PubMed; (m) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef CAS PubMed.
- (a) M. Yamashita, H. Horiguchi, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 7481 CrossRef CAS PubMed; (b) M. Yamashita, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 2337 CrossRef CAS PubMed; (c) K. Ueura, T. Satoh and M. Miura, J. Org. Chem., 2007, 72, 5362 CrossRef CAS PubMed.
- (a) T. Fukutani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 2867 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed.
- H. Wang, Y. Wang, H. Yang, C. Tan, Y. Jiang, Y. Zhao and H. Fu, Adv. Synth. Catal., 2015, 357, 489 CrossRef CAS.
- X. Liu, X. Li, H. Liu, Q. Guo, J. Lan, R. Wang and J. You, Org. Lett., 2015, 17, 2936 CrossRef CAS PubMed.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- (a) Y. Hoshino, Y. Shibata and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1577 CrossRef CAS; (b) Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2011, 50, 10917 CrossRef CAS PubMed; (c) F. Zhou, X. Han and X. Lu, Tetrahedron Lett., 2011, 52, 4681 CrossRef CAS; (d) D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326 CrossRef CAS PubMed; (e) D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474 CrossRef CAS PubMed.
- J. Wu, X. Cui, X. Mi, Y. Li and Y. Wu, Chem. Commun., 2010, 46, 6771 RSC.
- (a) Z. Fan, S. Song, W. Li, K. Geng, Y. Xu, Z.-H. Miao and A. Zhang, Org. Lett., 2015, 17, 310 CrossRef CAS PubMed; (b) Z. Fan, K. Wu, L. Xing, Q. Yao and A. Zhang, Chem. Commun., 2014, 50, 1682 RSC; (c) K. Wu, Z. Fan, Y. Xue, Q. Yao and A. Zhang, Org. Lett., 2014, 16, 42 CrossRef CAS PubMed; (d) L. Xing, Z. Fan, C. Hou, G. Yong and A. Zhang, Adv. Synth. Catal., 2014, 356, 972 CrossRef CAS.
- W. Li, Z. Fan, K. Geng, Y. Xu and A. Zhang, Org. Biomol. Chem., 2015, 13, 539 CAS.
- F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9982 CrossRef CAS PubMed.
- (a) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4688 CrossRef CAS PubMed; (b) G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124 CrossRef CAS PubMed; (c) A. Nakazaki, A. Mori, S. Kobayashi and T. Nishikawa, Tetrahedron Lett., 2012, 53, 7131 CrossRef CAS; (d) Y. Li, H. Zou, J. Gong, J. Xiang, T. Luo, J. Quan, G. Wang and Z. Yang, Org. Lett., 2007, 9, 4057 CrossRef CAS PubMed; (e) M. Fujita, O. Kitagawa, H. Lzawa, A. Dobashi, H. Fukaya and T. Taguchi, Tetrahedron Lett., 1999, 40, 1949 CrossRef CAS; (f) D. R. Kronenthal, C. Y. Han and M. K. Taylor, J. Org. Chem., 1982, 47, 2765 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00387c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |