
Copper(I) catalyzed C(sp2)–N bond formation: synthesis of pyrrolo[3,2-c]quinolinone derivatives†
Zhiguo
Zhang
*a,
Jingjing
Qian
a,
Guisheng
Zhang
*a,
Nana
Ma
a,
Qingfeng
Liu
a,
Tongxin
Liu
a,
Kai
Sun
ab and
Lei
Shi
a
aCollaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals, Key Laboratory of Green Chemical Media and Reactions, Ministry of Education, School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China. E-mail: zhangzg@htu.edu.cn; zgs6668@yahoo.com
bCollege of Chemistry and Chemical Engineering, Anyang Normal University, Anyang 455000, P. R. China
First published on 11th January 2016
Abstract
An intramolecular copper-catalyzed direct C(sp2)–H activation/C(sp2)–N bond formation reaction has been developed for the synthesis of pyrrolo[3,2-c]quinolinone derivatives under an oxygen atmosphere.
Natural products containing the pyrrolo[3,2-c]quinoline skeleton were first discovered in martinellic acid and martinelline in 1995.1 These tricyclic compounds, together with other synthetic pyrrolo[3,2-c]quinoline derivatives, have been demonstrated to possess significant biological activity as KYN-3-OHase inhibitors,2 Hedgehog signaling inhibitors,3 antitumor activity,4 gastric (H+/K+)-ATPase inhibitor,5 and others.6 Many synthetic approaches are available for the synthesis of substituted pyrrolo[3,2-c]quinolines. The two most common synthetic strategies are the cyclization of functionalized quinolines3,7 and the aza Diels–Alder reaction of N-phenylmethanimines with pyrroles.8 Beyond these specific methods, other general strategies for the construction of a tricyclic pyrroloquinoline ring system can be used, based on the Heck reaction,9 radical cyclisation,10 and 1,3-dipolar cycloadditions.11 However, a critical review of the literature shows that the construction of a quinolinone structural motif in pyrrolo[3,2-c]quinolinone derivatives has not been completed by means of C–H activation/C–N coupling reactions. We present here a facile method to prepare this type of tricyclic compounds in the presence of CuI under O2, in DMSO via the direct C(sp2)–H activation/C(sp2)–N coupling reactions (Fig. 1).
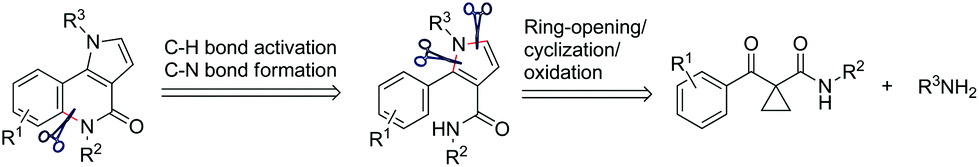 | ||
| Fig. 1 Retrosynthetic analysis for pyrrolo[3,2-c]quinolinones. | ||
The construction of C(sp2)–N bonds in aromatic compounds12 is an important transformation because it provides access to nitrogen-containing molecules of great interest in synthetic, biological, medicinal, materials sciences, coordination chemistry, and natural products chemistry.13 Metal-catalyzed C(sp2)–N bond formation reactions have been widely used in the construction of various five-, six-, and seven-membered N-containing heterocycles.14 Stoichiometric amounts of copper were first used for the construction of C(sp2)–N bonds by Ullmann in 1903.15 In the 1990s, Buchwald and Hartwig independently developed Pd- and Cu-catalyzed N-arylation reactions with the help of an appropriate diamine or phosphine ligands.16 Subsequently, considerable advances have been achieved and various metal salts (such as Cu, Pd, Ni, Rh, and Fe), in catalytic amounts, have been used for these reactions in the presence of various ligands.14a,17 However, they often need pre-prepared activated aryl (pseudo)halides (X–Ar, X = Cl, Br, I, OTf, OTS) to react with the amine nucleophiles, which reduces the scope and efficiency of the reaction.16c–f,18 In comparison, the formation of C(sp2)–N bond by direct C(sp2)–H activation/C(sp2)–N coupling of aromatic compounds with amines would be more practical.17a,c,19 Several groups have investigated the construction of 2-quinolinones via the intramolecular direct aromatic C(sp2)–H activation/C(sp2)–N bond formation reaction in the presence of metal catalysts. In 2010, Inamoto et al.20 reported that 3,3-diarylacrylamides could be smoothly transformed into 2-quinolinone compounds in the presence of PdCl2 and Cu(OAc)2 under O2 (Scheme 1, eqn (1)). Soon after, Cacchi and co-workers21 reported a copper-catalyzed approach for the construction of 2-quinolones from similar starting materials and the reaction selectively gave two products in the ratio of 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 to 99:1 (Scheme 1, eqn (2)). Recently, Fabis and co-workers17b focused on N-tosylcarboxamide as a transformable directing group for the Pd-catalyzed C(sp2)–H ortho-arylation reaction of N-tosylbenzamides with iodotoluenes, in which the intramolecular cyclization product 3-methyl-5-tosyl-8-(trifluoromethyl)phenanthridine-6(5H)-one was obtained as a special case in the synthesis of biarylcarboxamides (Scheme 1, eqn (3)).
:8 to 99:1 (Scheme 1, eqn (2)). Recently, Fabis and co-workers17b focused on N-tosylcarboxamide as a transformable directing group for the Pd-catalyzed C(sp2)–H ortho-arylation reaction of N-tosylbenzamides with iodotoluenes, in which the intramolecular cyclization product 3-methyl-5-tosyl-8-(trifluoromethyl)phenanthridine-6(5H)-one was obtained as a special case in the synthesis of biarylcarboxamides (Scheme 1, eqn (3)).
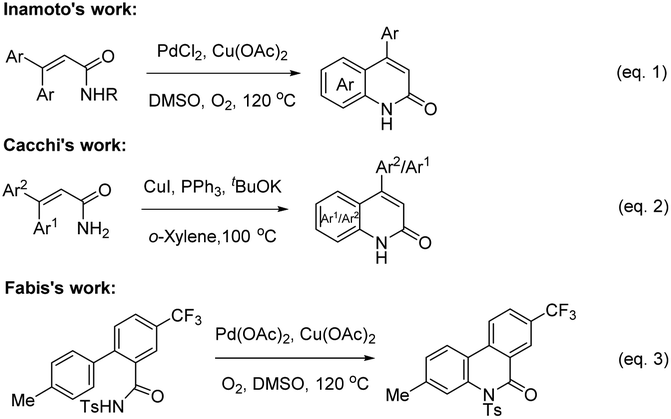 | ||
| Scheme 1 Metal-catalyzed the construction of the 2-quinolones skeleton. | ||
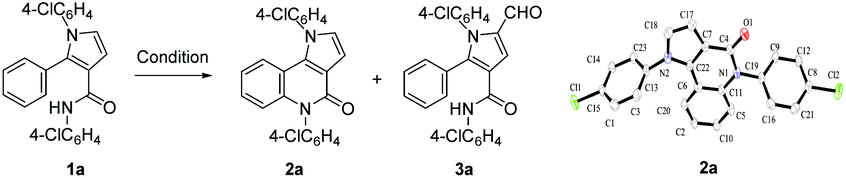
Very recently, we developed a facile ring-opening/cyclization/dehydrogenation domino reaction for the construction of multiple-substituted pyrroles by the reactions of doubly activated cyclopropanes with anilines in the presence of an iron salt.22 We have also found a series of derivatization reactions to form multi-substituted pyrroles.23 We are interested in further derivatization of readily available pyrroles for the construction of pyrrolo[3,2-c]quinolinones. We reasoned that the desired fused tricyclic pyrrolo[3,2-c]quinolinones could be synthesized via a metal-catalyzed intramolecular annulation by using the starting material of 2-aryl-1H-pyrrole-3-carboxamides (Fig. 1). Many more hetero-fused quinoline derivatives would be available through this route. Initially, compound 1a was selected as the model substrate to explore the optimal conditions for the ring-closure reaction. After many attempts, it was found that pyrrolo[3,2-c]quinolin-4-one (2a) could be obtained in 69% yield in the presence of 5 mol% CuI at 160 °C after 9 h (Table 1, entry 1). The structure of 2a was further confirmed by single-crystal X-ray diffraction analysis (Table 1).24 The yield of 2a decreased significantly if the temperature decreased (Table 1, entries 2 and 3). Further experiments showed that the yield of 2a could not be increased by increasing the amount of CuI (Table 1, entries 4 and 5). When performed under N2, the reaction did not afford the desired compound 2a and most of the starting material 1a was recovered, which indicated that dioxygen was involved in the reaction (Table 1, entry 6). Other copper salts including Cu(OAc)2, Cu(acac)2, and CuBr (Table 1, entries 7–9), and Cu powder proved to be less effective than CuI, especially for the Cu(OAc)2 and Cu(acac)2 catalysts, as they selectively gave the 2-formylpyrrole derivative 3a in 32% and 58% yield, respectively. CuCl and Cu2O showed a similar catalytic effect to CuI, but they needed longer reaction times (Table 1, entries 10 and 11). Other high boiling solvents such as DMF and xylene proved to be inefficient (Table 1, entries 13 and 14).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (equiv.) | Solvent | Temp/°C | Time/h | Yield/% |
| a Unless otherwise indicated, all reactions were carried out with 1a (0.2 mmol), CuI (0.05 equiv.) in DMSO (1.5 mL) at 160 °C under O2 (balloon, 1 atm). b The yield is an average of four reactions and less than 10% of 3a was isolated in each of the four reactions. c 88% of 1a was recovered. d 50% of 1a was recovered. e Reaction was performed under N2 and 92% of 1a was recovered. f 32% of 3a was obtained. g 58% of 3a was obtained. h 18% of 3a was obtained. i 65% of 1a was recovered. j 95% of 1a was recovered. | |||||
| 1 | CuI (0.05) | DMSO | 160 | 9 | 69b |
| 2 | CuI (0.05) | DMSO | 150 | 9 | 9c |
| 3 | CuI (0.05) | DMSO | 130 | 36 | 35d |
| 4 | CuI (0.2) | DMSO | 160 | 11 | 56% |
| 5 | CuI (0.5) | DMSO | 160 | 11 | 58% |
| 6 | CuI (0.05) | DMSO | 160 | 41 | 0e |
| 7 | Cu(OAc)2 (0.05) | DMSO | 160 | 4 | 25f |
| 8 | Cu(acac)2 (0.05) | DMSO | 160 | 37 | 0g |
| 9 | CuBr (0.05) | DMSO | 160 | 11 | 50h |
| 10 | CuCl (0.05) | DMSO | 160 | 16 | 72 |
| 11 | Cu2O (0.05) | DMSO | 160 | 28 | 75 |
| 12 | Cu (0.05) | DMSO | 160 | 18 | 53 |
| 13 | CuI (0.05) | DMF | 160 | 33 | 25i |
| 14 | CuI (0.05) | Xylene | Reflux | 12 | 0j |
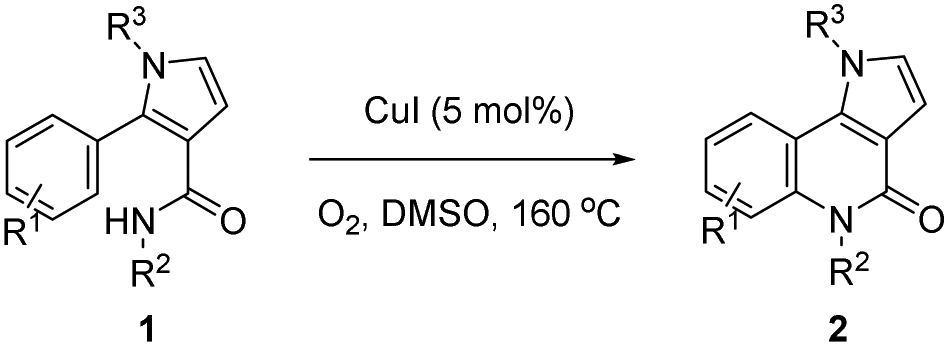
With the optimized conditions in hand (Table 1, entry 1), we proceeded to explore the scope and limitations of this ring-closure reaction using various starting materials such as 1. The results are summarized in Table 2. The substituents at the amide moiety were investigated first. It was found that the reactions of 1a–h bearing an aryl on the nitrogen afforded the corresponding pyrrolo[3,2-c]quinolinones 2a–h in 46–70% yields, and that the electronic nature (i.e., electron-donating or electron-withdrawing) and the position (i.e., ortho-, meta-, or para-position) of the substituents (e.g., –OMe, –Cl and –CO2Et) on the phenyl ring had little impact on the yields. However, benzyl and tertiary butyl alkyl groups on the nitrogen only gave the 2-formylpyrroles 3i in 70% and 3j in 40% yield, respectively, instead of the desired ring-closure products 2i and 2j. The effect of the substituent on the nitrogen of the pyrrole ring moiety was also examined. These experiments demonstrated that the yields of 2k (73%) and 2m (73%) with p-CO2Et phenyl and p-OMe phenyl substituents were slightly higher than 2l with a phenyl group on the nitrogen. The substrate 1n with a benzyl on the nitrogen gave 2n in 50% yield. However, tert-butyl substituted substrate 1o did not give the desired compound 2o and most of 1o was recovered. In addition, variations in the aryl at the α-position of the pyrrole ring were also taken into account. It seemed that the position of the substituent had a significant effect on the yield of 2. The substrates 1p, 1q and 1t with –OMe and –Cl substituents at the para-position of the aryl provided the products 2p (65%), 2q (71%) and 2t (67%) in approximately equal yields. In contrast, compounds 1r, 1s, and 1u with the –OMe or –Cl substituent at the ortho- or meta-position, mainly produced 2-formylation products 3r, 3s, and 3u along with an unidentified complex mixture of additional products. The product of the cyclization of 1r gave the target compound 2r regiospecifically with the para-position of the methoxyl group, in 23% yield.
|
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Time/h | Yield/% |
| a Unless otherwise indicated, all reactions were carried out with 1 (0.2 mmol), CuI (0.05 equiv.) in DMSO (1.5 mL) at 160 °C under O2 (balloon, 1 atm) and stopped when all of 1 had disappeared, and they produced less than 10% of the by-products 3. b 70% of 3i was obtained. c 40% 3j of was obtained. d 86% of 1o was recovered. e 14% of 3q was obtained. f The structure was determined by X-ray crystallography and 61% of 3r was obtained. g 68% of 3s was obtained. h 62% of 3u was obtained. | |||||
| 2a | H | 4-ClC6H4 | 4-ClC6H4 | 9 | 69 |
| 2b | H | 3-ClC6H4 | 4-ClC6H4 | 11 | 61 |
| 2c | H | 2-ClC6H4 | 4-ClC6H4 | 10 | 60 |
| 2d | H | 4-CO2EtC6H4 | 4-ClC6H4 | 12 | 63 |
| 2e | H | Ph | 4-ClC6H4 | 12 | 46 |
| 2f | H | 4-MeOC6H4 | 4-ClC6H4 | 12 | 70 |
| 2g | H | 3-MeOC6H4 | 4-ClC6H4 | 9 | 65 |
| 2h | H | 2-MeOC6H4 | 4-ClC6H4 | 8 | 53 |
| 2i | H | Bn | 4-ClC6H4 | 10 | 0b |
| 2j | H | t Bu | 4-ClC6H4 | 15 | 0c |
| 2k | H | 4-ClC6H4 | 4-CO2EtC6H4 | 18 | 73 |
| 2l | H | 4-ClC6H4 | Ph | 12 | 52 |
| 2m | H | 4-ClC6H4 | 2-MeOC6H4 | 9 | 73 |
| 2n | H | 4-ClC6H4 | Bn | 8 | 50 |
| 2o | H | 4-ClC6H4 | t Bu | 20 | 0d |
| 2p | 4-MeO | 4-ClC6H4 | Ph | 38 | 65 |
| 2q | 4-MeO | 4-ClC6H4 | 4-ClC6H4 | 10 | 71e |
| 2r | 3-MeO | Ph | 4-ClC6H4 | 11 | 23f |
| 2s | 2-MeO | Ph | 4-ClC6H4 | 31 | 0g |
| 2t | 4-Cl | Ph | 4-ClC6H4 | 20 | 67 |
| 2u | 3-Cl | Ph | 4-ClC6H4 | 11 | 0h |
In these transformations, a trace amount of by-products 4 were observed, and 4a was isolated and characterized by 1H NMR and 13C NMR spectroscopy, HRMS, and single-crystal X-ray diffraction analysis (Scheme 2). Interestingly, the product 2 would gradually disappear to form 4 when the reaction times were prolonged, which would give a direct method to produce valuable α-formyl pyrrolo[3,2-c]quinolinones 4 from pyrroles 1. As examples, three randomly selected reactions with starting materials 1 afforded the desired compounds 4a, 4e, and 4l in 31–35% yields when the reactions were prolonged until the intermediates 2 had disappeared. Although the yields were moderate, these preliminary results are meaningful given the advantages of the one-pot tandem reactions.
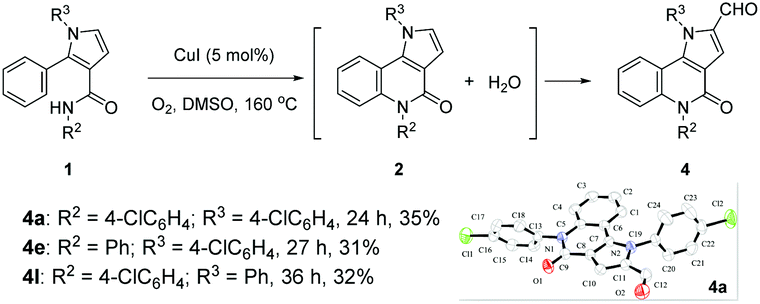 | ||
| Scheme 2 Examples of one-pot cascade reactions of 1 to α-formyl pyrrolo[3,2-c]quinolinones 4. | ||
During the transformation of 1a to 4a, it was observed that compound 2a was the single product early in the reaction (6 h), and later the formylated pyrrole 3a formed more gradually, and finally the α-formyl pyrrolo[3,2-c]quinolinone 4a along with an un-isolated mixture (mainly consisting of 3a and the β-formyl pyrrolo[3,2-c]quinolinone 4a′) were produced at the end (24 h). This observation indicates that the water formed in situ is necessary for the formylation, which draws a carbon from the solvent DMSO.23a,b This was further confirmed by the reactions of pure 2a (Scheme 3). The reaction did not occur under standard conditions; however, it afforded the formylated isomers 4a (Rf = 0.25, petroleum ether/EtOAc 2:1, 61%) and 4a′ (Rf = 0.50, petroleum ether/EtOAc 2:1, 28%), as characterized by 1H NMR and 13C NMR spectroscopy, HRMS, and by single-crystal X-ray diffraction analysis, when 10 equiv. water was added to the reaction mixture. The preliminary study revealed the feasibility of further modifying the target compounds 2 with an active aldehyde group.
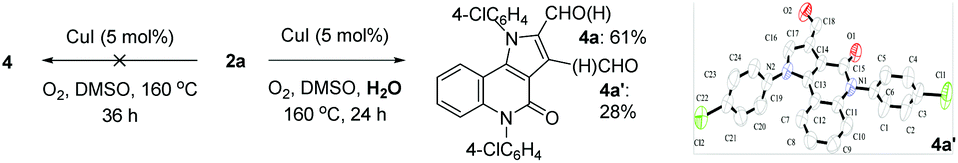 | ||
| Scheme 3 α- and β-formylation reaction of 2a. | ||
To clarify the reaction mechanism, three separate reactions were conducted by the stoichiometric addition of an electron-transfer scavenger (1,4-dinitrobenzene), a radical clock (diallyl ether), and a radical inhibitor (hydroquinone).25 It was observed that the reaction still proceeded smoothly to afford the desired product 2a along with 3a. These observed results suggest that a radical process is not involved in this transformation. In addition, control experiments showed that CuI and Cu(OAc)2 could catalyze the transformation under O2 (Table 1, entries 1 and 7), but no product was detected under N2 (Table 1, entry 6). These results suggest that a Cu(III) species might be involved in this transformation. On the basis of these control experiments and the previous literature,17b,20,21,25a,26 a plausible pathway for this pyrrolo[3,2-c]quinolinone synthesis is outlined in Scheme 4. Initially, a reactive Cu(III) intermediate A is formed by the reaction of Cu(I) and substrate 1 and O2in situ27via an electrophilic metallation or a C–H bond activation.28 Reductive elimination delivers the product 2 with concurrent formation of Cu(I). Formylation products 3 and 4 are generated via the aerobic Cu(I)-catalyzed Pummerer-like reaction,29 and DMSO serving as the one carbon donor for the –CHO.23a,b,30
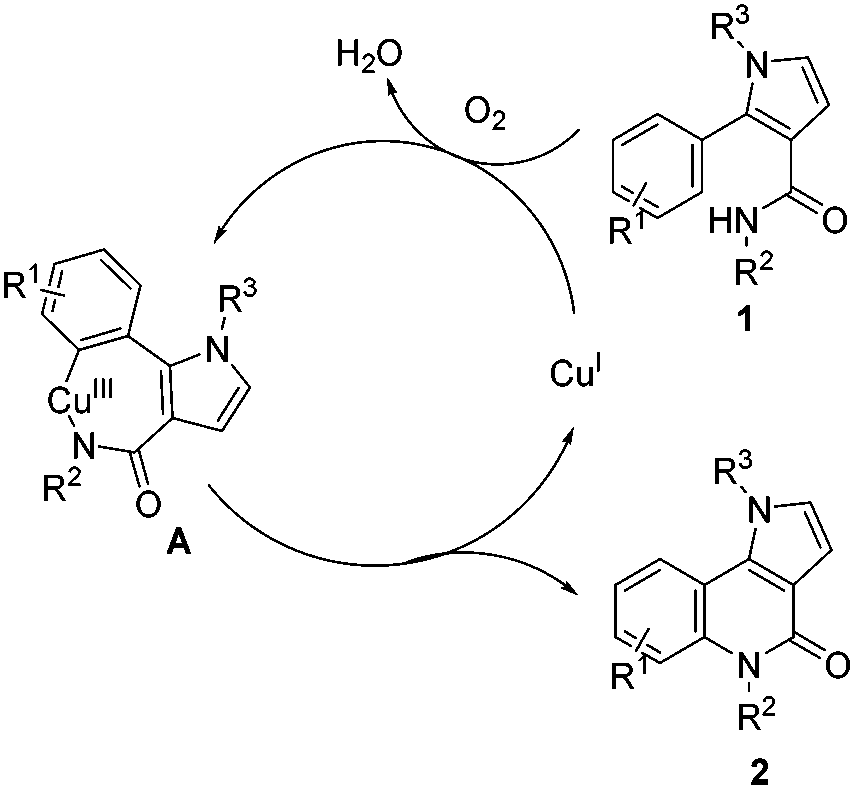 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, we have described the direct C(sp2)–H functionalization and intramolecular amide arylation for the efficient construction of substituted pyrrolo[3,2-c]quinolinones. The method provides an alternative, novel, efficient, and valuable route to the pyrrolo[3,2-c]quinolinones. Preliminary investigations on the preparation of valuable formylated pyrrolo[3,2-c]quinolinones via this method have also been carried out. Further studies of the application of the Cu(I)-catalyzed intramolecular C(sp2)–H amination process to the synthesis of other nitrogen atom-based heterocycles are currently underway.Acknowledgements
We thank the NSFC (21172056, 21272057 and 21372065), PCSIRT (IRT1061), China Postdoctoral Science Foundation funded project (2013M530339 and 2015M572109), and Key Project of Henan Educational Committee (14A150053).Notes and references
- K. M. Witherup, R. W. Ransom, A. C. Graham, A. M. Bernard, M. J. Salvatore, W. C. Lumma, P. S. Anderson, S. M. Pitzenberger and S. L. Varga, J. Am. Chem. Soc., 1995, 117, 6682–6685 CrossRef CAS.
- F. Heidempergher, P. Pevarello, A. Pillan, V. Pinciroli, A. Della Torre, C. Speciale, M. Marconi, M. Cini, S. Toma, F. Greco and M. Varasi, Farmaco, 1999, 54, 152–160 CrossRef CAS PubMed.
- T. Ohashi, Y. Oguro, T. Tanaka, Z. Shiokawa, S. Shibata, Y. Sato, H. Yamakawa, H. Hattori, Y. Yamamoto, S. Kondo, M. Miyamoto, H. Tojo, A. Baba and S. Sasaki, Bioorg. Med. Chem., 2012, 20, 5496–5506 CrossRef CAS PubMed.
- P. Helissey, S. Giorgi-Renault, J. Renault and S. Cros, Chem. Pharm. Bull., 1989, 37, 2413–2416 CrossRef CAS PubMed.
- T. H. Brown, R. J. Ife, D. J. Keeling, S. M. Laing, C. A. Leach, M. E. Parsons, C. A. Price, D. R. Reavill and K. J. Wiggall, J. Med. Chem., 1990, 33, 527–533 CrossRef CAS PubMed.
- P. Helissey, H. Parrot-Lopez, J. Renault and S. Cros, Eur. J. Med. Chem., 1987, 22, 366–368 CrossRef CAS.
- A. I. S. Almeida, A. M. S. Silva and J. A. S. Cavaleiro, Synlett, 2011, 2955–2958 CAS.
- (a) D. A. Powell and R. A. Batey, Org. Lett., 2002, 4, 2913–2916 CrossRef CAS PubMed; (b) S. Roy and O. Reiser, Angew. Chem., Int. Ed., 2012, 51, 4722–4725 CrossRef CAS PubMed.
- E. K. Yum, S. K. Kang, S. S. Kim, J.-K. Choi and H. G. Cheon, Bioorg. Med. Chem. Lett., 1999, 9, 2819–2822 CrossRef CAS PubMed.
- A. Shirai, O. Miyata, N. Tohnai, M. Miyata, D. J. Procter, D. Sucunza and T. Naito, J. Org. Chem., 2008, 73, 4464–4475 CrossRef CAS PubMed.
- B. B. Snider, Y. Ahn and S. M. O'Hare, Org. Lett., 2001, 3, 4217–4220 CrossRef CAS PubMed.
- (a) J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC; (b) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC.
- (a) D. J. Brown, Quinoxalines, in Chem. Heterocycl. Compd. Supplement II, John Wiley & Sons, Inc, 2004, vol. 61 Search PubMed; (b) Aromaticity in Heterocyclic Compounds in Topics in Heterocyclic Chemistry, volume ed. T. M. Krygowski and M. K. Cyrański, Springer-Verlag, Berlin, Heidelberg, 2009 Search PubMed.
- (a) Q. Liao, X. Yang and C. Xi, J. Org. Chem., 2014, 79, 8507–8515 CrossRef CAS PubMed; (b) J. Li, S. Benard, L. Neuville and J. Zhu, Org. Lett., 2012, 14, 5980–5983 CrossRef CAS PubMed.
- F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 2382–2384 CrossRef.
- (a) J. F. Hartwig, Synlett, 1997, 329–340 CrossRef CAS; (b) J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852–860 CrossRef CAS; (c) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS; (d) J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef CAS; (e) J. F. Hartwig, Pure Appl. Chem., 1999, 71, 1417–1423 CrossRef CAS; (f) B. H. Yang and S. L. Buchwald, J. Organomet. Chem., 1999, 576, 125–146 CrossRef CAS.
- (a) X. Li, L. He, H. Chen, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3636–3646 CrossRef CAS PubMed; (b) F. Péron, C. Fossey, T. Cailly and F. Fabis, Org. Lett., 2012, 14, 1827–1829 CrossRef PubMed; (c) D. G. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802–8805 CrossRef CAS PubMed.
- (a) P. Sang, Y. Xie, J. Zou and Y. Zhang, Org. Lett., 2012, 14, 3894–3897 CrossRef CAS PubMed; (b) N. Xia and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 337–339 CrossRef CAS PubMed; (c) X. Diao, L. Xu, W. Zhu, Y. Jiang, H. Wang, Y. Guo and D. Ma, Org. Lett., 2011, 13, 6422–6425 CrossRef CAS PubMed.
- (a) X. Wang, Y. Jin, Y. Zhao, L. Zhu and H. Fu, Org. Lett., 2012, 14, 452–455 CrossRef CAS PubMed; (b) X. Li, L. He, H. Chen, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3636–3646 CrossRef CAS PubMed; (c) Q. Shuai, G. Deng, Z. Chua, D. S. Bohle and C.-J. Li, Adv. Synth. Catal., 2010, 352, 632–636 CrossRef CAS; (d) G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932–1934 CrossRef CAS PubMed.
- K. Inamoto, T. Saito, K. Hiroya and T. Doi, J. Org. Chem., 2010, 75, 3900–3903 CrossRef CAS PubMed.
- R. Berrino, S. Cacchi, G. Fabrizi and A. Goggiamani, J. Org. Chem., 2012, 77, 2537–2542 CrossRef CAS PubMed.
- Z. Zhang, W. Zhang, J. Li, Q. Liu, T. Liu and G. Zhang, J. Org. Chem., 2014, 79, 11226–11233 CrossRef CAS PubMed.
- (a) Z. Zhang, Q. Tian, J. Qian, Q. Liu, T. Liu, L. Shi and G. Zhang, J. Org. Chem., 2014, 79, 8182–8188 CrossRef CAS PubMed; (b) J. J. Qian, Z. G. Zhang, Q. F. Liu, T. X. Liu and G. S. Zhang, Adv. Synth. Catal., 2014, 356, 3119–3124 CrossRef CAS; (c) Z. Zhang, D. Wang, B. Wang, Q. Liu, T. Liu, W. Zhang, B. Yuan, Z. Zhao, D. Han and G. Zhang, Tetrahedron, 2013, 69, 9063–9067 CrossRef CAS.
- CCDC 1056410 (2a), 1057211 (2q), 1056498 (4a), and 1056576 (4a′).
- (a) W. Zhou, Y. Liu, Y. Yang and G.-J. Deng, Chem. Commun., 2012, 48, 10678–10680 RSC; (b) Q. Qi, Q. Shen and L. Lu, J. Am. Chem. Soc., 2012, 134, 6548–6551 CrossRef CAS PubMed; (c) Please see ESI† for more information.
- X. X. Guo, D. W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed.
- For recent reviews on metal-catalyzed reactions using O2 as oxidant, see: (a) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed; (b) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC; (c) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464–3484 RSC; (d) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
- (a) H. Wang, Y. Wang, C. Peng, J. Zhang and Q. Zhu, J. Am. Chem. Soc., 2010, 132, 13217–13219 CrossRef CAS PubMed; (b) A. E. King, L. M. Huffman, A. Casitas, M. Costas, X. Ribas and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 12068–12073 CrossRef CAS PubMed.
- R. Pummerer, Ber. Dtsch. Chem. Ges., 1909, 42, 2282–2291 CrossRef CAS.
- (a) Y. Lv, Y. Li, T. Xiong, W. Pu, H. Zhang, K. Sun, Q. Liu and Q. Zhang, Chem. Commun., 2013, 49, 6439–6441 RSC; (b) Y.-F. Wang, F.-L. Zhang and S. Chiba, Synthesis, 2012, 1526–1534 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, 1H and 13C NMR spectra of all compounds. CCDC 1056410 (2a), 1057211 (2q), 1056498 (4a), and 1056576 (4a′). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00417a |
| This journal is © the Partner Organisations 2016 |