
Direct isoperfluoropropylation of arenediazonium salts with hexafluoropropylene†
Xiaojun
Wang
ab,
Yajun
Li
b,
Yuwei
Guo
ab,
Zhentong
Zhu
bc,
Yongming
Wu
*b and
Weiguo
Cao
*ab
aDepartment of Chemistry, Shanghai University, 99 Shangda Road, Shanghai 200444, China. E-mail: wgcao@staff.shu.edu.cn
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: ymwu@sioc.ac.cn
cSchool of Chemical and Environmental Engineering, Shanghai Institute of Technology, 100 Haiquan Road, Shanghai 201418, China
First published on 29th December 2015
Abstract
An efficient copper-promoted isoperfluoropropylation of aryl diazonium salts is described. The reaction occurs under mild conditions using commercially available hexafluoropropylene (HFP) as the starting material. In addition, a one-pot direct diazotization and isoperfluoropropylation protocol was developed. The method allows facile conversion of various arylamines into isoperfluoropropylarenes with good functional group compatibility using HFP.
Introduction
Incorporating fluorine-containing groups into organic compounds has a significant influence on their physicochemical properties and biological activities. Among the various organofluorine molecules, perfluoroalkylarenes play an increasingly important role in pharmaceuticals, agrochemicals and materials due to the low polarizability, high lipophilicity, strong electron-withdrawing nature and prominent metabolic stability of perfluoroalkyl groups.1 To date, significant progress has been made in introducing the trifluoromethyl group into aromatic rings.2 However, methods for introducing a higher analogue of perfluoroalkyl groups (such as C2F5, C3F7etc.) into an aromatic ring are rare.3 To the best of our knowledge, there are only few methods available for the synthesis of isoperfluoropropylarenes, although these compounds have been widely used in the synthesis of pesticides, organocatalysts, and functional materials (Fig. 1).4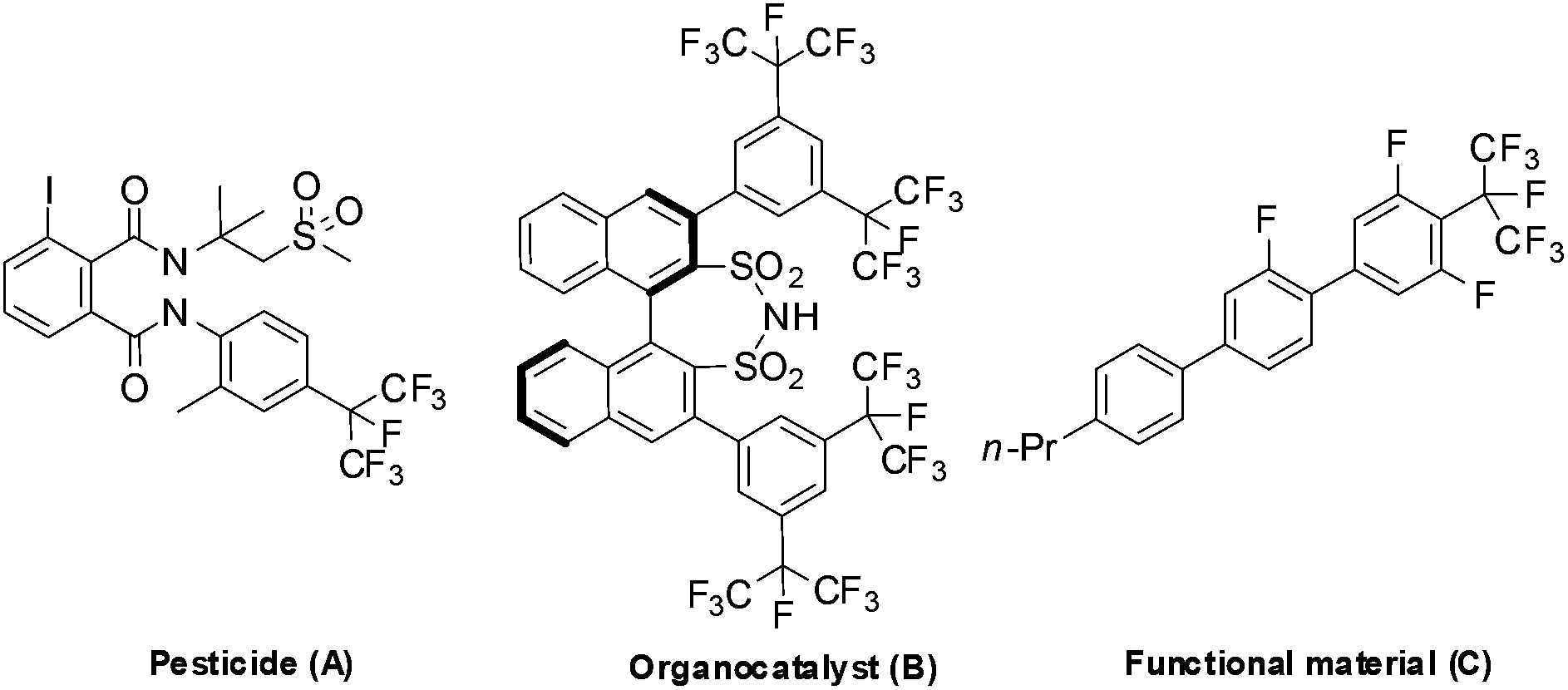 | ||
| Fig. 1 Isoperfluoropropylated substances. | ||
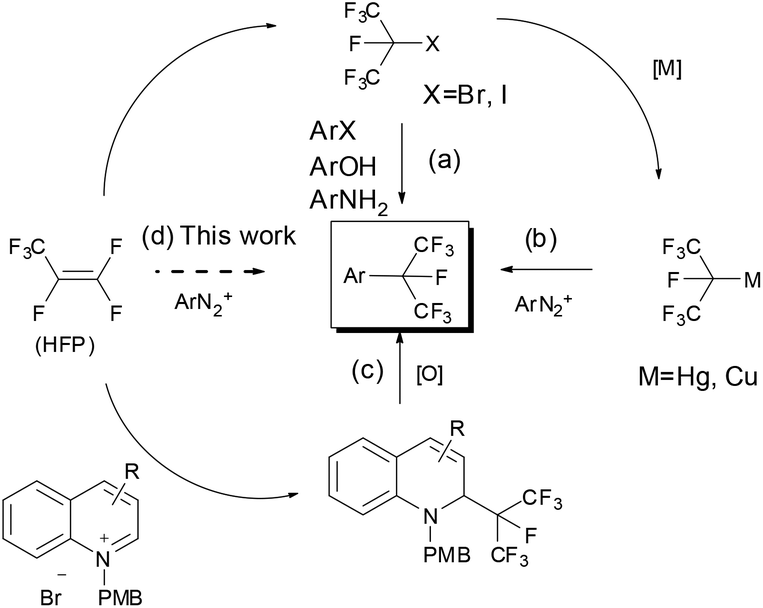
Traditional methods for preparing isoperfluoropropylarenes require several isoperfluoropropylation reagents. The most commonly used reagents are isoperfluoropropyl halides, which incorporate the isoperfluoropropyl (i-C3F7) group into aromatic rings upon treatment with phenols and arylamines via sulfinatodehalogenation reactions5 or with aryl halides via Ullmann reactions (Scheme 1, path a).6 On the other hand, a number of isoperfluoropropylmetal (MC3F7-i) reagents (such as i-C3F7Li, i-C3F7MgBr, i-C3F7ZnI, (i-C3F7)2Hg, (i-C3F7)2Cd etc.) have been synthesized, but very few of them are suitable for the synthesis of isoperfluoropropylarenes due to their instability or toxicity.7 Recently, by using a pre-prepared sensitive i-C3F7Cu(CH3CN) complex that was synthesized from an isoperfluoropropyl halide with copper at elevated temperatures, Chen's group has developed a method of converting arenediazonium salts into isoperfluoropropylarenes (Scheme 1, path b).3g Makosza et al. have reported an alternative strategy by introducing i-C3F7 into heterocyclic rings using the isoperfluoropropyl carbanion generated by HFP and KF. But this method needs an additional oxidation step to obtain aromatic heterocyclic compounds (Scheme 1, path c).8
 | ||
| Scheme 1 Methods for the synthesis of isoperfluoropropylarenes categorised by isoperfluoropropylation reagents. | ||
Recently, novel Sandmeyer-type trifluoromethylation,9 trifluoromethylthiolation10 and difluoromethylthiolation11 of arenediazonium salts have been reported. Generally, the mechanism of the Sandmeyer reaction is believed to proceed via radical intermediates.9a,c,d,11,12 It is plausible that single-electron transfer (SET) from Cu(I) species to the diazonium group forms a diazo radical and Cu(II) intermediate. The resulting diazo radical then releases nitrogen to form an aryl radical, which further reacts with the Cu(II) intermediate to generate the final trifluoromethylated (trifluoromethylthiolated/difluoromethylthiolated) products. The trifluoromethylating Sandmeyer reactions were discovered nearly simultaneously by the groups of Fu, Gooßen and Wang in 2013.9a–c In the above examples, the CF3 source is Umemoto's reagent in Fu's method and Ruppert–Prakash reagent (TMSCF3) in Gooßen's protocol and Wang's protocols. By the fluoroform-derived CuCF3, Grushin's group also had realized trifluoromethylation of arenediazonium salts in aqueous medium.9d Despite the progress in the Sandmeyer-type fluoroalkylation, the known routes to isoperfluoropropylarenes still rely mainly on toxic isoperfluoropropyl halide agents. So it is urgent to develop alternative solutions for more practical applications. Several attempts for isoperfluoropropylation of aryl iodides or arynes have been made by using MC3F7-i derived from i-C3F7H, TMSC3F7-i or isoperfluoropropyl halides, but the effects are unsatisfactory.3d,e,13 In sight of the successful examples of introducing i-C3F7 into aromatic rings and inspired by the fact that fluoroalkylmetals may act as the key intermediate in Sandmeyer fluoroalkylation, we hypothesized that isoperfluoropropyl copper species could be generated in situ in the presence of isoperfluoropropyl silver reagent and cuprous salt, which may react with arenediazonium to form the isoperfluoropropylated compounds (Scheme 2).
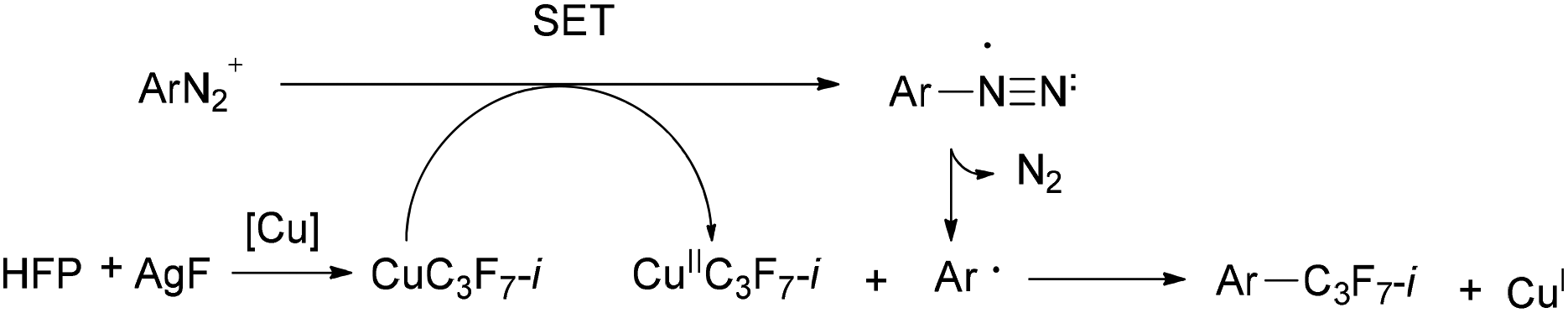 | ||
| Scheme 2 Proposed mechanism of Sandmeyer isoperfluoropropylation of diazonium salts. | ||
It is worth noting that HFP is the starting material for the synthesis of isoperfluoropropyl halides and isoperfluoropropylmetal reagents, which act as isoperfluoropropylation reagents (vide supra). According to the literature,14 when silver fluoride is introduced into a solution of HFP in acetonitrile, a reasonably stable isoperfluoropropyl silver reagent was formed. Considering both the stability and toxicity of isoperfluoropropylmetals, we envisioned that isoperfluoropropyl silver could act as the ideal agent for introducing i-C3F7 into aromatic rings. In fact, we have recently reported that this reagent is active for isoperfluoropropylation of arylboronic acids.15 Encouraged by this result, we aimed to explore the possibility for isoperfluoropropylation of arenediazonium salts directly from HFP, an inexpensive and readily available agent (Scheme 1, path d).
Results and discussion
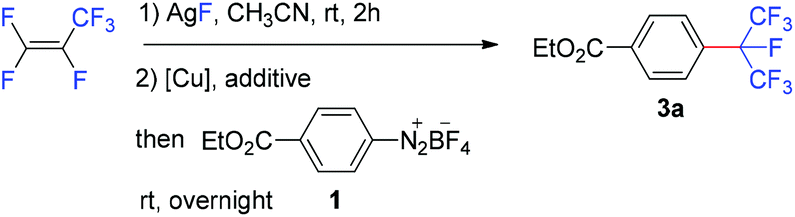
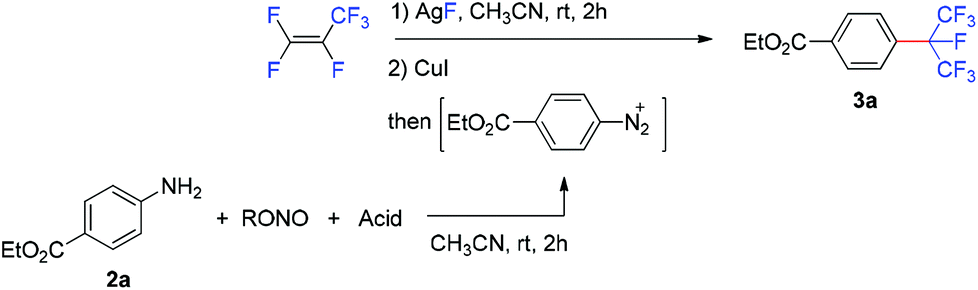
The reaction of 4-(ethoxycarbonyl)benzenediazonium tetrafluoroborate (1) with the isoperfluoropropyl silver reagent derived from HFP in the presence of copper or copper salt has been investigated (Table 1). The direct reaction of 1 with the isoperfluoropropyl silver reagent generated in situ at room temperature resulted in no desired product, ethyl 4-isoperfluoropropyl-benzoate (3a) (Table 1, entry 1). Under the identical conditions, when copper powder (1.6 equivalents) was added, 3a was formed in 30% yield after 24 hours (based on 19F NMR analysis) (entry 2). This result indicated that copper is essential for the formation of the desired product. Other copper species were screened (entries 3–9) and the results suggested that cuprous iodide was the best choice, affording 3a in 77% yield. It was also found that the addition of phosphine or nitrogen ligands had a disadvantageous impact on the reaction (entries 9–15).|
|
|||
|---|---|---|---|
| Entry | [Cu] | Additive | Yieldb (%) |
| a Reaction conditions: HFP (excess, balloon, 1 atm), AgF (0.2 mmol), [Cu] (0.16 mmol), additive (0.16 mmol), 1 (0.1 mmol), CH3CN (1.5 + 1.0 mL), under a N2 atmosphere. b Yield determined by 19F NMR analysis versus PhCF3 as an internal standard. c P(o-tol)3 = tris(o-tolyl)phosphine. d Bphen = bathophenanthroline. e 0.4 equivalent. | |||
| 1 | — | — | 0 |
| 2 | Cu | — | 30 |
| 3 | CuBr2 | — | 27 |
| 4 | CuCl | — | 46 |
| 5 | CuSCN | — | 29 |
| 6 | CuCN | — | 15 |
| 7 | CuOTf·2MeCN | — | 19 |
| 8 | CuI | — | 77 |
| 9 | CuI·P(OEt)3 | — | 51 |
| 10 | CuI | P(o-tol)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
48 |
| 11 | CuI | biPy | 24 |
| 12 | CuI | TMEDA | 12 |
| 13 | CuI | BPhend | 23 |
| 14 | CuI | PPh3 | 8 |
| 15 | CuI | Xantphose | 59 |
We then examined the possibility of developing a one-pot protocol for this reaction by using in situ generated arenediazonium salt from the corresponding anilines instead of the pre-prepared arenediazonium tetrafluoroborates. The solution of diazonium salt generated from the reaction of ethyl 4-aminobenzoate (2a) and tert-butyl nitrite in acetonitrile was added into the solution containing the isoperfluoropropyl silver reagent generated in situ and cuprous iodide. However, the isoperfluoropropylated product (3a) was not found (Table 2, entry 1). Fortunately, when sulfuric acid was added in the diazotization step, the desired product was obtained in 52% yield based on 19F NMR spectroscopy (entry 2),3g,9c,16 implying that the counterions of arenediazonium salts had a significant impact on this reaction. A further study indicated that when p-toluenesulfonic acid (pTSA) was employed, the product yield was improved to 75% (entry 6), while other acids had less effect (entries 3–5). In addition, the use of isopropyl nitrite reduced the yield to 64% (entry 7). It was found that this reaction is sensitive to the amount of tert-butyl nitrite; extra tert-butyl nitrite may have a negative effect on the reaction (entry 9).
|
|
|||
|---|---|---|---|
| Entry | Acid | RONO | Yieldb (%) |
| a Reaction conditions: HFP (excess, balloon, 1 atm), AgF (0.2 mmol), CuI (0.16 mmol), 2a (0.1 mmol), RONO (0.12 mmol), acid (0.15 mmol), CH3CN (1.5 mL + 1.5 mL), under a N2 atmosphere. b Yield determined by 19F NMR analysis versus PhCF3 as an internal standard. c 1.0 equiv. tBuONO. d 1.35 equiv. tBuONO. | |||
| 1 | — | t BuONO | 0 |
| 2 | H2SO4 (98.3%) | t BuONO | 52 |
| 3 | CF3COOH | t BuONO | 67 |
| 4 | CH3SO3H | t BuONO | 71 |
| 5 | CF3SO3H | t BuONO | 58 |
| 6 | pTSA | t BuONO | 75 |
| 7 | pTSA | i PrONO | 64 |
| 8 | pTSA | t BuONOc | 57 |
| 9 | pTSA | t BuONOd | 66 |
After establishing this one-pot diazotization and isoperfluoropropylation method, the scope of the reaction with various arylamines was explored (Scheme 3). It showed that aromatic amines bearing either electron-donating or -withdrawing groups reacted smoothly to form the corresponding isoperfluoropropylarenes in moderate to good yields. The method is compatible with a broad array of functional groups, including ether, ester, amide, keto, cyano, nitro etc. Substrates containing fluoro, chloro, bromo or iodo substituents (3l–3r) were well tolerated which might be of potential for further transformation. Remarkably, the oxidation sensitive vinyl group also tolerated the reaction conditions (3h). Although i-C3F7 is a very bulky group, anilines bearing ortho-substituents, such as iodo and bulky phenyl, gave the corresponding products 3o and 3t in yields comparable to those obtained in the reactions with the para-substituted analogues (3q and 3s). In addition, substrates with unprotected carbonyl functionalities afforded the desired products in good yields (3f and 3g) and various heterocycles were also isoperfluoropropylated successfully (3v and 3w). However, ortho-nitroaniline failed to generate the isoperfluoropropylated product, probably due to the coordination of the ortho nitro group to the metal center.
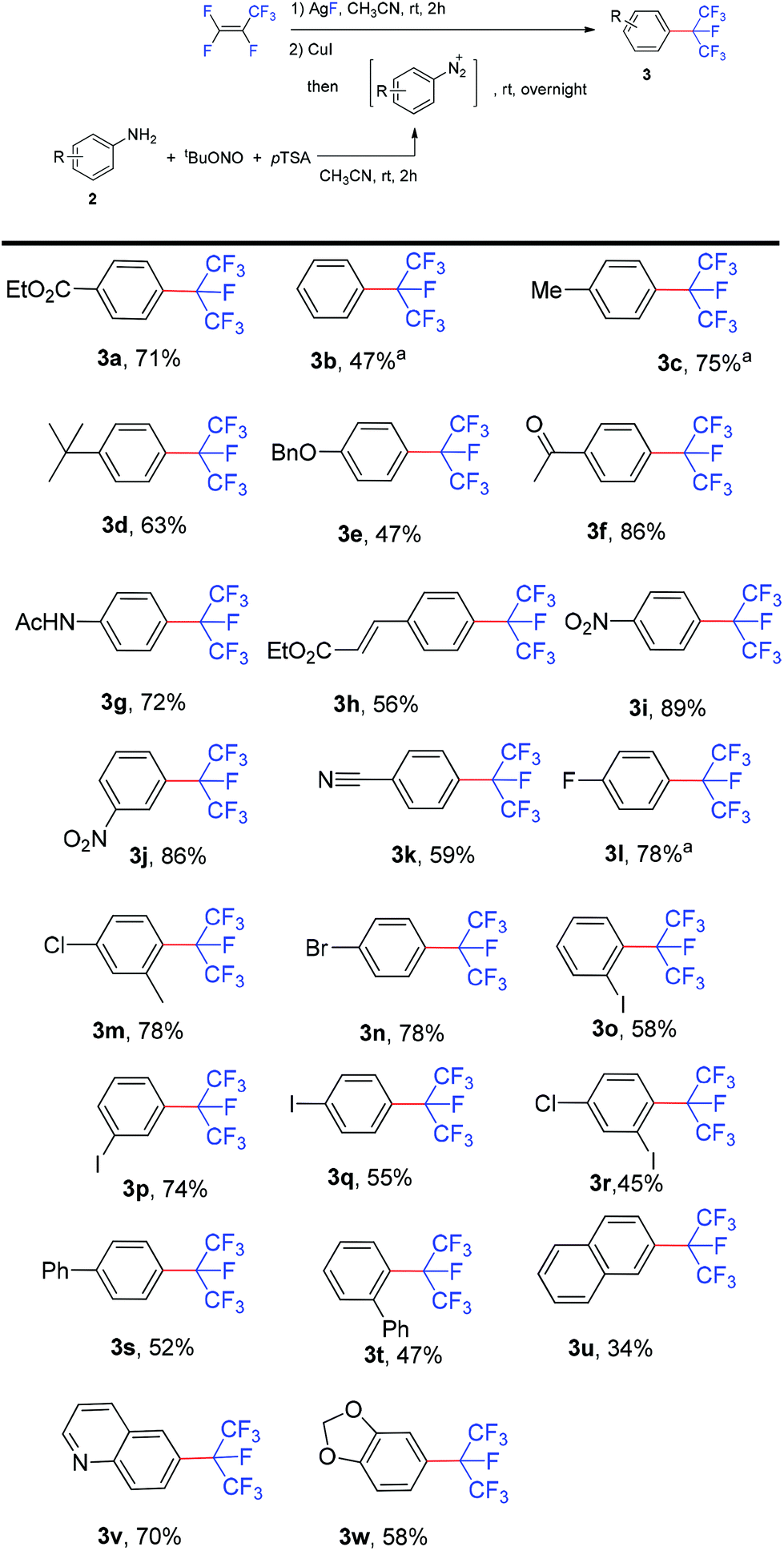 | ||
| Scheme 3 Scope of the reaction. Reaction conditions: HFP (excess, balloon, 1 atm), AgF (0.6 mmol), CuI (0.48 mmol), 2 (0.3 mmol), tBuONO (0.36 mmol), pTSA (0.45 mmol), CH3CN (3 mL + 3 mL). The yield of the isolated product is given. aYield determined by 19F NMR analysis versus PhCF3 as an internal standard. | ||
Conclusions
In summary, we have developed a copper-promoted isoperfluoropropylation reaction for converting arylamines to isoperfluoropropylarenes and heteroarenes by using the readily accessible hexafluoropropylene as the starting material. The transformation can proceed in a one-pot protocol forming isoperfluoropropylarenes from aromatic amines with good functional group tolerance. This method provides an attractive approach to valuable isoperfluoropropylarenes. Further investigations on the synthetic application and the mechanism of the transformation are currently underway in our laboratory.Experimental section
Typical procedure for the direct isoperfluoropropylation of arenediazonium salts with hexafluoropropylene
In a nitrogen-filled glove box, an oven-dried 20 mL crimp cap vessel (1) with a Teflon-coated stirrer bar was charged with silver fluoride (76.2 mg, 0.60 mmol) and was brought under an atmosphere of dry nitrogen. To this vessel, 3 mL of anhydrous acetonitrile and hexafluoropropylene (balloon, excess) were added, and the mixture was stirred at room temperature under ordinary pressure in the dark until silver fluoride precipitate disappeared. This process takes about two hours and the isoperfluoropropyl silver is generated. In the process of this reaction, in a nitrogen-filled glove box, an oven-dried 20 mL crimp cap vessel (2) with a Teflon-coated stirrer bar was charged with p-toluenesulfonic acid (77.4 mg, 0.45 mmol) and was brought under an atmosphere of dry nitrogen. To this vessel, 2a (49.5 mg, 0.3 mmol), 3 mL of anhydrous acetonitrile and tert-butyl nitrite (37.1 mg, 0.36 mmol) were added. The reaction mixture was stirred at ambient temperature for 2 h to generate the corresponding diazonium salt. After these procedures, the reaction mixtures in crimp cap vessels (1) and (2) were added in sequence via a syringe into an third oven-dried 20 mL crimp cap vessel with a Teflon-coated stirrer bar charging with cuprous iodide (91.4 mg, 0.48 mmol) under nitrogen. The new reaction mixture was stirred at ambient temperature overnight. The resulting mixture was diluted with Et2O (10 mL), then filtered through a short pad of Celite and rinsed with diethyl ether. The resulting organic solution was added into water (10 mL) and extracted with ethyl Et2O (3 × 10 mL). The organic layer was dried over MgSO4, filtered and concentrated. The residue was further purified by flash chromatography on silica gel to give the desired product.Acknowledgements
This work was supported by the National Basic Research Program of China (2012CB821600) and the National Natural Science Foundation of China (no. 21172239).Notes and references
- (a) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) K. Uneyama, Organofluorine Chemistry, Blackwell, Oxford, 2006 Search PubMed; (c) K. Mueller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef CAS PubMed; (d) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed.
- For selective reports, see: (a) F. Swart, Bull. Acad. R. Med. Belg., 1892, 24, 309 Search PubMed; (b) V. C. R. McLoughlin and J. Thrower, Tetrahedron, 1969, 25, 5921 CrossRef CAS; (c) C. P. Zhang, Z. L. Wang, Q. Y. Chen, C. T. Zhang, Y. C. Gu and J. C. Xiao, Angew. Chem., Int. Ed., 2011, 50, 1896 CrossRef CAS PubMed; (d) D. A. Nagib and D. W. C. Macmillan, Nature, 2011, 480, 224 CrossRef CAS PubMed; (e) A. Zanardi, M. A. Novikov, E. Martin, J. B. Buchholz and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 20901 CrossRef CAS PubMed; (f) L. Chu and F. L. Qing, J. Am. Chem. Soc., 2012, 134, 1298 CrossRef CAS PubMed; (g) Y. Zeng, L. Zhang, Y. Zhao, C. Ni, J. Zhao and J. Hu, J. Am. Chem. Soc., 2013, 135, 2955 CrossRef CAS PubMed; (h) S. Seo, J. B. Taylor and M. F. Greaney, Chem. Commun., 2013, 49, 6385 RSC; (i) Z. Gonda, S. Kovács, C. Wéber, T. Gáti, A. Mészáros, A. Kotschy and Z. Novák, Org. Lett., 2014, 16, 4268 CrossRef CAS PubMed; (j) M. Shang, S. Z. Sun, H. L. Wang, B. N. Laforteza, H. X. Dai and J. Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 10439 CrossRef CAS PubMed; (k) X. Li, J. Zhao, L. Zhang, M. Hu, L. Wang and J. Hu, Org. Lett., 2015, 17, 298 CrossRef CAS PubMed; (l) J. Zheng, J. H. Lin, X. Y. Deng and J. C. Xiao, Org. Lett., 2015, 17, 532 CrossRef CAS PubMed.
- (a) H. Morimoto, T. Tsubogo, N. D. Litvinas and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 3793 CrossRef CAS PubMed; (b) Q. Qi, Q. Shen and L. Lu, J. Am. Chem. Soc., 2012, 134, 6548 CrossRef CAS PubMed; (c) N. D. Litvinas, P. S. Fier and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 536 CrossRef CAS PubMed; (d) A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2013, 135, 12584 CrossRef CAS PubMed; (e) Y. Zeng and J. Hu, Chem. – Eur. J., 2014, 20, 6866 CrossRef CAS PubMed; (f) M. G. Mornino, P. S. Fier and J. F. Hartwig, Org. Lett., 2014, 16, 1744 CrossRef PubMed; (g) D. F. Jiang, C. Liu, Y. Guo, J. C. Xiao and Q. Y. Chen, Eur. J. Org. Chem., 2014, 6303 CrossRef CAS.
- (a) M. Tohnishi, H. Nakao, T. Furuya, A. Seo, H. Kodama, K. Tsubata, S. Fujioka, H. Kodama, T. Hirooka and T. Nishimatsu, J. Pestic. Sci., 2005, 30, 354 CrossRef CAS; (b) A. Conte and H. Kuehne, US, 20070185058, 2007 Search PubMed; (c) J. Guin, C. Rabalako and B. List, Angew. Chem., Int. Ed., 2012, 51, 8859 CrossRef CAS PubMed; (d) O. Kazuo, JP, 2007308483, 2007 Search PubMed.
- W. Y. Huang and F. H. Wu, Isr. J. Chem., 1999, 39, 167 CrossRef CAS.
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed.
- (a) R. D. Chambers, W. K. R. Musgrave and J. Savory, J. Chem. Soc., 1962, 1993 RSC; (b) W. T. Miller and M. B. Freedman, J. Am. Chem. Soc., 1963, 85, 180 CrossRef CAS; (c) P. E. Aldrich, E. G. Howard, W. J. Linn, W. J. Middleton and W. H. Sharkry, J. Org. Chem., 1963, 28, 184 CrossRef CAS; (d) B. L. Dyatkin, S. R. Sterlin, B. I. Martynov, E. I. Mysov and I. L. Knunyants, Tetrahedron, 1971, 27, 2843 CrossRef CAS; (e) H. Lange and D. Naumann, J. Fluorine Chem., 1984, 26, 1 CrossRef CAS.
- R. Loska and M. Makosza, J. Org. Chem., 2007, 72, 1354 CrossRef CAS PubMed.
- (a) J. J. Dai, C. Fang, B. Xiao, J. Yi, J. Xu, Z. J. Liu, X. Lu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 8436 CrossRef CAS PubMed; (b) X. Wang, Y. Xu, F. Mo, G. Ji, D. Qiu, J. Feng, Y. Ye, S. Zhang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 10330 CrossRef CAS PubMed; (c) G. Danoun, B. Bayarmagnai, M. F. Grünberg and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 7972; (d) A. Lishchynskyi, G. Berthon and V. V. Grushin, Chem. Commun., 2014, 50, 10237 RSC.
- G. Danoun, B. Bayarmagnai, M. F. Grünberg and L. J. Gooßen, Chem. Sci., 2014, 5, 1312 RSC.
- J. Wu, Y. Gu, X. Leng and Q. Shen, Angew. Chem., Int. Ed., 2015, 54, 7648 CrossRef CAS PubMed.
- (a) J. K. Kochi, J. Am. Chem. Soc., 1957, 79, 2942 CrossRef CAS; (b) C. Galli, Chem. Rev., 1988, 88, 765 CrossRef CAS.
- H. Kato, K. Hirano, D. Kurauchi, N. Toriumi and M. Uchiyama, Chem. – Eur. J., 2015, 21, 3895 CrossRef CAS PubMed.
- W. T. Miller Jr. and R. J. Burnard, J. Am. Chem. Soc., 1968, 90, 7367 CrossRef.
- Y. Li, X. Wang, Z. Zhu, Y. Guo, Y. Wu and Y. Gong, Chem. Commun., 2016, 52, 796 RSC.
- B. Bayarmagnai, C. Matheis, E. Risto and L. J. Goossen, Adv. Synth. Catal., 2014, 356, 2343 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00388a |
| This journal is © the Partner Organisations 2016 |