
Reactivity and mechanistic insight into the cross coupling reaction between isochromans and β-keto esters through C–H bond activation under visible light irradiation†
Ming
Xiang
,
Qing-Yuan
Meng
,
Xue-Wang
Gao
,
Tao
Lei
,
Bin
Chen
,
Chen-Ho
Tung
and
Li-Zhu
Wu
*
Key Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry & University of Chinese Academy of Sciences, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: lzwu@mail.ipc.ac.cn
First published on 10th February 2016
Abstract
The visible light catalytic cross coupling reaction from two different C–H bonds provides an efficient protocol for C–H bond activation and C–C bond construction. The application of the oxidative photoredox strategy to couple C–H bonds next to an oxygen atom with other C–H bonds is more challenging because of the more positive oxidation potential of ethers than that of amines. Here, we take advantage of organic dye 9-mesityl-10-methylacridinium perchlorate (Acr+-Mes ClO4−) as the photosensitizer and BrCCl3 as the terminal oxidant to achieve the addition of β-keto esters to oxonium species, directly generated from isochromans, leading to the formation of alkylation products in the α-position of an oxygen atom under visible light irradiation.
Introduction
The dramatic development of visible light catalysis in the past several years is very closely related to the single-electron redox abilities of photosensitizers which absorb visible light.1 Due to their excellent photophysical properties, transition metal complexes, like ruthenium, platinum or iridium polypyridyl complexes,2 have so far been frequently utilized as photosensitizers. However, the high cost, potential toxicity and limited availability of noble metals have often limited their further utilization on a larger scale.3 As an alternative, organic dyes (Fig. 1) have advantages over the metal complexes with regard to lower cost, less toxicity and more convenient modifications to tune redox potentials,4 which have been recently used in visible light photoredox catalysis.3–5 In 2011, for example, König and Zeitler employed eosin Y as an effective photosensitizer to successfully realize the asymmetric intermolecular α-alkylation of aldehydes.6 Our group exerted eosin Y or bodipy to accomplish the cross coupling reactions of N-aryltetrahydroisoquinolines with various nucleophiles.7 In the present work, we make use of an electron donor–acceptor-linked dyad, 9-mesityl-10-methylacridinium ion (Acr+-Mes),8 to active the C–H bond next to an oxygen atom for a new C–C bond formation.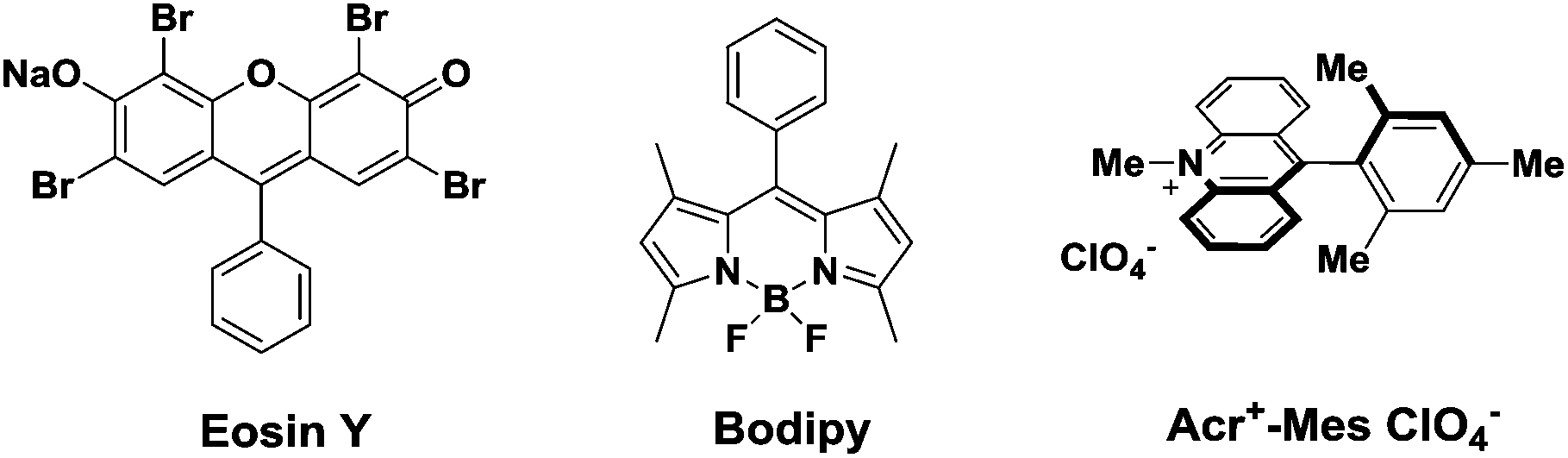 | ||
| Fig. 1 Molecular structures of common organic dyes. | ||
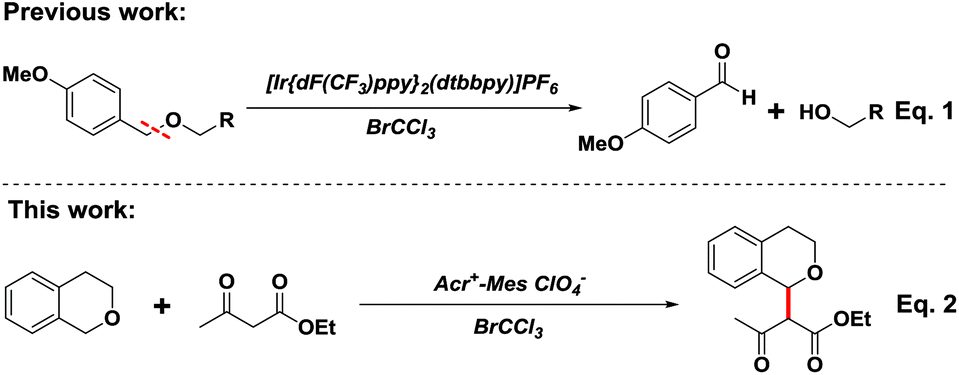
The construction of a C–C bond is a very essential and vital part of organic synthesis.9 The cross coupling from two different C–H bonds provides one of the ideal protocols to this end, because it not only forms new C–C bonds and reduces synthetic steps by avoiding the prefunctionalization of starting materials, but also displays atom-economical and environmentally benign characteristics.10 The formation of C–C bonds via the functionalization of C–H bonds adjacent to a nitrogen atom has been extensively studied under photocatalytic conditions.11 However, the visible light catalytic cross coupling of C–H bonds next to an oxygen atom with other C–H bonds is very limited due to the higher oxidation potential of ethers compared with amines.12 Oxonium species generated by the two-electron oxidation of ethers were expected to work as reactive intermediates for ether functionalization,13 and several examples involving oxocarbenium ions formed by oxidative photoredox catalysis have been reported.14 For instance, Stephenson et al. first disclosed a highly efficient approach to the removal of the para-methoxybenzyl group by hydrolysis of oxonium species employing an iridium complex as the photosensitizer and BrCCl3 as the terminal oxidant (Scheme 1, eqn (1)).14a Later, Cheng et al. achieved a similar result by the combination of eosin Y and H2O2,14b and Li et al. transformed alkyl benzyl ethers to alkyl esters via a tandem hydrolysis and esterification procedure.14c Very recently, our group demonstrated that the formation of a C–C bond next to an O atom could be achieved under photocatalytic oxidant-free conditions via an oxonium intermediate.15 Based on these results, we questioned whether oxonium species could be captured by nucleophiles to generate coupling products under oxidative photocatalytic conditions. Considering that the oxidation byproducts (CHCl3 and HBr) of BrCCl3 are easily separated from the reaction system, we herein design a new combination, Acr+-Mes cooperated with oxidant BrCCl3 (Scheme 1, eqn (2)), which successfully executes our plan.
 | ||
| Scheme 1 Utilization of oxonium species by oxidative photocatalysis. | ||
Results and discussion
Initially to explore the above transformation, we selected isochroman (1a) and ethyl acetoacetate (2a) as substrates for our experiment with various bases under different solvent conditions (Table 1). The reaction of 1a with 2a in the presence of Acr+-Mes ClO4− (5 mol%), 2 equivalents of 2,6-dimethylpyridine and BrCCl3 in anhydrous MeCN under an ambient argon atmosphere was performed, and after 24 h irradiation by blue LEDs (λ = 450 ± 10 nm), the desired cross coupling product 3a was obtained in 45% yield with 65% conversion of 2a (entry 1). Among the bases screened (entries 1–3), Na2HPO4 (entry 2) provided the optimal efficiency, and Na2CO3 (entry 3) could also facilitate this reaction despite a slightly lower yield. The solvent screening suggested that replacing MeCN with DCM (entry 4) or THF (entry 5) did not improve the reaction yield. As transitional metal salts may have the ability to activate a nucleophile and benzyl C–H bond,9,10 several readily accessible and inexpensive copper salts were then added. To our delight, a catalytic amount of Cu(OTf)2 proved to be the best choice for the model cross coupling reaction, which afforded 3a in an excellent yield of 87% with an almost complete conversion of 2a (96%) (entry 8). CuCl2 (entry 6) or CuBr2 (entry 7) as an alternative copper salt could also give 3a in a moderate yield. Notably, control experiments revealed that a very low yield was obtained in the absence of the base Na2HPO4 (entry 9). The selective removal of light, BrCCl3 or photosensitizer (Acr+-Mes ClO4−) showed that each of them was necessary for the successful oxidative coupling between 1a and 2a (entry 10).|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Metal salt. | Conversion 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
Yield 3ab (%) |
| a Isochroman (1.0 mmol), ethyl acetoacetate (0.5 mmol), Acr+-Mes (0.025 mmol, 5 mol%), BrCCl3 (1.0 mmol), base (1.0 mmol) and metal salt. (0.05 mmol, 10 mol%) in the solvent (2 mL) were irradiated by blue LEDs for 24 hours under an argon atmosphere. b Determined by 1H NMR spectroscopy using an internal standard. c General conditions, but with no Acr+-Mes, BrCCl3 or light. n.d. = not determined. | |||||
| 1 | 2,6-Dimethylpyridine | MeCN | None | 65 | 45 |
| 2 | Na2HPO4 | MeCN | None | 78 | 76 |
| 3 | Na2CO3 | MeCN | None | 68 | 57 |
| 4 | Na2HPO4 | DCM | None | 64 | 20 |
| 5 | Na2HPO4 | THF | None | 74 | 49 |
| 6 | Na2HPO4 | MeCN | CuCl2 | 98 | 79 |
| 7 | Na2HPO4 | MeCN | CuBr2 | 98 | 76 |
| 8 | Na2HPO4 | MeCN | Cu(OTf)2 | 96 | 87 |
| 9 | None | MeCN | Cu(OTf)2 | 74 | 27 |
| 10c | Na2HPO4 | MeCN | Cu(OTf)2 | n.d. | <10 |

With the optimized reaction conditions in hand, we first sought to define the scope of β-keto esters, and the corresponding results are listed in Scheme 2. A variety of aliphatic and electronically varied aromatic β-keto esters were found to be good substrates for the reaction with a diastereomeric ratio of 1:1–1:1.9 (Scheme 2, 3a–3h). Due to the chloro-functionality of the product 3g, obtained in a relatively high yield, it could serve as a potential intermediate for further organic synthesis. tert-Butyl acetoacetate, however, showed little difference in the reactivity, giving rise to a cross coupling product in 28% yield, probably due to the decomposition of product 3e. More importantly, other nucleophiles, such as the 1,3-dicarbonyl compound (2,4-pentanedione) and benzoylnitromethane, reacted smoothly under the standard conditions, and the desired products were achieved in moderate to good yields (Scheme 2, 3i and 3j).
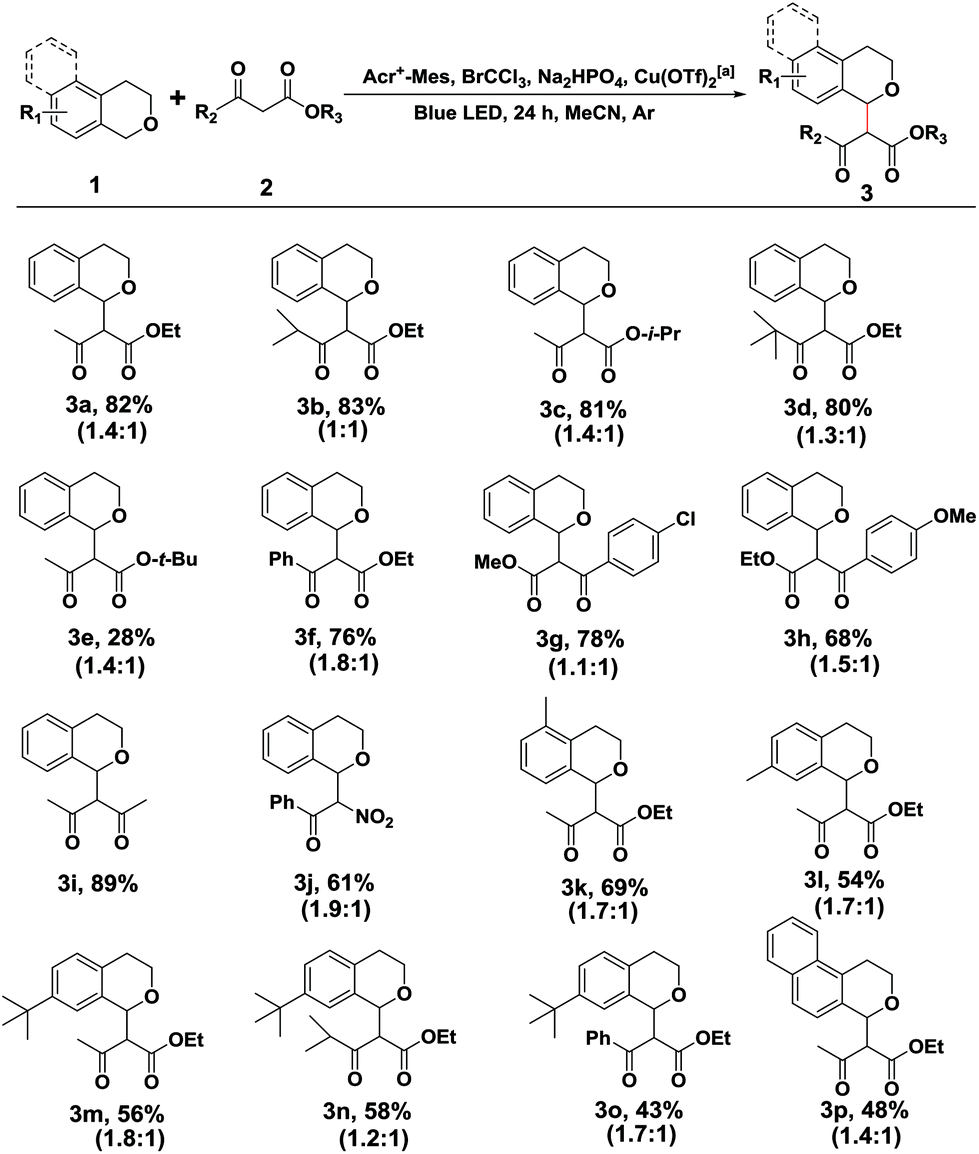 | ||
| Scheme 2 Scope of substrates: variants of isochroman or β-keto esters. aIsochromans (1.0 mmol), β-keto esters (0.5 mmol), Acr+-Mes (0.025 mmol, 5 mol%), BrCCl3 (1.0 mmol), Na2HPO4 (1.0 mmol), Cu(OTf)2 (0.05 mmol, 10 mol%) in the MeCN (2 mL) were irradiated by blue LEDs for 24 hours under an argon atmosphere. The ratios of two diastereomers were determined by 1H NMR spectra and indicated in brackets. | ||
Subsequently, the substituent effect of isochroman, including methyl, tert-butyl, methoxy or chloro, on the cross coupling reaction was investigated. Alkylsubstituents on the aromatic ring of isochroman participated in the reaction with reduced yields, probably due to the increased steric bulk (Scheme 2, 3k–3o). However, no reaction took place when a methoxy or chloro substituent at C7 of isochromans was applied to the photocatalytic reaction. Then, 1,4-dihydro-2H-benzo[f]isochromene was found to be an alternative substrate, although the yield of product 3p dropped to 48% with part of the substrate remaining. Finally, when the acylic substrate (1-(methoxymethyl)-4-methylbenzene) was treated with 2a, 4-methylbenzaldehyde was isolated as the main product, and no cross coupling product was observed with most of the substrate being consumed. This observation was consistent with previous reports.14 Since an acylic oxonium ion was less stable, trace water in the reaction system may quench it and block the route of nucleophilic addition.
It should be pointed out that the reaction cannot tolerate strong electron donating or withdrawing groups toward the isochroman coupling partner. The former observation could be explained by Floreancig's mechanistic studies of DDQ-mediated ether functionalizations.16 The substrate with a lower oxidation potential was found to possess a higher bond dissociation energy of the scissile C–H bond, so the methoxy substituent isochroman variant almost remained unchanged after the reaction. The latter substrate cannot be oxidated by an acridinium photosensitizer because of a higher oxidant potential of the electron-poor aromatic group.
To shed light on the primary process for the visible light-driven reaction, the oxidation potential (Eox) of isochroman (1a) and the reduction potential (Ered) of BrCCl3 were determined to be 2.03 V and −0.84 V vs. SCE in MeCN (Fig. S1a and S1b†). The Eox value of 1a is lower than the one-electron reduction potential of the electron-transfer state Acr˙-Mes˙+ (2.06 V vs. SCE in MeCN),17 so the electron transfer from 1a to Acr˙-Mes˙+ is thermodynamically feasible, whereas the electron transfer from Acr˙-Mes˙+ (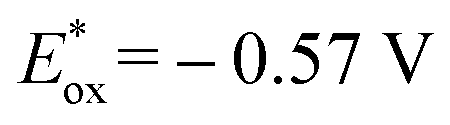 vs. SCE in MeCN)17 to BrCCl3 is energetically unfavourable. However, this unfavourable step is irreversible due to the decomposition of BrCCl3,18 and may be the rate-determining step of the reaction.
vs. SCE in MeCN)17 to BrCCl3 is energetically unfavourable. However, this unfavourable step is irreversible due to the decomposition of BrCCl3,18 and may be the rate-determining step of the reaction.
Furthermore, electron paramagnetic resonance (EPR) was conducted to detect the change of reaction intermediates in the photocatalytic cross coupling reaction. The measurement was carried out in an argon-saturated MeCN solution of Acr+-Mes with or without 1a (or BrCCl3). The sample cavity was first irradiated by a high-pressure mercury lamp for 2 min at 233 K. After the irradiation, the sample cell was immediately cooled to 123 K and the ESR spectra were recorded. The signal at g = 2.0036 (Fig. 2 solid line) is assigned to Acr˙-Mes+˙ due to the superposition of the ESR signals of the radical cation of the mesitylene moiety and the acridinyl radical moiety.19 Because the electron transfer from 1a to Acr˙-Mes+˙ is energetically favorable, the change of EPR signals (Fig. 2a) might result from two aspects: (1) the signal of Acr˙-Mes+˙ is quenched by the addition of 1a, resulting in a reduction in the intensity of the original signal; (2) Acr˙-Mes˙+ accepts an electron from 1a to generate Acr˙-Mes, whose signal is at g = 2.0035 (Fig. 2b).20 On the other hand, Acr˙-Mes+˙ delivers an electron to BrCCl3 to afford ˙CCl3 and Br−, so the obvious new signal at g = 2.0092 is from ˙CCl3, which is consistent with the reported value (Fig. 2d).21 These observations again demonstrate that the single electron transfer process proceeds not only between Acr˙-Mes˙+ and 1a, but also between Acr˙-Mes˙+ and BrCCl3.
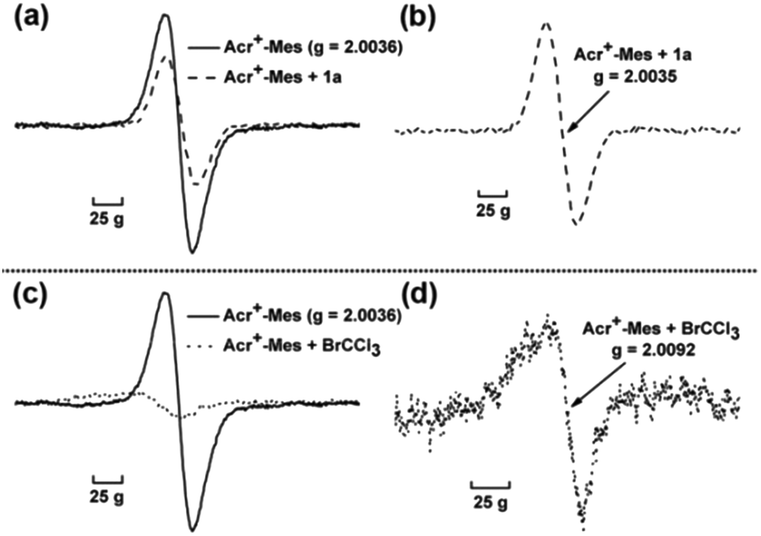 | ||
| Fig. 2 EPR spectra observed after irradiation of an argon-saturated MeCN solution of Acr+-Mes (0.0125 M) at 233 K and then measured at 123 K. (a) In the absence and presence of isochroman (0.5 M); (b) enlarged graph in the situation of adding isochroman (c) in the absence and presence of BrCCl3 (0.5 M); (d) enlarged graph in the situation of adding BrCCl3. | ||
A likely mechanism of this photocatalytic cross coupling reaction is outlined in Fig. 3. The first step involves photoinduced intramolecular electron transfer from the Mes moiety to the singlet excited state of the Acr+ moiety to generate Acr˙-Mes˙+, an extremely long-lived (e.g., 2 h at 203 K) electron-transfer state.8a The Mes˙+ moiety can oxidize isochroman (1a) to generate the arene radical cation (4), whereas Acr˙ can reduce BrCCl3 to afford Br− and ˙CCl3. The photosensitizer (Acr+-Mes) is then regenerated. The trichloromethyl radical ˙CCl3 (5) subsequently abstracts a hydrogen atom from the intermediate (4) to produce the oxocarbenium ion (6) along with CHCl3 generation. Notably, the trichloromethane CHCl3 in the reaction system was observed by 1H NMR and GC-MS analysis (Fig. S2†), and the kinetic isotope effect value was determined from intermolecular competition experiments to be kH/kD = 3.6 (Fig. 3) on the basis of analysing the 1H NMR spectra of the isolated products (Fig. S3†). The obtained results indicated that benzylic C–H bond cleavage is probably involved in the reaction process. Finally, the resulting oxonium species react with the nucleophile (2a), activated by Cu(OTf)2, to provide the desired C–C bond coupling product (3a).
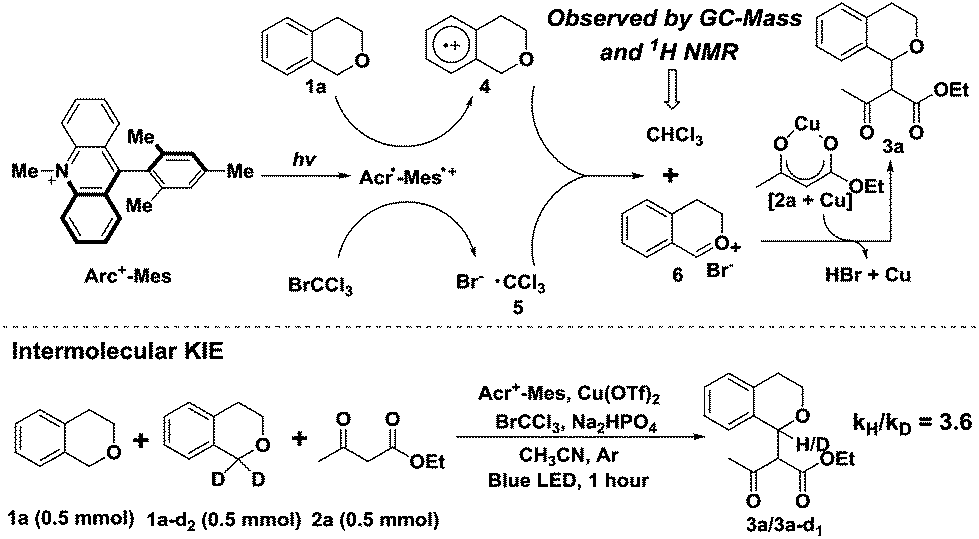 | ||
| Fig. 3 Proposed mechanism. | ||
Conclusions
In summary, a novel acridinium catalyzed utilization of oxocarbenium ions, directly obtained from isochromans, is developed. This photocatalytic method is a practical synthetic tool for the functionalization of a C(sp3)–H bond adjacent to the O atom of ethers and α-alkylation products were obtained in moderate to good yields. Mechanistic studies give rich information about the reaction route. Electrochemistry experiments demonstrate that reductive quenching is thermodynamically feasible, whereas the unfavourable oxidation of the electron-transfer state Acr˙-Mes˙+ is probably the rate-determining step in this transformation. Furthermore, EPR and KIE studies reveal that BrCCl3, after accepting an electron from Acr˙-Mes˙+, can generate ˙CCl3, which would abstract a hydrogen atom from the benzylic position. Additional investigation to explore the synthetic applications of the oxonium species by this photocatalytic protocol is underway in our group.Acknowledgements
Financial support for this research from the Ministry of Science and Technology of China (2013CB834804, 2013CB834505 and 2014CB239402), the National Natural Science Foundation of China (21390404, 91427303 and 21402217), the Key Research Programme of the Chinese Academy of Sciences (KGZD-EW-T05), and the Chinese Academy of Sciences is gratefully acknowledged.Notes and references
- For recent reviews on photoredox catalysis, see: (a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef CAS PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (d) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (e) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (f) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- (a) D. Zhang, L.-Z. Wu, L. Zhou, X. Han, Q.-Z. Yang, L.-P. Zhang and C.-H. Tung, J. Am. Chem. Soc., 2004, 126, 3440 CrossRef CAS PubMed; (b) K. Feng, R.-Y. Zhang, L.-Z. Wu, B. Tu, M.-L. Peng, L.-P. Zhang, D. Zhao and C.-H. Tung, J. Am. Chem. Soc., 2006, 128, 14685 CrossRef CAS PubMed; (c) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; (d) A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464 CrossRef CAS PubMed; (e) J. J. Zhong, Q. Y. Meng, G. X. Wang, Q. Liu, B. Chen, K. Feng, C. H. Tung and L. Z. Wu, Chem. – Eur. J., 2013, 19, 6443 CrossRef CAS PubMed; (f) Q.-Y. Meng, T. Lei, L.-M. Zhao, C.-J. Wu, J.-J. Zhong, X.-W. Gao, C.-H. Tung and L.-Z. Wu, Org. Lett., 2014, 16, 5968 CrossRef CAS PubMed.
- D. P. Hari and B. Konig, Chem. Commun., 2014, 50, 6688 RSC.
- (a) S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561 RSC; (b) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059 RSC.
- (a) D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169 CrossRef CAS; (b) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2013, 4, 355 CrossRef.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS PubMed.
- (a) Q. Liu, Y.-N. Li, H.-H. Zhang, B. Chen, C.-H. Tung and L.-Z. Wu, Chem. – Eur. J., 2012, 18, 620 CrossRef CAS PubMed; (b) J.-J. Zhong, Q.-Y. Meng, B. Liu, X.-B. Li, X.-W. Gao, T. Lei, C.-J. Wu, Z.-J. Li, C.-H. Tung and L.-Z. Wu, Org. Lett., 2014, 16, 1988 CrossRef CAS PubMed; (c) X.-Z. Wang, Q.-Y. Meng, J.-J. Zhong, X.-W. Gao, T. Lei, L.-M. Zhao, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2015, 51, 11256 RSC.
- (a) S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS PubMed; (b) H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 15999 CrossRef CAS PubMed; (c) D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577 CrossRef CAS PubMed.
- (a) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS PubMed; (b) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed.
- (a) C.-J. Li, Acc. Chem. Res., 2008, 42, 335 CrossRef PubMed; (b) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed.
- (a) A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464 CrossRef CAS PubMed; (b) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef CAS PubMed; (c) P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672 CrossRef CAS PubMed; (d) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338 CrossRef CAS PubMed.
- J. Jin and D. W. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565 CrossRef CAS PubMed.
- (a) Y. Zhang and C.-J. Li, Angew. Chem., Int. Ed., 2006, 45, 1949 CrossRef CAS PubMed; (b) Y. Zhang and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 4242 CrossRef CAS PubMed; (c) Z. Li, R. Yu and H. Li, Angew. Chem., Int. Ed., 2008, 47, 7497 CrossRef CAS PubMed.
- (a) J. W. Tucker, J. M. R. Narayanam, P. S. Shah and C. R. J. Stephenson, Chem. Commun., 2011, 47, 5040 RSC; (b) Z. Liu, Y. Zhang, Z. Cai, H. Sun and X. Cheng, Adv. Synth. Catal., 2015, 357, 589 CrossRef CAS; (c) P. Lu, T. Hou, X. Gu and P. Li, Org. Lett., 2015, 17, 1954 CrossRef CAS PubMed.
- M. Xiang, Q. Y. Meng, J. X. Li, Y. W. Zheng, C. Ye, Z. J. Li, B. Chen, C. H. Tung and L. Z. Wu, Chem. – Eur. J., 2015, 21, 18080 CrossRef CAS PubMed.
- H. H. Jung and P. E. Floreancig, Tetrahedron, 2009, 65, 10830 CrossRef CAS PubMed.
- K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601 RSC.
- K. Ohkubo, R. Iwata, S. Miyazaki, T. Kojima and S. Fukuzumi, Org. Lett., 2006, 8, 6079 CrossRef CAS PubMed.
- S. Fukuzumi, A. Itoh, T. Suenobu and K. Ohkubo, J. Phys. Chem. C, 2014, 118, 24188 CAS.
- S. Fukuzumi, K. Doi, A. Itoh, T. Suenobu, K. Ohkubo, Y. Yamada and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15572 CrossRef CAS PubMed.
- P. F. Fitzpatrick and J. J. Villafranca, J. Am. Chem. Soc., 1985, 107, 5022 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all compounds. See DOI: 10.1039/c5qo00412h |
| This journal is © the Partner Organisations 2016 |