
Controlled TfOH- or AuCl-catalyzed cycloisomerization and tandem hydrolytic defluorination of 1,2-allenyl perfluoroalkyl ketones†
Can
Xue
,
Xin
Huang
,
Shangze
Wu
,
Chunling
Fu
* and
Shengming
Ma
*
Laboratory of Molecular Recognition and Synthesis, Department of Chemistry, Zhejiang University, Hangzhou 310027, Zhejiang, People's Republic of China. E-mail: masm@sioc.ac.cn
First published on 30th March 2016
Abstract
An efficient TfOH- or AuCl-catalyzed cycloisomerization reaction of easily available n-perfluoroalkyl 1,2-allenyl ketones occurring smoothly to form 2-perfluoroalkyl furans has been reported. In the presence of TfOH and H2O, a corresponding tandem hydrolytic defluorination afforded multi-substituted furan-2-yl n-perfluoroalkyl ketones.
Introduction
As one of the ubiquitous heterocyclic cores, furan plays a significant role in organic synthesis,1 and also broadly exists in natural products2 and useful materials.3 Many efforts have been made to construct intricate furan derivatives,4–10among which transition metal-catalyzed (such as Ag, Au, Pd, Rh, etc.) cycloisomerization of allenones6–10 (Scheme 1a) was found to be apposite due to the substituent-loading capability of the unique substrates. On the other hand, due to their extensive application in the field of functional materials and the importance of organofluorine compounds,11 the development of synthetic methodologies for generating perfluoroalkyl substituted heterocycles is of current interest. In our previous work, we reported a TfOH-catalyzed12 direct formation of furanyl perfluoroalkyl ketones from easily available n-perfluoroalkyl allenones13 (Scheme 1b).14 In this study, we wish to describe a TfOH- or AuCl-catalyzed controlled cycloisomerization of perfluoroalkyl allenones, affording perfluoroalkyl furans first, which may be converted to corresponding hydrolytically defluorinated ketones in the presence of TfOH and water. | ||
| Scheme 1 Cyclization reactions of ordinary allenones and perfluoroalkyl allenones. | ||
Results and discussion
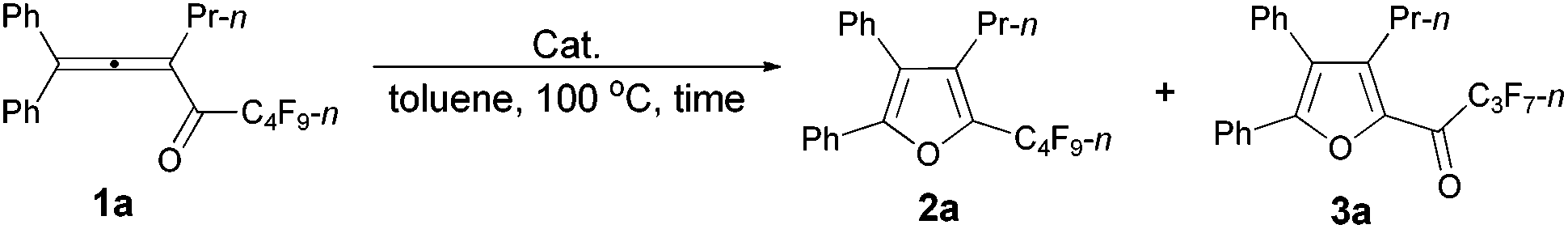
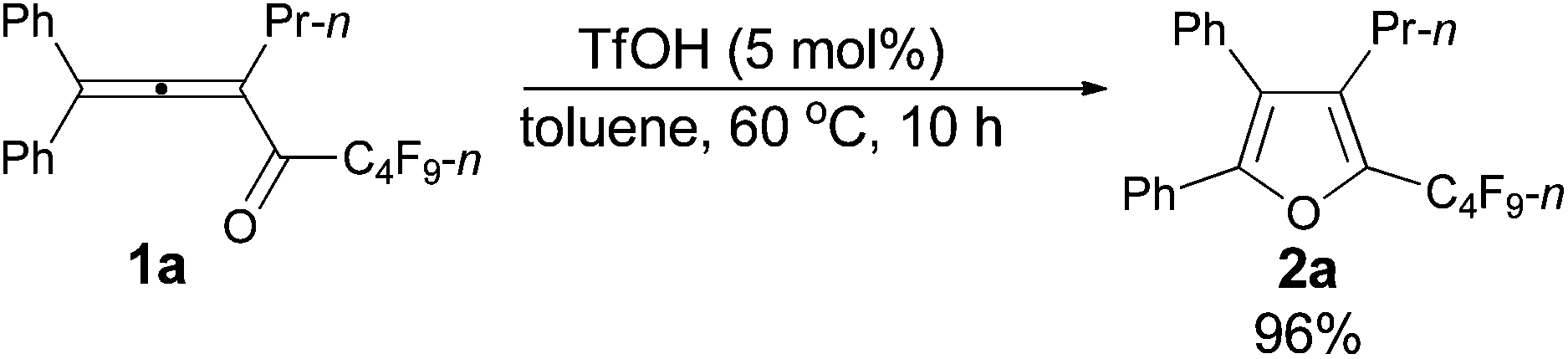
We started to explore the cycloisomerization reaction with fully-substituted perfluoroalkyl allenone 1a as the model substrate. No reaction occurred when AuCl was used as the catalyst (entry 1, Table 1), while AuCl3 produced perfluoroalkyl furan 2a in only 46% NMR yield (entry 2, Table 1). It was interesting to observe that aged triflic acid could catalyze the conversion of perfluoroalkyl substituted allenone 1a to form furan 2a together with furanyl ketone 3a (entry 3, Table 1). To our delight, furan 2a was formed in 96% NMR yield as the sole product (entry 5, Table 1) with a catalytic amount of newly purchased TfOH (5 mol%) in toluene at 60 °C for 10 h.|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Time (h) | NMR yieldb (%) | Recovery of 1ab (%) | |
| 2a | 3a | ||||
| a The reactions were conducted on a 0.2 mmol scale in toluene (2 mL). b Determined using 1H NMR analysis with CH2Br2 as the internal standard. c Aged reagent, may contain water. d 1 equiv. H2O was added with newly purchased TfOH. e The reaction was conducted at 60 °C and mesitylene was used instead of CH2Br2 as the internal standard. | |||||
| 1 | AuCl (10) | 21 | 0 | 0 | 82 |
| 2 | AuCl3 (10) | 19 | 46 | 0 | 51 |
| 3 | TfOHc (10) | 2 | 35 | 59 | 0 |
| 4 | TfOHd (10) | 2.5 | 0 | 98 | 0 |
| 5e | TfOH (5) | 10 | 96 | 0 | 0 |
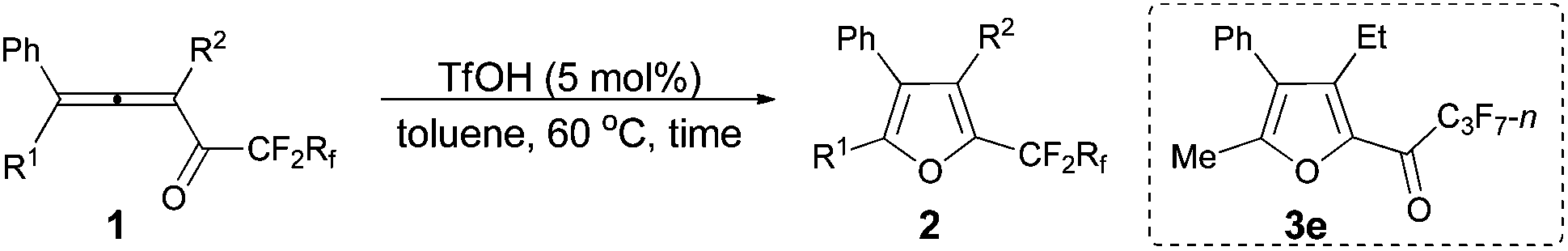
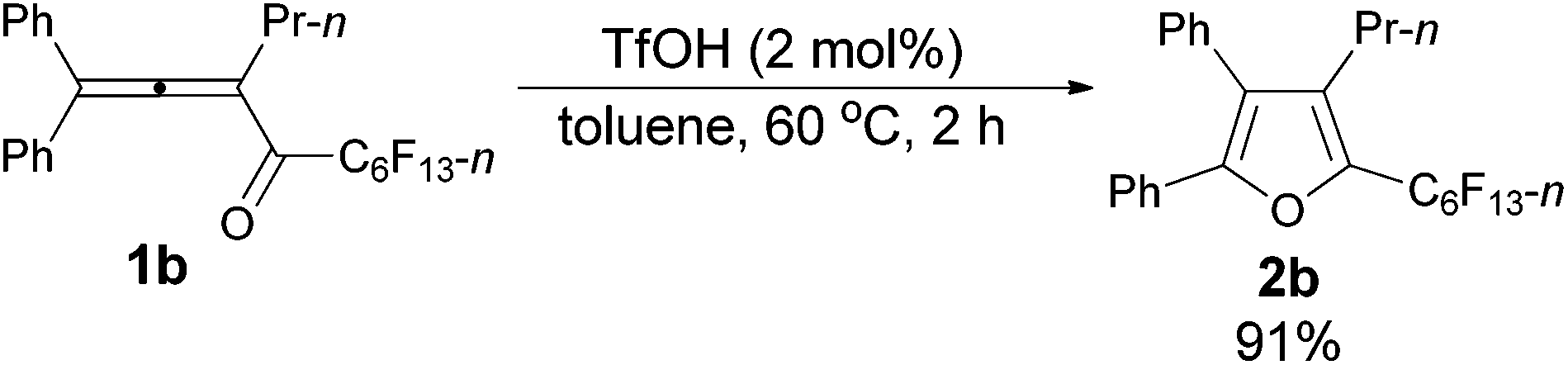
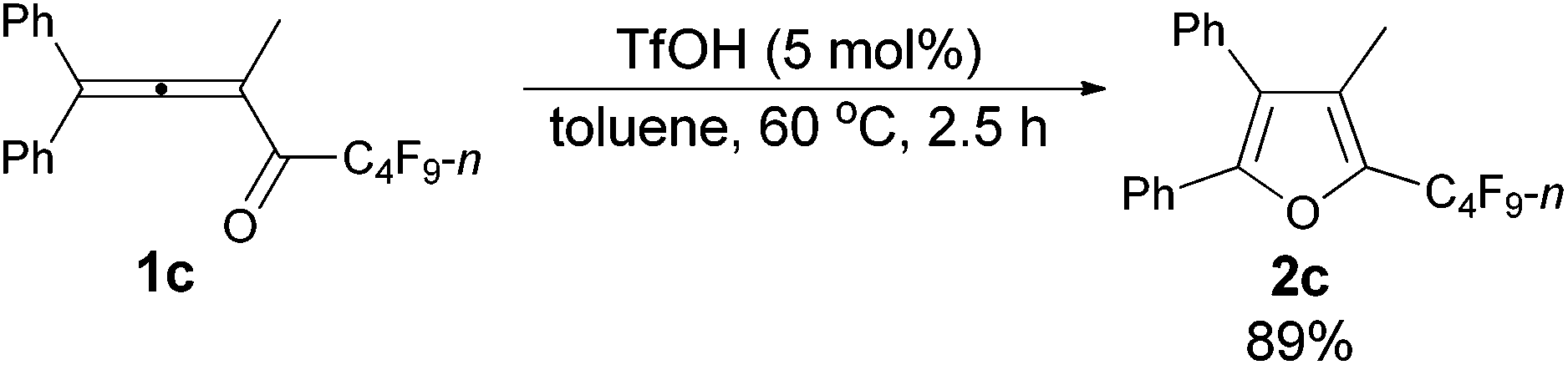
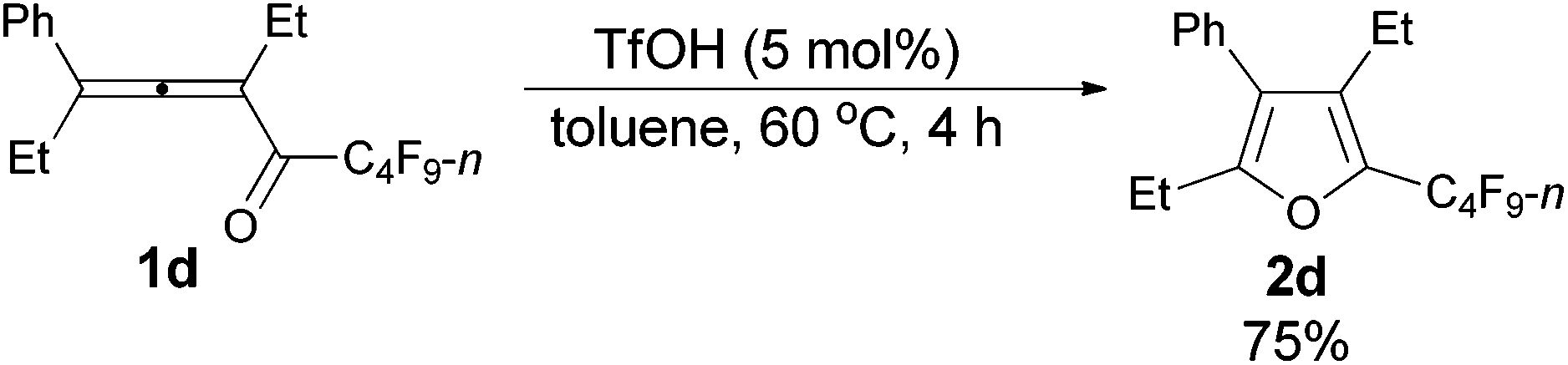
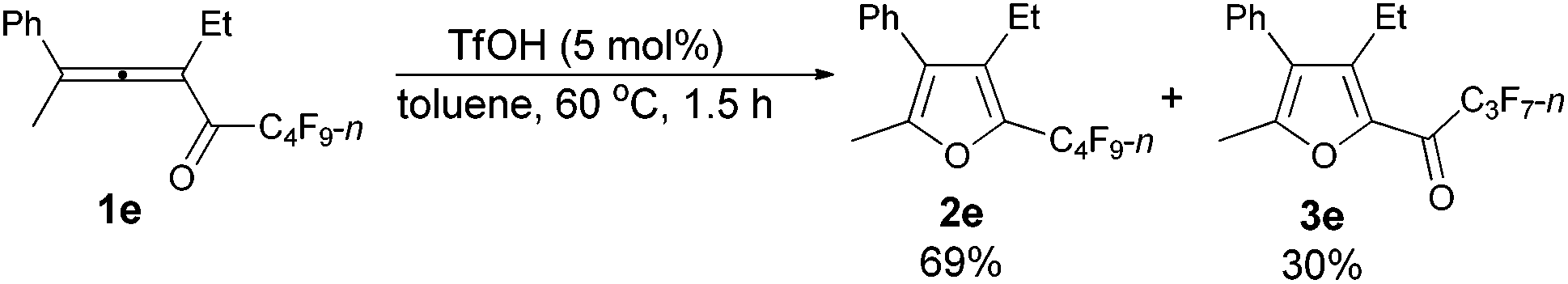
The reaction scope was then explored using the optimized reaction conditions presented in entry 5 of Table 1. For the 1,1-diphenyl-substituted allenyl perfluoroalkyl ketones 1a–1c, no matter whether R3 = propyl or methyl, with Rf = n-perfluoropropyl or pentyl, the corresponding products 2a–2c could be formed exclusively (entries 1–3, Table 2). Although 2d could be obtained as a single product in good yield (entry 4, Table 2), on changing R1 from Et to Me, furan 2e (69%) was formed together with ketone 3e14 (30%) (entry 5, Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Rf | Time (h) | Yield of 2b (%) |
| a Conditions: substrate (0.5 mmol), TfOH (5 mol%) and toluene (5 mL), at 60 °C. b Isolated yield. c The reaction was conducted on a 0.2 mmol scale. d TfOH (2 mol%) was used and the reaction was conducted on a 0.3 mmol scale. e 3e was also afforded in 30% isolated yield. | |||||
| 1c | Ph | n-Pr | n-C3F7 (1a) | 10 | 96 (2a) |
| 2d | Ph | n-Pr | n-C5F11 (1b) | 2 | 91 (2b) |
| 3 | Ph | Me | n-C3F7 (1c) | 2.5 | 89 (2c) |
| 4 | Et | Et | n-C3F7 (1d) | 4 | 75 (2d) |
| 5 | Me | Et | n-C3F7 (1e) | 1.5 | 69 (2e)e |
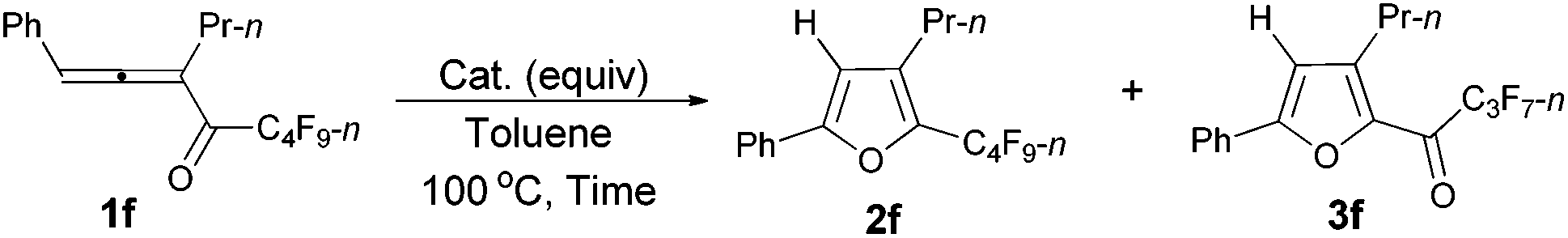
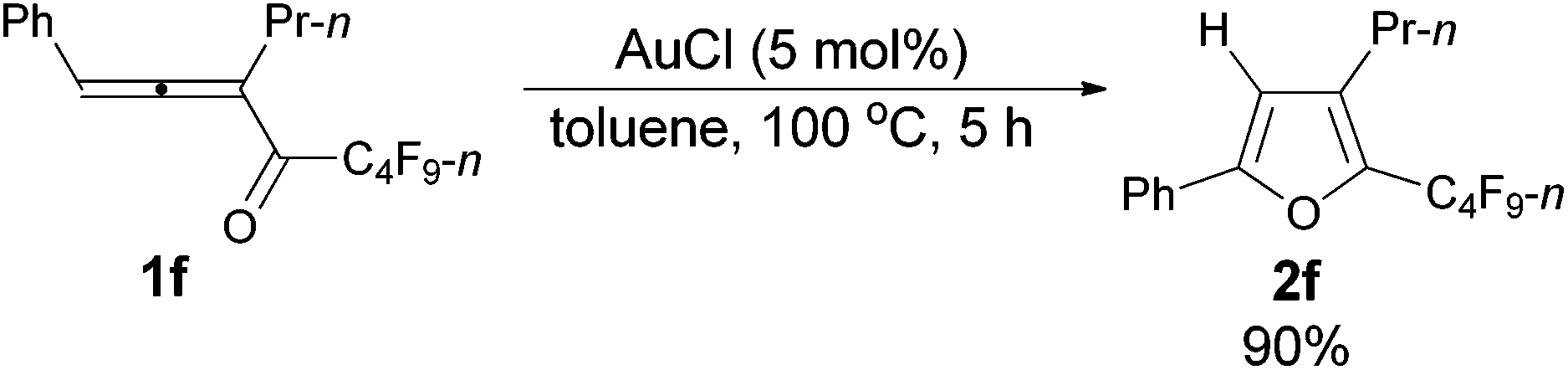
However, when we attempted to extend this cycloisomerization to 1-mono-substituted allenyl ketone 1f, only 9% of furan 2f was afforded with 87% recovery of 1f (entry 1, Table 3). When trifluoromethanesulfonates such as Cu(OTf)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9f,g and AgOTf9f,g were applied as the catalyst, there was still 88% and 56% recovery of 1f, respectively (entries 2 and 3, Table 3). Interestingly, 2f was obtained efficiently with a catalytic amount of AuCl in toluene at 100 °C (entry 4, Table 3). It should be mentioned that AuCl has been identified as a catalyst for the selective cycloisomerization of allenones for the first time.9i Then a series of other cheaper Lewis acids was screened: although 43% of 2f could be produced when FeCl3 was used (entry 6, Table 3), only 11% and 9% of 2f were detected using NMR under the catalysis of SnCl4 and BF3·2H2O (entries 7 and 8, Table 3), respectively; BF3·Et2O and CuI9a failed to catalyze the reaction (entries 9 and 10, Table 3); while with TiCl4, complete decomposition of 1f occurred (entry 11, Table 3). Thus, the reaction conditions presented in entry 4 of Table 3 were defined as the standard conditions for further study.
9f,g and AgOTf9f,g were applied as the catalyst, there was still 88% and 56% recovery of 1f, respectively (entries 2 and 3, Table 3). Interestingly, 2f was obtained efficiently with a catalytic amount of AuCl in toluene at 100 °C (entry 4, Table 3). It should be mentioned that AuCl has been identified as a catalyst for the selective cycloisomerization of allenones for the first time.9i Then a series of other cheaper Lewis acids was screened: although 43% of 2f could be produced when FeCl3 was used (entry 6, Table 3), only 11% and 9% of 2f were detected using NMR under the catalysis of SnCl4 and BF3·2H2O (entries 7 and 8, Table 3), respectively; BF3·Et2O and CuI9a failed to catalyze the reaction (entries 9 and 10, Table 3); while with TiCl4, complete decomposition of 1f occurred (entry 11, Table 3). Thus, the reaction conditions presented in entry 4 of Table 3 were defined as the standard conditions for further study.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (equiv.) | Time (h) | NMR yieldb (%) | Recovery of 1fb (%) | |
| 2f | 3f | ||||
| a The reactions were conducted on a 0.1 mmol scale in toluene (1 mL). b Determined using 1H NMR analysis with CH2Br2 as the internal standard and the isolated yield is shown in the brackets. c The reaction was conducted at 60 °C. d The reaction was conducted on a 0.5 mmol scale. e The reaction was conducted on a 0.2 mmol scale. | |||||
| 1c | TfOH (0.05) | 11 | 9 | 2 | 87 |
| 2 | Cu(OTf)2 (0.1) | 11 | 10 | 2 | 88 |
| 3 | AgOTf (0.1) | 11 | 42 | 2 | 56 |
| 4d | AuCl (0.05) | 5 | 98 (90) | 0 | 0 |
| 5e | AuCl3 (0.05) | 9 | 96 | 0 | 0 |
| 6 | FeCl3 (0.1) | 24 | 43 | 0 | 55 |
| 7 | SnCl4 (1.0) | 14 | 11 | 5 | 54 |
| 8 | BF3·2H2O (3.0) | 48 | 9 | 3 | 81 |
| 9 | BF3·Et2O (3.0) | 27 | 0 | 5 | 84 |
| 10 | CuI (0.1) | 19 | 0 | 0 | 88 |
| 11 | TiCl4 (1.0) | 22 | Complicated mixture of products | 0 | |
With the optimized conditions in hand we then studied the generality of the AuCl-catalyzed cycloisomerization reaction using tri-substituted allenyl perfluorobutyl ketones 1f–1l. The electronic effects of the substituent groups of the aryl group have almost no effect on the conversion (entries 4 and 5, Table 4). A heterocyclic 2-thienyl group and an acyclic alkyl group could be compatible as well (entries 6 and 7, Table 4).
|
|
||||
|---|---|---|---|---|
| Entry | R | R3 | Time (h) | Yield of 2b (%) |
| a Conditions: substrate (0.5 mmol), AuCl (5 mol%) and toluene (5 mL), at 100 °C. b Isolated yield. c The reaction was carried out on a 0.2 mmol scale in toluene (2 mL). d AuCl (10 mol%) was used. e Purity is 94.0%. | ||||
| 1 | Ph | n-Pr (1f) | 5 | 90 (2f) |
| 2 | Ph | Et (1g) | 3 | 93 (2g) |
| 3 | Ph | Me (1h) | 5 | 92 (2h) |
| 4c | p-MeC6H4 | n-Pr (1i) | 12 | 98 (2i) |
| 5 | p-FC6H4 | n-Pr (1j) | 3 | 90 (2j) |
| 6c,d | 2-Thienyl | n-Pr (1k) | 16 | 88 (2k) |
| 7c | n-C7H15 | n-Pr (1l) | 11 | 56e (2l) |
As shown in entry 3 of Table 1, besides the normal cycloisomerization product furan 2a, a corresponding hydrolytic defluorination product ketone 3a14 could be obtained in 59% NMR yield as well. Thus, we attempted to form 3a from 2a in the presence of acid and water. To our delight, TfOH performed the best among the various acids used. Moreover, 99% of 3a could be generated with 1.5 equiv. of water (entry 2, Table 5).
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | H2O (equiv.) | NMR yield of 3ab (%) | Recovery of 2ab (%) |
| a The reactions were conducted on a 0.1 mmol scale in toluene (1 mL) under a N2 atmosphere. b Determined using 1H NMR analysis with CH2Br2 as the internal standard. c The reaction was conducted on a 0.2 mmol scale in toluene (2 mL) under a N2 atmosphere. d The dosage of the catalyst was 10 mol%. e The dosage of the catalyst was 1.0 equiv. | ||||
| 1 | TfOH | 1.0 | 97 | 0 |
| 2c | TfOH | 1.5 | 99 | 0 |
| 3 | TfOH | 2.0 | 98 | 0 |
| 4 | TfOH | 5.0 | 94 | 0 |
| 5d | TFA | 1.5 | 0 | 94 |
| 6e | TsOH·H2O | 1.5 | 39 | 61 |
Using the optimized reaction conditions presented in entry 2 of Table 5, we studied the scope of this hydrolytic defluorination reaction. When R1 = Ph, R2 = acyclic alkyl, and Rf = n-C3F7 or n-C5F11, the ketone products 3a–3c could all be obtained with excellent yields (entries 1–3, Table 6). R1 and R2 could both be alkyl groups: 3e was produced with a 71% isolated yield (entry 4, Table 6).
Furthermore, when tri-substituted furan 2f was treated with TfOH (5 mol%) and H2O (1.5 equiv.), the corresponding ketone 3f could also be formed with a good yield (eqn (1)).
 | (1) |
Considering that 2f could be efficiently prepared from 1f with AuCl as the catalyst (entry 4, Table 3) and that the acid-catalyzed reaction of 2f would form 3f (eqn (1)), a two-step process was thus established (Table 7): the addition of 1.5 equiv. of H2O to the reaction vessel afforded 3f exclusively in the highest yield (entry 2, Table 7).
|
|
||||
|---|---|---|---|---|
| Entry | H2O (equiv.) | Time 1/time 2 (h) | NMR yieldb (%) | |
| 2f | 3f | |||
| a The reactions were conducted on a 0.1 mmol scale in toluene (1.0 mL). b Determined using 1H NMR analysis with CH2Br2 as the internal standard. c A complicated mixture of products was obtained. | ||||
| 1 | 1 | 3.5/24 | 4 | 63 |
| 2 | 1.5 | 12/8 | 0 | 84 |
| 3 | 2 | 3.5/3 | 0 | 79 |
| 4 | 3 | 12/8 | 6 | 61 |
| 5 | 5 | 4/5 | 7 | 75 |
| 6c | 10 | 4/5 | Trace | 50 |
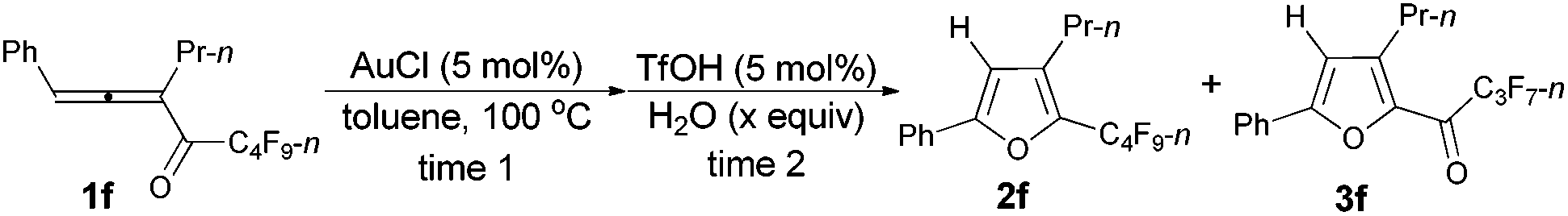
With a reliable procedure in hand, we next turned our attention to studying the scope of the two-step one-pot transformation: with Ar = Ph, the tandem reactions could all proceed efficiently, no matter whether R1 was a propyl, an ethyl, or a methyl group (entries 1–4, Table 8); the substituent of the aryl group had almost no significant effect on the reaction (entries 5 and 6, Table 8); a 2-thienyl group may also be compatible, albeit with a lower yield (entry 7, Table 8). To demonstrate the practicality of the procedure, a gram scale (2.50 mmol) reaction of 1f was also conducted, providing the product in good yield (78%) as shown in entry 2, Table 8.
|
|
||||
|---|---|---|---|---|
| a Conditions: substrate (0.5 mmol), AuCl (5 mol%) and toluene (5 mL), at 100 °C for time 1, then TfOH (5 mol%) and H2O (1.5 equiv.) were added. b Isolated yield. c The reaction was conducted on a 1.0 mmol scale. d The reaction was carried out on a gram scale. e The reaction was carried out on a 0.2 mmol scale. | ||||
| Entry | Ar | R1 | Time 1/time 2 (h) | Yield of 3b (%) |
| 1c | Ph | n-Pr (1f) | 3/3 | 76 (3f) |
| 2d | Ph | n-Pr (1f) | 3/3.5 | 78 (3f) |
| 3 | Ph | Et (1g) | 13/10 | 86 (3g) |
| 4c | Ph | Me (1h) | 3.5/13 | 86 (3h) |
| 5 | p-MeC6H4 | n-Pr (1i) | 11/5 | 88 (3i) |
| 6 | p-FC6H4 | n-Pr (1j) | 12/10 | 82 (3j) |
| 7e | 2-Thienyl | n-Pr (1k) | 13/10 | 43 (3k) |
Conclusions
In conclusion, we have developed an efficient controlled cycloisomerization of allenyl perfluoroalkyl ketones under the catalysis of AuCl (for 3-monoaryl allenones) or TfOH (for fully-substituted allenones) to afford 2-perfluoroalkyl furans. In the presence of water and a catalytic amount of TfOH, these furans could be efficiently transformed into corresponding furan-2-yl perfluoroalkyl ketones. Further studies in this area are still ongoing in our laboratory.Experimental section
General
The perfluoroalkyl allenones were prepared according to the literature procedures.13 Toluene was distilled from Na wire/benzophenone. TfOH (Alfa) was stored in a glovebox, and transferred with a microsyringe. The other commercially available chemicals were purchased and used without additional purification. The reactions were performed under an atmosphere of nitrogen using standard Schlenk tubes unless otherwise stated. Petroleum ether with a boiling point range from 30 to 60 °C was used. Flash-column chromatography was carried out on silica gel H (10–40 μm). 1H NMR spectra (300 MHz) were recorded using TMS as an internal standard (δ 0 ppm). 13C NMR spectra (75 MHz) were recorded using CDCl3 as an internal standard (δ 77.00 ppm). 19F NMR spectra (282 MHz) were recorded using CFCl3 as an internal standard (δ 0 ppm). IR spectra were recorded with a Perkin-Elmer 983G instrument. Mass spectrometry was performed with an HP 5989A system. High-resolution mass spectrometry was conducted with a Finnigan MAT 8430 or Bruker APEXIII instrument. The structures of 2d, 2e, 2g, 2k, and 2l were established through NOE studies.TfOH-catalyzed cycloisomerization reactions of fully-substituted allenyl perfluoroalkyl ketones 1a–1e
Typical procedure I: to a dried Schlenk tube, 1,1-diphenylhexa-1,2-dien-3-yl perfluorobutyl ketone 1a (0.0961 g, 0.20 mmol)/rinsed with toluene (2 mL) and TfOH (0.9 μL, d = 1.695 g mL−1, 0.0015 g, 0.01 mmol) were added at room temperature under a nitrogen atmosphere. The resulting reaction mixture was placed in a pre-heated oil bath at 60 °C and stirred for 10 h, and monitored using TLC. After being cooled to room temperature, the crude reaction mixture was transferred to a round bottom flask. After removing the solvent via rotary evaporation, column chromatography on silica gel (chromatography eluent: petroleum ether) afforded 2a (0.0924 g, 96%) as a waxy solid: 1H NMR (300 MHz, CDCl3) δ 7.49–7.33 (m, 5 H, ArH), 7.33–7.26 (m, 2 H, ArH), 7.25–7.18 (m, 3 H, ArH), 2.42 (t, J = 8.0 Hz, 2 H, CH2), 1.42–1.28 (m, 2 H, CH2), 0.78 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 151.2, 134.4 (t, J = 31.7 Hz), 132.54, 132.49, 130.1, 129.7, 129.0, 128.4, 128.3, 128.1, 126.0, 124.4, 25.0, 23.6, 13.9; 19F NMR (282 MHz, CDCl3) δ −81.4 (t, J = 9.7 Hz, 3 F), −110.5 to −110.8 (m, 2 F), −123.6 to −123.8 (m, 2 F), −126.5 to −126.7 (m, 2 F); IR (neat) ν (cm−1) 3078, 3029, 2981, 2951, 2917, 1642, 1610, 1494, 1448, 1432, 1405, 1315, 1294, 1268, 1213, 1157, 1132, 1095, 1057; MS (70 eV, EI) m/z (%) 480 (M+, 50.59), 311 (100); HRMS calcd for C23H17F9O (M+): 480.1136, found: 480.1124.
The following compounds 2b–2e were prepared following typical procedure I.
The reaction of 1,1-diphenylhexa-1,2-dien-3-yl n-perfluorohexyl ketone 1b (0.1735 g, 0.30 mmol) with TfOH (99%, 0.54 μL, d = 1.695 g mL−1, 0.0009 g, 0.006 mmol) in toluene (3 mL) at 60 °C for 2 h afforded 2b (0.1574 g, 91%) (chromatography eluent: petroleum ether) as a solid: m.p. 53.4–55.2 °C (recrystallization solvent: n-hexane/ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 7.53–7.37 (m, 5 H, ArH), 7.37–7.30 (m, 2 H, ArH), 7.29–7.10 (m, 3 H, ArH), 2.45 (tt, J1 = 7.8 Hz, J2 = 1.8 Hz, 2 H, CH2), 1.46–1.30 (m, 2 H, CH2), 0.81 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 151.2, 134.5 (t, J = 32.2 Hz), 132.6, 132.5, 130.1, 129.7, 129.0, 128.4, 128.3, 128.1, 126.0, 124.4, 25.1, 23.6, 13.8; 19F NMR (282 MHz, CDCl3) δ −81.3 (t, J = 9.7 Hz, 3 F), −110.5 (t, J = 12.3 Hz, 2 F), −122.2 to −122.6 (m, 2 F), −122.6 to −122.9 (m, 2 F), −123.0 to −123.5 (m, 2 F), −126.5 to −126.8 (m, 2 F); IR (KBr) ν (cm−1) 3064, 3033, 2967, 2937, 2877, 1623, 1605, 1583, 1561, 1505, 1482, 1467, 1447, 1402, 1382, 1362, 1333, 1239, 1095, 1073, 1057, 1029; MS (70 eV, EI) m/z (%) 581 (31.20), 580 (M+, 100); Elemental analysis calcd for C25H17F13O: C, 51.74; H, 2.95; found: C, 51.74; H, 3.12.
The reaction of 1,1-diphenylbuta-1,2-dien-3-yl n-perfluorobutyl ketone 1c (0.2265 g, 0.50 mmol) with TfOH (99%, 2.3 μL, d = 1.695 g mL−1, 0.0039 g, 0.026 mmol) in toluene (5 mL) at 60 °C for 2.5 h afforded 2c (0.2020 g, 89%) (chromatography eluent: petroleum ether/ethyl acetate = 100/1) as a solid: m.p. 52.3–53.2 °C (recrystallization solvent: n-hexane/ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 7.58–7.02 (m, 10 H, ArH), 2.03 (s, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 151.0, 134.5 (t, J = 31.8 Hz), 132.1, 130.1, 129.7, 129.0, 128.4, 128.3, 128.0, 127.6, 126.1, 124.7, 8.5; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 9.7 Hz, 3 F), −111.2 to −111.6 (m, 2 F), −123.9 to −124.4 (m, 2 F), −126.5 to −126.9 (m, 2 F); IR (KBr) ν (cm−1) 3064, 3033, 2936, 1629, 1605, 1584, 1563, 1505, 1481, 1447, 1402, 1351, 1335, 1235, 1203, 1135, 1110, 1081, 1030, 1006; MS (70 eV, EI) m/z (%) 452 (M+, 48.75), 283 (100); Elemental analysis calcd for C21H13F9O: C, 55.76; H, 2.90; found: C, 55.96; H, 3.19.
The reaction of 3-phenylhepta-3,4-dien-5-yl n-perfluorobutyl ketone 1d (0.2090 g, 0.50 mmol) with TfOH (99%, 2.3 μL, d = 1.695 g mL−1, 0.0039 g, 0.026 mmol) in toluene (5 mL) at 60 °C for 4 h afforded 2d (0.1569 g, 75%) (chromatography eluent: petroleum ether) as a liquid: 1H NMR (300 MHz, CDCl3) δ 7.50–7.31 (m, 3 H, ArH), 7.31–7.20 (m, 2 H, ArH), 2.59 (q, J = 7.5 Hz, 2 H, CH2), 2.50 (q, J = 7.4 Hz, 2 H, CH2), 1.18 (t, J = 7.5 Hz, 3 H, CH3), 0.93 (t, J = 7.5 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 156.6, 133.1 (t, J = 32.0 Hz), 132.3, 132.2, 129.7, 128.6, 127.5, 123.2, 19.9, 16.5, 15.0, 12.6; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 10.4 Hz, 3 F), −111.0 (t, J = 10.6 Hz, 2 F), −123.9 to −124.3 (m, 2 F), −126.6 to −127.0 (m, 2 F); IR (neat) ν (cm−1) 3035, 2979, 2941, 2883, 1630, 1576, 1496, 1466, 1408, 1378, 1353, 1324, 1276, 1236, 1135, 1091, 1062, 1004; MS (70 eV, EI) m/z (%) 418 (M+, 66.28), 249 (100); HRMS calcd for C18H15F9O (M+): 418.0979, found: 418.0977.
The reaction of 2-phenylhexa-2,3-dien-4-yl perfluorobutyl ketone 1e (0.2025 g, 0.50 mmol) with TfOH (99%, 2.3 μL, d = 1.695 g mL−1, 0.0039 g, 0.026 mmol) in toluene (5 mL) at 60 °C for 1.5 h afforded 2e (0.1389 g, 69%) and 3e (0.0567 g, 30%) (chromatography eluent: petroleum ether, then petroleum ether/ethyl acetate = 200/1).
2e (liquid, less polar): 1H NMR (300 MHz, CDCl3) δ 7.49–7.32 (m, 3 H, ArH), 7.29–7.20 (m, 2 H, ArH), 2.51 (q, J = 7.5 Hz, 2 H, CH2), 2.26 (s, 3 H, CH3), 0.93 (t, J = 7.5 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 151.7, 133.2 (t, J = 31.8 Hz), 132.4, 132.2, 129.6, 128.6, 127.5, 123.9, 16.5, 15.0, 12.1; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 9.7 Hz, 3 F), −110.9 (t, J = 10.7 Hz, 2 F), −123.7 to −124.1 (m, 2 F), −126.6 to −126.9 (m, 2 F); IR (neat) ν (cm−1) 3084, 3061, 3036, 2978, 2941, 2883, 1633, 1578, 1497, 1467, 1445, 1411, 1354, 1279, 1235, 1135, 1091, 1055, 1012; MS (70 eV, EI) m/z (%) 404 (M+, 52.43), 235 (100); HRMS calcd for C17H13F9O (M+): 404.0823, found: 404.0815.
3e (liquid, more polar): 1H NMR (300 MHz, CDCl3) δ 7.51–7.33 (m, 3 H, ArH), 7.29–7.21 (m, 2 H, ArH), 2.78 (q, J = 7.5 Hz, 2 H, CH2), 2.38 (s, 3 H, CH3), 1.10 (t, J = 7.4 Hz, 3 H, CH3); 19F NMR (282 MHz, CDCl3) δ −81.0 (t, J = 9.0 Hz, 3 F), −117.6 to −118.1 (m, 2 F), −126.2 to −126.6 (m, 2 F).
AuCl-catalyzed cycloisomerization of 3-monoaryl-substituted allenyl perfluorobutyl ketones 1f–1l
Typical procedure II: to a dried Schlenk tube, anhydrous AuCl (97%, 0.0062 g, 0.026 mmol), 1-phenylhexa-1,2-dien-3-yl n-perfluorobutyl ketone 1f (0.2022 g, 0.50 mmol), and anhydrous toluene (5 mL) were added at room temperature under a N2 atmosphere. The resulting mixture was then placed in a pre-heated oil bath at 100 °C and stirred for 5 h, and monitored using TLC. After being cooled to room temperature, the crude reaction mixture was filtered through a short column of silica gel and eluted with 60 mL of Et2O. After removing the solvent via rotary evaporation, column chromatography on silica gel (chromatography eluent: petroleum ether) afforded 2f (0.1818 g, 90%) as an oil: 1H NMR (300 MHz, CDCl3) δ 7.73–7.63 (m, 2 H, ArH), 7.46–7.27 (m, 3 H, ArH), 6.62 (s, 1 H,
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 2.56 (tt, J1 = 7.5 Hz, J2 = 1.8 Hz, 2 H, CH2), 1.74–1.56 (m, 2 H, CH2), 0.98 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 155.9, 134.5 (t, J = 32.3 Hz), 132.6, 129.4, 128.8, 128.7, 124.4, 107.9, 26.3, 23.2, 13.7; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 10.0 Hz, 3 F), −110.7 (t, J = 10.6 Hz, 2 F), −123.9 to −124.3 (m, 2 F), −126.5 to −126.9 (m, 2 F); IR (neat) ν (cm−1) 3066, 3038, 2965, 2933, 2876, 1948, 1877, 1802, 1750, 1621, 1605, 1584, 1551, 1487, 1451, 1397, 1350, 1313, 1235, 1135, 1095, 1057; MS (70 eV, EI) m/z (%) 404 (M+, 30.78), 235 (100); HRMS calcd for C17H13F9O (M+): 404.0823, found: 404.0836.
CH), 2.56 (tt, J1 = 7.5 Hz, J2 = 1.8 Hz, 2 H, CH2), 1.74–1.56 (m, 2 H, CH2), 0.98 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 155.9, 134.5 (t, J = 32.3 Hz), 132.6, 129.4, 128.8, 128.7, 124.4, 107.9, 26.3, 23.2, 13.7; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 10.0 Hz, 3 F), −110.7 (t, J = 10.6 Hz, 2 F), −123.9 to −124.3 (m, 2 F), −126.5 to −126.9 (m, 2 F); IR (neat) ν (cm−1) 3066, 3038, 2965, 2933, 2876, 1948, 1877, 1802, 1750, 1621, 1605, 1584, 1551, 1487, 1451, 1397, 1350, 1313, 1235, 1135, 1095, 1057; MS (70 eV, EI) m/z (%) 404 (M+, 30.78), 235 (100); HRMS calcd for C17H13F9O (M+): 404.0823, found: 404.0836.
The following compounds 2g–2l were prepared following typical procedure II.
The reaction of 1-phenylpenta-1,2-dien-3-yl n-perfluorobutyl ketone 1g (0.1951 g, 0.50 mmol) with AuCl (99%, 0.0056 g, 0.024 mmol) in toluene (5 mL) at 100 °C for 3 h afforded 2g (0.1808 g, 93%) (chromatography eluent: petroleum ether) as a liquid: 1H NMR (300 MHz, CDCl3) δ 7.73–7.61 (m, 2 H, ArH), 7.46–7.27 (m, 3 H, ArH), 6.64 (s, 1 H,
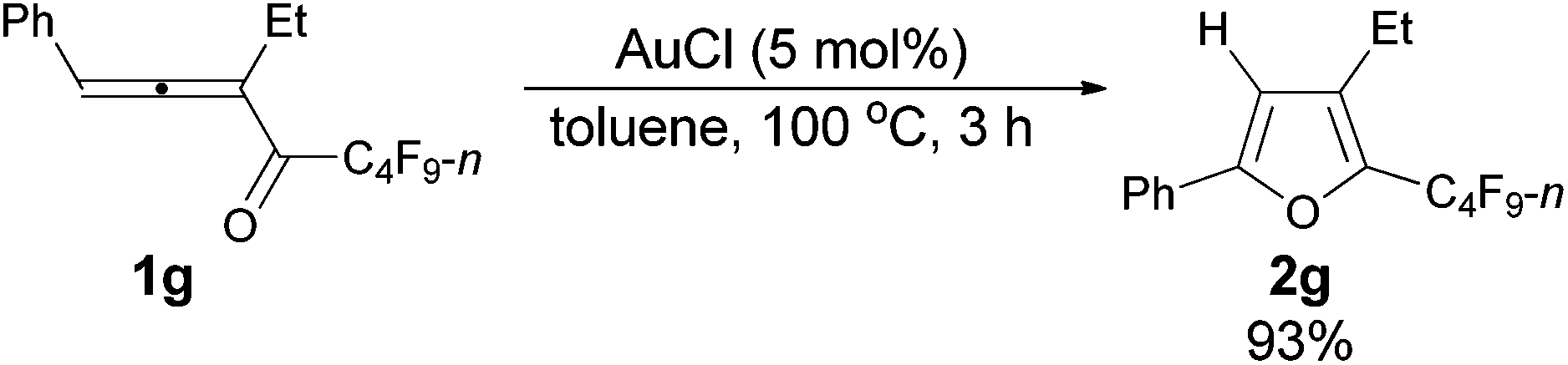 CH), 2.71–2.50 (m, 2 H, CH2), 1.23 (t, J = 7.7 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 156.0, 134.2, 134.0 (t, J = 31.7 Hz), 129.5, 128.8, 128.7, 124.4, 107.6, 17.8, 14.5; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 9.9 Hz, 3 F), −111.1 (t, J = 10.6 Hz, 2 F), −124.1 to −124.4 (m, 2 F), −126.6 to −127.1 (m, 2 F); IR (neat) ν (cm−1) 3067, 3039, 2980, 2942, 2884, 1621, 1605, 1585, 1551, 1487, 1466, 1452, 1398, 1380, 1351, 1282, 1235, 1135, 1091, 1003; MS (70 eV, EI) m/z (%) 390 (M+, 37.26), 221 (100); HRMS calcd for C16H11F9O (M+): 390.0666, found: 390.0664.
CH), 2.71–2.50 (m, 2 H, CH2), 1.23 (t, J = 7.7 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 156.0, 134.2, 134.0 (t, J = 31.7 Hz), 129.5, 128.8, 128.7, 124.4, 107.6, 17.8, 14.5; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 9.9 Hz, 3 F), −111.1 (t, J = 10.6 Hz, 2 F), −124.1 to −124.4 (m, 2 F), −126.6 to −127.1 (m, 2 F); IR (neat) ν (cm−1) 3067, 3039, 2980, 2942, 2884, 1621, 1605, 1585, 1551, 1487, 1466, 1452, 1398, 1380, 1351, 1282, 1235, 1135, 1091, 1003; MS (70 eV, EI) m/z (%) 390 (M+, 37.26), 221 (100); HRMS calcd for C16H11F9O (M+): 390.0666, found: 390.0664.
The reaction of 1-phenylbuta-1,2-dien-3-yl n-perfluorobutyl ketone 1h (0.1891 g, 0.50 mmol) with AuCl (97%, 0.0062 g, 0.026 mmol) in toluene (5 mL) at 100 °C for 5 h afforded 2h (0.1749 g, 92%) (chromatography eluent: petroleum ether) as a liquid: 1H NMR (300 MHz, CDCl3) δ 7.74–7.60 (m, 2 H, ArH), 7.46–7.26 (m, 3 H, ArH), 6.56 (s, 1 H,
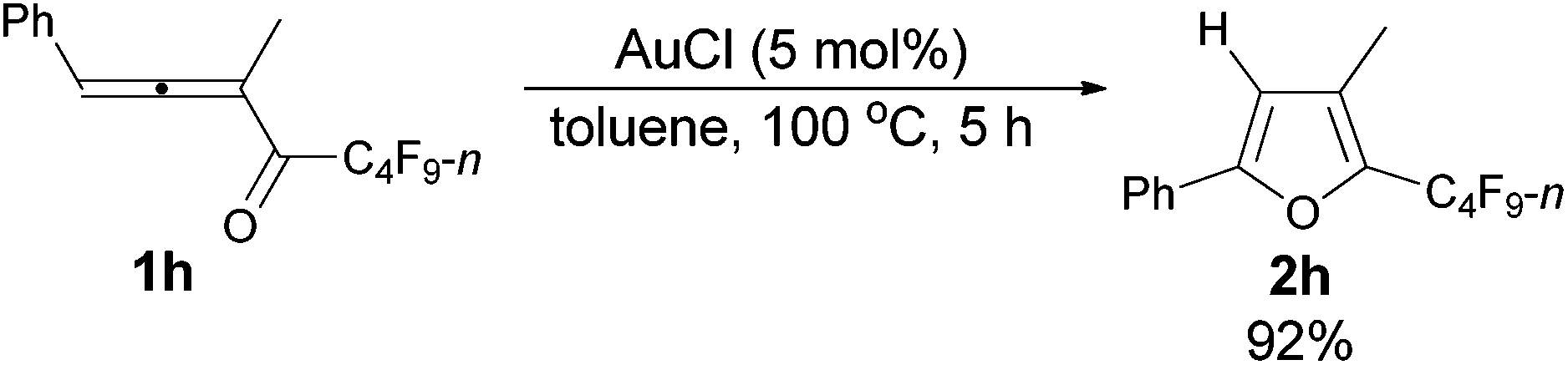 CH), 2.21 (s, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 155.8, 134.8 (t, J = 31.9 Hz), 129.4, 128.8, 128.7, 127.6, 124.4, 109.4, 10.0; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 9.4 Hz, 3 F), −111.2 to −111.6 (m, 2 F), −124.2 to −124.6 (m, 2 F), −126.6 to −127.1 (m, 2 F); IR (neat) ν (cm−1) 3067, 3039, 2936, 1949, 1878, 1802, 1751, 1626, 1606, 1585, 1551, 1488, 1451, 1401, 1384, 1350, 1235, 1135, 1087, 1056, 1027, 1005; MS (70 eV, EI) m/z (%) 376 (M+, 47.65), 207 (100); HRMS calcd for C15H9F9O (M+): 376.0510, found: 376.0515.
CH), 2.21 (s, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 155.8, 134.8 (t, J = 31.9 Hz), 129.4, 128.8, 128.7, 127.6, 124.4, 109.4, 10.0; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 9.4 Hz, 3 F), −111.2 to −111.6 (m, 2 F), −124.2 to −124.6 (m, 2 F), −126.6 to −127.1 (m, 2 F); IR (neat) ν (cm−1) 3067, 3039, 2936, 1949, 1878, 1802, 1751, 1626, 1606, 1585, 1551, 1488, 1451, 1401, 1384, 1350, 1235, 1135, 1087, 1056, 1027, 1005; MS (70 eV, EI) m/z (%) 376 (M+, 47.65), 207 (100); HRMS calcd for C15H9F9O (M+): 376.0510, found: 376.0515.
The reaction of 1-(4′-methylphenyl)hexa-1,2-dien-3-yl perfluorobutyl ketone 1i (0.0832 g, 0.20 mmol) with AuCl (97%, 0.0023 g, 0.010 mmol) in toluene (2 mL) at 100 °C for 12 h afforded 2i (0.0812 g, 98%) (chromatography eluent: petroleum ether) as a liquid: 1H NMR (300 MHz, CDCl3) δ 7.56 (d, J = 8.1 Hz, 2 H, ArH), 7.20 (d, J = 8.1 Hz, 2 H, ArH), 6.56 (s, 1 H,
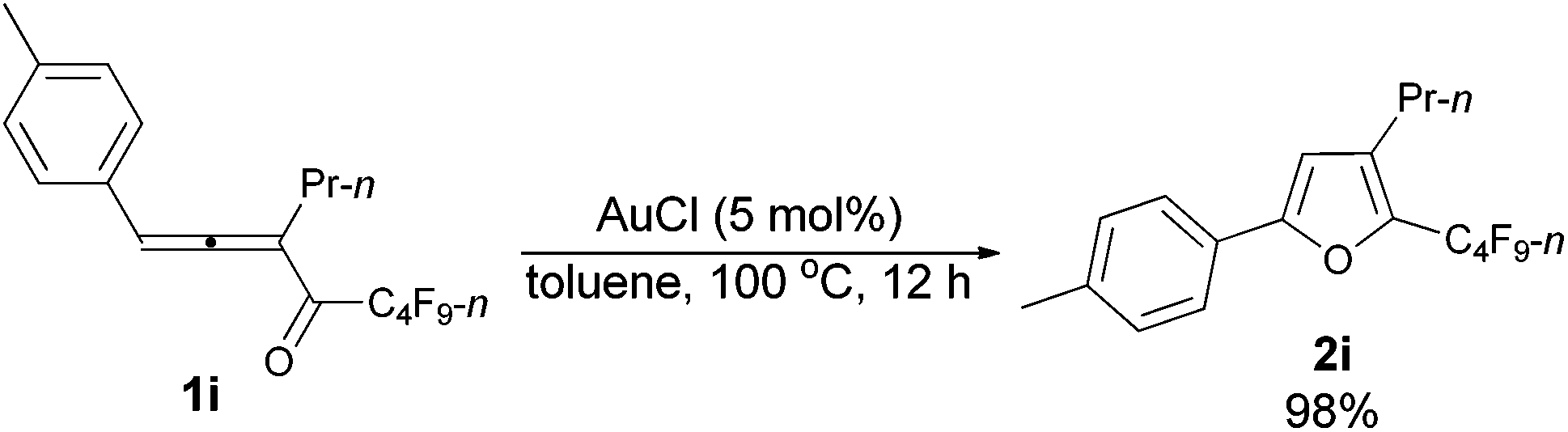 CH), 2.55 (t, J = 7.7 Hz, 2 H, CH2), 2.37 (s, 3 H, CH3), 1.74–1.54 (m, 2 H, CH2), 0.97 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 156.1, 138.7, 134.0 (t, J = 32.3 Hz), 132.6, 129.5, 126.7, 124.3, 107.2, 26.3, 23.2, 21.3, 13.7; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 9.4 Hz, 3 F), −110.8 (t, J = 11.6 Hz, 2 F), −124.0 to −124.3 (m, 2 F), −126.6 to −126.9 (m, 2 F); IR (neat) ν (cm−1) 3029, 2967, 2935, 2876, 1906, 1621, 1581, 1557, 1498, 1468, 1417, 1393, 1350, 1315, 1235, 1135, 1094, 1053; MS (70 eV, EI) m/z (%) 418 (M+, 20.78), 249 (100); HRMS calcd for C18H15F9O (M+): 418.0979, found: 418.0978.
CH), 2.55 (t, J = 7.7 Hz, 2 H, CH2), 2.37 (s, 3 H, CH3), 1.74–1.54 (m, 2 H, CH2), 0.97 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 156.1, 138.7, 134.0 (t, J = 32.3 Hz), 132.6, 129.5, 126.7, 124.3, 107.2, 26.3, 23.2, 21.3, 13.7; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 9.4 Hz, 3 F), −110.8 (t, J = 11.6 Hz, 2 F), −124.0 to −124.3 (m, 2 F), −126.6 to −126.9 (m, 2 F); IR (neat) ν (cm−1) 3029, 2967, 2935, 2876, 1906, 1621, 1581, 1557, 1498, 1468, 1417, 1393, 1350, 1315, 1235, 1135, 1094, 1053; MS (70 eV, EI) m/z (%) 418 (M+, 20.78), 249 (100); HRMS calcd for C18H15F9O (M+): 418.0979, found: 418.0978.
The reaction of 1-(4′-fluorophenyl)hexa-1,2-dien-3-yl n-perfluorobutyl ketone 1j (0.2120 g, 0.50 mmol) with AuCl (97%, 0.0061 g, 0.026 mmol) in toluene (5 mL) at 100 °C for 3 h afforded 2j (0.1909 g, 90%) (chromatography eluent: petroleum ether) as a liquid: 1H NMR (300 MHz, CDCl3) δ 7.63 (dd, J1 = 8.4 Hz, J2 = 5.4 Hz, 2 H, ArH), 7.08 (t, J = 8.7 Hz, 2 H, ArH), 6.54 (s, 1 H,
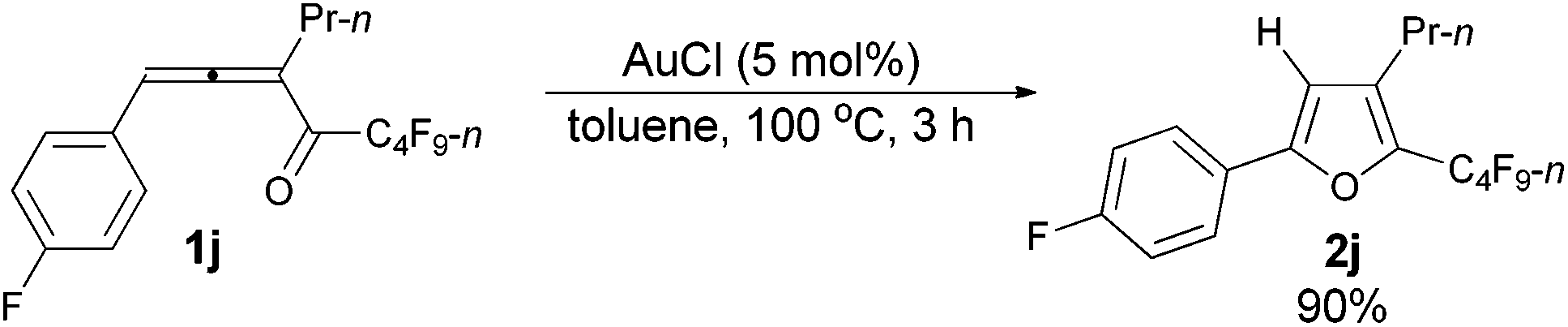 CH), 2.55 (t, J = 7.5 Hz, 2 H, CH2), 1.72–1.56 (m, 2 H, CH2), 0.98 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 163.0 (d, J = 247.4 Hz), 155.1, 134.6 (t, J = 31.6 Hz), 132.8, 126.3 (d, J = 8.1 Hz), 125.9 (d, J = 2.3 Hz), 115.9 (d, J = 21.9 Hz), 107.6, 26.3, 23.2, 13.6; 19F NMR (282 MHz, CDCl3) δ −81.4 to −81.6 (m, 3 F), −110.6 to −111.0 (m, 2 F), −112.4 to −112.7 (m, 1 F), −123.9 to −124.3 (m, 2 F), −126.5 to −126.9 (m, 2 F); IR (neat) ν (cm−1) 3052, 2969, 2937, 2878, 1891, 1621, 1607, 1559, 1496, 1469, 1421, 1392, 1351, 1235, 1135, 1097, 1054, 1014; MS (70 eV, EI) m/z (%) 422 (M+, 65.02), 253 (100); HRMS calcd for C17H12F10O (M+): 422.0728, found: 422.0730.
CH), 2.55 (t, J = 7.5 Hz, 2 H, CH2), 1.72–1.56 (m, 2 H, CH2), 0.98 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 163.0 (d, J = 247.4 Hz), 155.1, 134.6 (t, J = 31.6 Hz), 132.8, 126.3 (d, J = 8.1 Hz), 125.9 (d, J = 2.3 Hz), 115.9 (d, J = 21.9 Hz), 107.6, 26.3, 23.2, 13.6; 19F NMR (282 MHz, CDCl3) δ −81.4 to −81.6 (m, 3 F), −110.6 to −111.0 (m, 2 F), −112.4 to −112.7 (m, 1 F), −123.9 to −124.3 (m, 2 F), −126.5 to −126.9 (m, 2 F); IR (neat) ν (cm−1) 3052, 2969, 2937, 2878, 1891, 1621, 1607, 1559, 1496, 1469, 1421, 1392, 1351, 1235, 1135, 1097, 1054, 1014; MS (70 eV, EI) m/z (%) 422 (M+, 65.02), 253 (100); HRMS calcd for C17H12F10O (M+): 422.0728, found: 422.0730.
The reaction of 1-(2-thienyl)hexa-1,2-dien-3-yl n-perfluorobutyl ketone 1k (0.0828 g, 0.20 mmol) with AuCl (97%, 0.0049 g, 0.020 mmol) in toluene (2 mL) at 100 °C for 16 h afforded 2k (0.0727 g, 88%) (chromatography eluent: petroleum ether) as a liquid: 1H NMR (300 MHz, CDCl3) δ 7.38–7.26 (m, 2 H, ArH), 7.05 (dd, J1 = 4.8 Hz, J2 = 3.9 Hz, 1 H, ArH), 6.46 (s, 1 H,
 CH), 2.53 (t, J = 7.5 Hz, 2 H, CH2), 1.71–1.55 (m, 2 H, CH2), 0.97 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 151.5, 134.0 (t, J = 32.0 Hz), 132.7, 132.1, 127.8, 125.8, 124.5, 107.8, 26.3, 23.2, 13.6; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 10.2 Hz, 3 F), −110.7 (t, J = 11.0 Hz, 2 F), −123.9 to −124.2 (m, 2 F), −126.6 to −126.9 (m, 2 F); IR (neat) ν (cm−1) 3110, 3081, 2967, 2935, 2877, 1792, 1620, 1575, 1495, 1468, 1424, 1400, 1350, 1235, 1135, 1094, 1051, 1019; MS (70 eV, EI) m/z (%) 410 (M+, 32.52), 241 (100); HRMS calcd for C15H11F9OS (M+): 410.0387, found: 410.0387.
CH), 2.53 (t, J = 7.5 Hz, 2 H, CH2), 1.71–1.55 (m, 2 H, CH2), 0.97 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 151.5, 134.0 (t, J = 32.0 Hz), 132.7, 132.1, 127.8, 125.8, 124.5, 107.8, 26.3, 23.2, 13.6; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 10.2 Hz, 3 F), −110.7 (t, J = 11.0 Hz, 2 F), −123.9 to −124.2 (m, 2 F), −126.6 to −126.9 (m, 2 F); IR (neat) ν (cm−1) 3110, 3081, 2967, 2935, 2877, 1792, 1620, 1575, 1495, 1468, 1424, 1400, 1350, 1235, 1135, 1094, 1051, 1019; MS (70 eV, EI) m/z (%) 410 (M+, 32.52), 241 (100); HRMS calcd for C15H11F9OS (M+): 410.0387, found: 410.0387.
The reaction of trideca-4,5-dien-4-yl n-perfluorobutyl ketone 1l (0.0854 g, 0.20 mmol) with AuCl (99%, 0.0023 g, 0.01 mmol) in toluene (2 mL) at 100 °C for 11 h afforded 2l (0.0513 g, purity of 94.0% determined using CH2Br2 as the internal standard, 56% yield) (chromatography eluent: petroleum ether) as an oil: 1H NMR (300 MHz, CDCl3) δ 5.98 (s, 1 H,
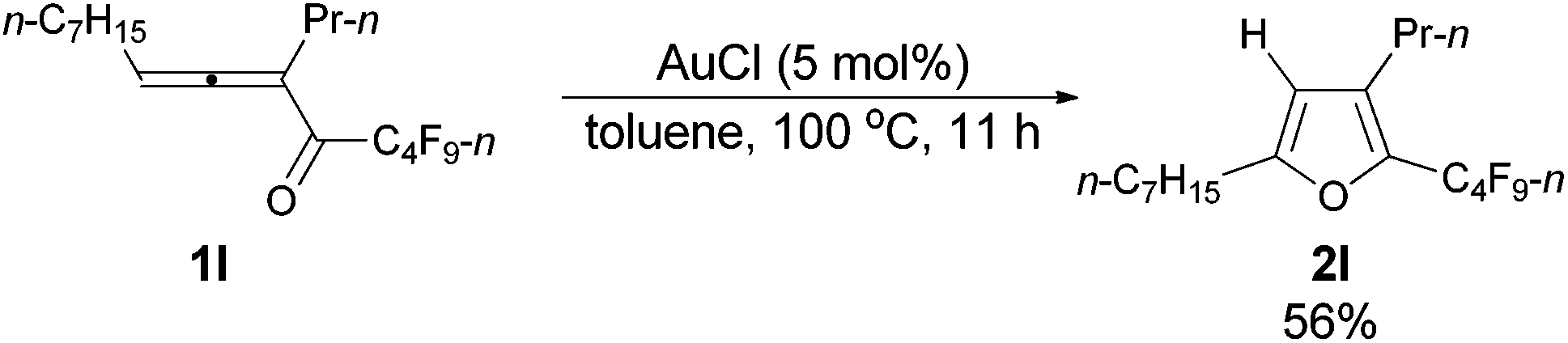 CH), 2.59 (t, J = 7.7 Hz, 2 H, CH2), 2.52–2.42 (m, 2 H, CH2), 1.68–1.50 (m, 4 H, 2 × CH2), 1.35–1.23 (m, 8 H, 4 × CH2), 0.93 (t, J = 7.4 Hz, 3 H, CH3), 0.88 (t, J = 6.8 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 159.4, 133.2 (t, J = 31.9 Hz), 131.5, 108.5, 31.7, 29.0, 28.9, 27.9, 27.6, 26.3, 23.2, 22.6, 14.0, 13.7; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 9.9 Hz, 3 F), −110.8 (t, J = 11.0 Hz, 2 F), −124.3 to −124.5 (m, 2 F), −126.8 to −127.0 (m, 2 F); IR (neat) ν (cm−1) 2962, 2930, 2859, 1700, 1625, 1563, 1468, 1403, 1351, 1235, 1135, 1092, 1048; MS (70 eV, EI) m/z (%) 426 (M+, 26.24), 257 (100); HRMS calcd for C18H23F9O (M+): 426.1605, found: 426.1602.
CH), 2.59 (t, J = 7.7 Hz, 2 H, CH2), 2.52–2.42 (m, 2 H, CH2), 1.68–1.50 (m, 4 H, 2 × CH2), 1.35–1.23 (m, 8 H, 4 × CH2), 0.93 (t, J = 7.4 Hz, 3 H, CH3), 0.88 (t, J = 6.8 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 159.4, 133.2 (t, J = 31.9 Hz), 131.5, 108.5, 31.7, 29.0, 28.9, 27.9, 27.6, 26.3, 23.2, 22.6, 14.0, 13.7; 19F NMR (282 MHz, CDCl3) δ −81.5 (t, J = 9.9 Hz, 3 F), −110.8 (t, J = 11.0 Hz, 2 F), −124.3 to −124.5 (m, 2 F), −126.8 to −127.0 (m, 2 F); IR (neat) ν (cm−1) 2962, 2930, 2859, 1700, 1625, 1563, 1468, 1403, 1351, 1235, 1135, 1092, 1048; MS (70 eV, EI) m/z (%) 426 (M+, 26.24), 257 (100); HRMS calcd for C18H23F9O (M+): 426.1605, found: 426.1602.
Hydrolytic defluorination reactions of fully substituted furans 2a–2e
Typical procedure III: to a dried Schlenk tube, furan 2a (0.0962 g, 0.20 mmol)/rinsed with toluene (2 mL), TfOH (0.9 μL, d = 1.695 g mL−1, 0.0015 g, 0.01 mmol), and H2O (5.4 μL, d = 1.0 g mL−1, 0.0054 g, 0.3 mmol) were added at room temperature under a nitrogen atmosphere. The resulting reaction mixture was placed in a pre-heated oil bath at 100 °C and stirred for 12 h, and monitored using TLC. After being cooled to room temperature, the crude reaction mixture was transferred to a round bottom flask with Et2O (3 × 10 mL). After removing the solvent via rotary evaporation, column chromatography on silica gel (chromatography eluent: petroleum ether/ethyl acetate = 100/1) afforded 3a14 (0.0889 g, 97%) as a solid: m.p. 54.4–56.5 °C (n-hexane/ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 7.53–7.43 (m, 5 H, ArH), 7.35–7.24 (m, 5 H, ArH), 2.76–2.63 (m, 2 H, CH2), 1.57–1.41 (m, 2 H, CH2), 0.86 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.5 (t, J = 25.8 Hz), 154.5, 145.9, 143.4, 131.4, 130.0, 129.7, 129.2, 128.9, 128.7, 128.5, 127.0, 126.8, 26.6, 22.6, 14.0; 19F NMR (282 MHz, CDCl3) δ −80.9 (t, J = 9.6 Hz, 3 F), −117.7 to −117.9 (m, 2 F), −126.1 to −126.3 (m, 2 F).
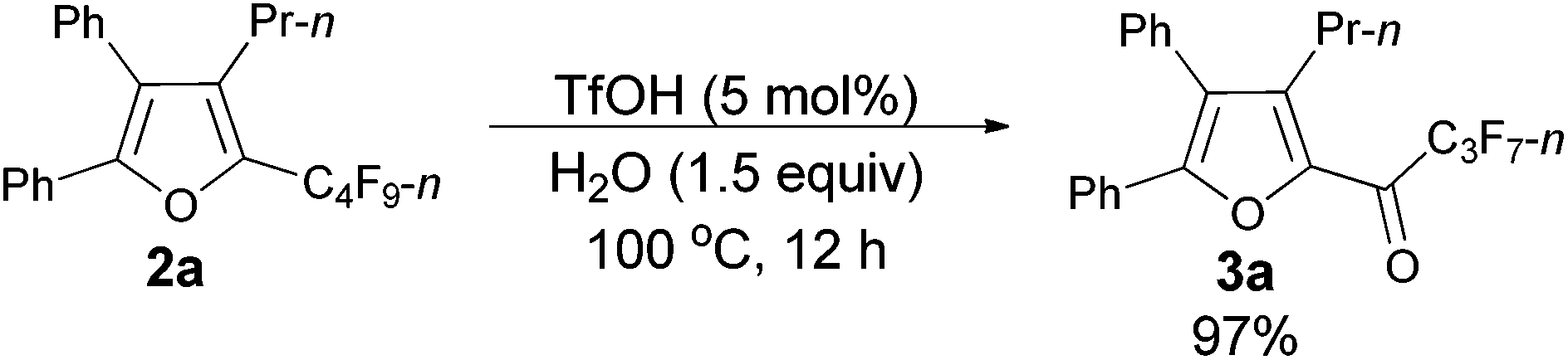
The following compounds 3b–3e were prepared following typical procedure III.
The reaction of furan 2b (0.1162 g, 0.20 mmol) with TfOH (99%, 0.9 μL, d = 1.695 g mL−1, 0.0015 g, 0.01 mmol), and H2O (5.4 μL, d = 1.0 g mL−1, 0.0054 g, 0.3 mmol) in toluene (2 mL) at 100 °C for 10 h afforded 3b14 (0.1059 g, 95%) (chromatography eluent: petroleum ether/ethyl acetate = 100/1) as an oil: 1H NMR (300 MHz, CDCl3) δ 7.53–7.42 (m, 5 H, ArH), 7.36–7.25 (m, 5 H, ArH), 2.76–2.62 (m, 2 H, CH2), 1.57–1.41 (m, 2 H, CH2), 0.86 (t, J = 7.5 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.5 (t, J = 26.1 Hz), 154.5, 146.0, 143.4, 131.4, 130.0, 129.7, 129.2, 128.9, 128.7, 128.5, 127.0, 126.8, 26.6, 22.6, 14.0; 19F NMR (282 MHz, CDCl3) δ −81.2 (tt, J1 = 9.7 Hz, J2 = 2.4 Hz, 3 F), −116.8 to −117.1 (m, 2 F), −121.7 to −122.1 (m, 2 F), −122.6 to −123.0 (m, 2 F), −126.4 to −126.8 (m, 2 F).
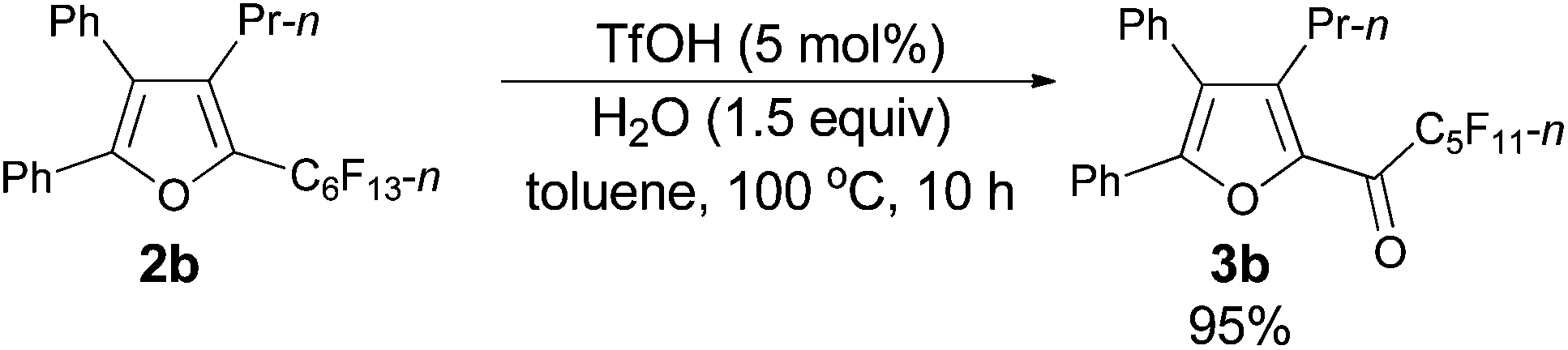
The reaction of furan 2c (0.0903 g, 0.20 mmol) with TfOH (99%, 0.9 μL, d = 1.695 g mL−1, 0.0015 g, 0.01 mmol), and H2O (5.4 μL, d = 1.0 g mL−1, 0.0054 g, 0.3 mmol) in toluene (2 mL) at 100 °C for 10 h afforded 3c14 (0.0812 g, 95%) (chromatography eluent: petroleum ether/ethyl acetate = 100/1) as a solid: m.p. 55.3–56.7 °C (recrystallization solvent: n-hexane/ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 7.55–7.44 (m, 5 H, ArH), 7.37–7.25 (m, 5 H, ArH), 2.33 (s, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.9 (t, J = 26.0 Hz), 154.5, 143.6, 141.2, 131.2, 129.9, 129.8, 129.2, 128.9, 128.7, 128.5, 127.2, 126.9, 10.9; 19F NMR (282 MHz, CDCl3) δ −80.9 (t, J = 9.0 Hz, 3 F), −117.8 to −118.1 (m, 2 F), −126.1 to −126.3 (m, 2 F).
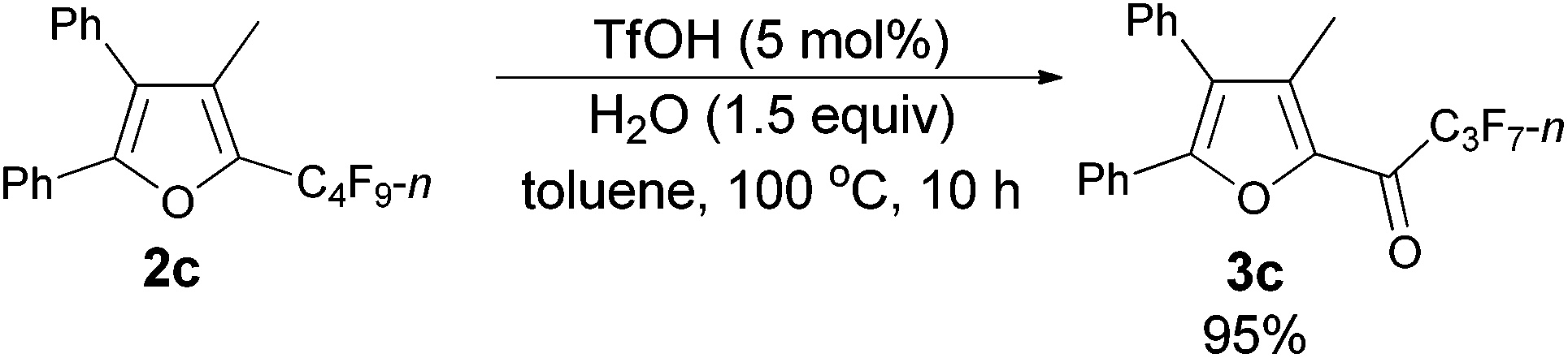
The reaction of furan 2e (0.0809 g, 0.20 mmol) with TfOH (99%, 0.9 μL, d = 1.695 g mL−1, 0.0015 g, 0.01 mmol), and H2O (5.4 μL, d = 1.0 g mL−1, 0.0015 g, 0.3 mmol) in toluene (2 mL) at 100 °C for 3 h afforded 3e14 (0.0543 g, 71%) (chromatography eluent: petroleum ether/ethyl acetate = 100/1) as an oil: 1H NMR (300 MHz, CDCl3) δ 7.51–7.37 (m, 3 H, ArH), 7.28–7.22 (m, 2 H, ArH), 2.78 (q, J = 7.5 Hz, 2 H, CH2), 2.38 (s, 3 H, CH3), 1.10 (t, J = 7.5 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.1 (t, J = 26.0 Hz), 156.9, 146.2, 143.4, 130.8, 129.5, 128.8, 128.0, 127.1, 18.3, 13.5, 13.0; 19F NMR (282 MHz, CDCl3) δ −81.0 (t, J = 8.9 Hz, 3 F), −117.8 to −117.9 (m, 2 F), −126.3 to −126.4 (m, 2 F).
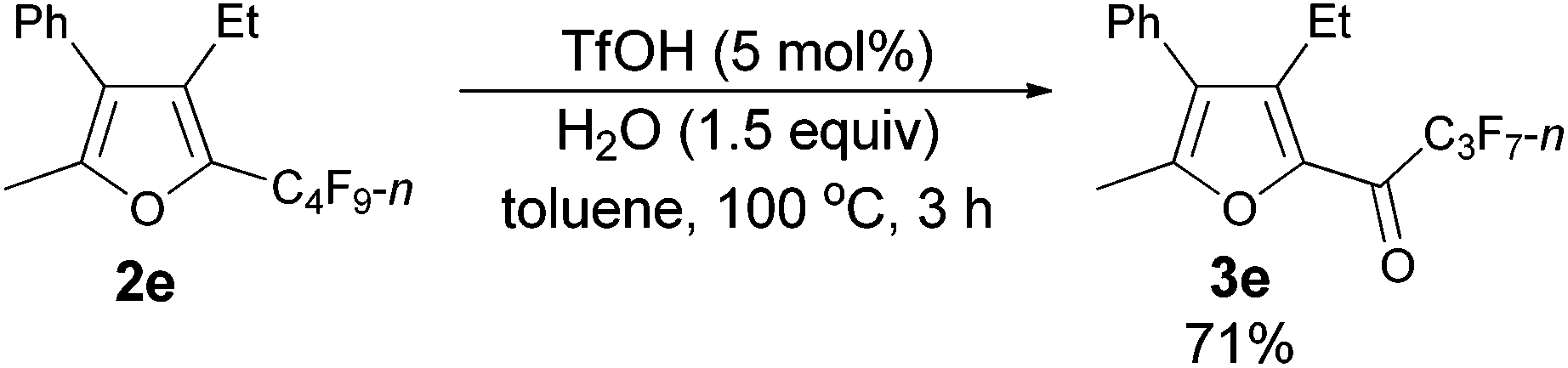
Two-step one-pot tandem cycloisomerization/hydrolytic defluorination reactions of 3-monoaryl-substituted allenyl perfluorobutyl ketones 1f–1k
Typical procedure IV: to a dried Schlenk tube, anhydrous AuCl (97%, 0.0121 g, 0.051 mmol), 1-phenylhexa-1,2-dien-3-yl n-perfluorobutyl ketone 1f (0.4035 g, 1.00 mmol), and anhydrous toluene (5 mL) were added at room temperature under a N2 atmosphere. The resulting mixture was then placed in a pre-heated oil bath at 100 °C and stirred. After 3 h, the cycloisomerization reaction was over, as monitored using TLC. TfOH (99%, 4.5 μL, d = 1.695 g mL−1, 0.0076 g, 0.050 mmol) and H2O (27 μL, d = 1.0 g mL−1, 0.0270 g, 1.50 mmol) were added to the above crude reaction mixture, and the resulting mixture was stirred at 100 °C for another 3 h, and monitored using TLC. After being cooled to room temperature, the reaction mixture was transferred to a round bottom flask. After removing the solvent via rotary evaporation, column chromatography on silica gel (chromatography eluent: petroleum ether, then petroleum ether/ethyl acetate = 500/1) afforded 3f14 (0.2910 g, 76%) as a liquid: 1H NMR (300 MHz, CDCl3) δ 7.84–7.71 (m, 2 H, ArH), 7.51–7.36 (m, 3 H, ArH), 6.82 (s, 1 H,
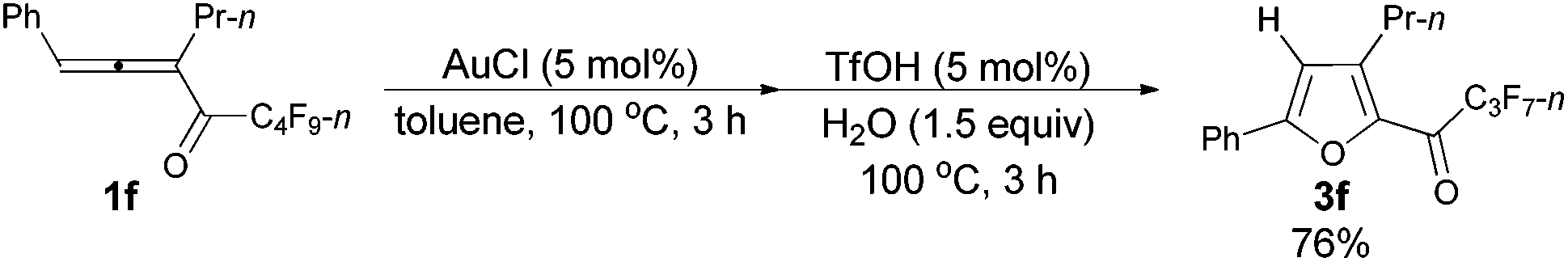 CH), 2.90 (t, J = 7.7 Hz, 2 H, CH2), 1.80–1.59 (m, 2 H, CH2), 1.01 (t, J = 7.5 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.4 (t, J = 25.8 Hz), 159.1, 146.9, 143.9, 130.3, 129.1, 128.5, 125.4, 110.4, 28.4, 22.3, 13.8 (d, J = 2.1 Hz); 19F NMR (282 MHz, CDCl3) δ −80.9 to −81.1 (m, 3 F), −118.0 (q, J = 8.8 Hz, 2 F), −126.3 (s, 2 F).
CH), 2.90 (t, J = 7.7 Hz, 2 H, CH2), 1.80–1.59 (m, 2 H, CH2), 1.01 (t, J = 7.5 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.4 (t, J = 25.8 Hz), 159.1, 146.9, 143.9, 130.3, 129.1, 128.5, 125.4, 110.4, 28.4, 22.3, 13.8 (d, J = 2.1 Hz); 19F NMR (282 MHz, CDCl3) δ −80.9 to −81.1 (m, 3 F), −118.0 (q, J = 8.8 Hz, 2 F), −126.3 (s, 2 F).
The following compounds 3f–3k were prepared following typical procedure IV.
The reaction of 1-phenylhexa-1,2-dien-3-yl n-perfluorobutyl ketone 1f (1.0102 g, 2.50 mmol) with AuCl (97%, 0.0301 g, 0.126 mmol) in toluene (12.5 mL) at 100 °C for 3 h afforded crude product 2f. Then TfOH (99%, 11.2 μL, d = 1.695 g mL−1, 0.0188 g, 0.125 mmol) and H2O (67.5 μL, d = 1.0 g mL−1, 0.0675 g, 3.75 mmol) were added and reacted at 100 °C for another 3.5 h to afford 3f14 (0.7448 g, 78%) (chromatography eluent: petroleum ether/ethyl acetate = 500/1) as an oil: 1H NMR (300 MHz, CDCl3) δ 7.83–7.74 (m, 2 H, ArH), 7.54–7.39 (m, 3 H, ArH), 6.83 (s, 1 H,
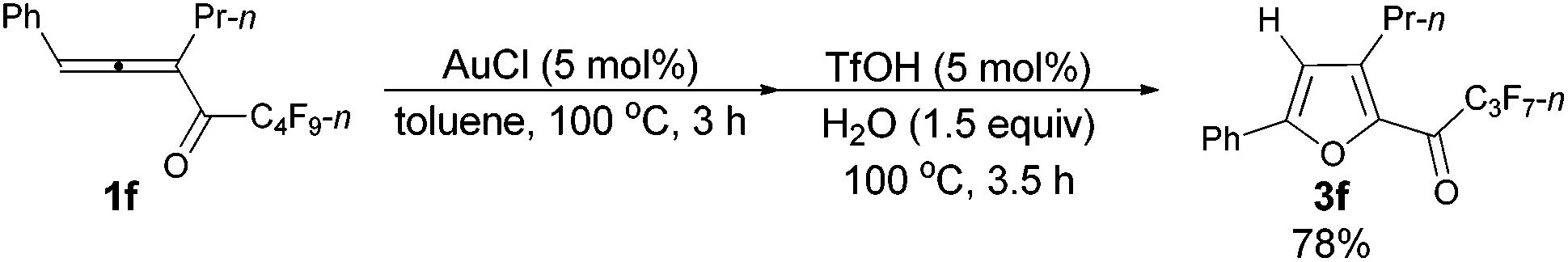 CH), 2.91 (t, J = 7.7 Hz, 2 H, CH2), 1.80–1.63 (m, 2 H, CH2), 1.02 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.4 (t, J = 25.9 Hz), 159.0, 146.8, 143.9, 130.2, 129.1, 128.5, 125.4, 110.3, 28.4, 22.3, 13.8; 19F NMR (282 MHz, CDCl3) δ −81.0 (t, J = 8.9 Hz, 3 F), −118.0 (q, J = 9.5 Hz, 2 F), −126.3 (s, 2 F).
CH), 2.91 (t, J = 7.7 Hz, 2 H, CH2), 1.80–1.63 (m, 2 H, CH2), 1.02 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.4 (t, J = 25.9 Hz), 159.0, 146.8, 143.9, 130.2, 129.1, 128.5, 125.4, 110.3, 28.4, 22.3, 13.8; 19F NMR (282 MHz, CDCl3) δ −81.0 (t, J = 8.9 Hz, 3 F), −118.0 (q, J = 9.5 Hz, 2 F), −126.3 (s, 2 F).
The reaction of 1-phenylpenta-1,2-dien-3-yl perfluorobutyl ketone 1g (0.1945 g, 0.50 mmol) with AuCl (97%, 0.0061 g, 0.026 mmol) in toluene (5 mL) at 100 °C for 13 h afforded crude product 2g. Then TfOH (99%, 2.3 μL, d = 1.695 g mL−1, 0.0039 g, 0.026 mmol) and H2O (13.5 μL, d = 1.0 g mL−1, 0.0135 g, 0.75 mmol) were added and reacted at 100 °C for another 10 h to afford 3g14 (0.1581 g, 86%) (chromatography eluent: petroleum ether/ethyl acetate = 500/1) as a solid: m.p. 40.2–41.2 °C (recrystallization solvent: n-hexane/ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 7.89–7.69 (m, 2 H, ArH), 7.59–7.33 (m, 3 H, ArH), 6.86 (s, 1 H,
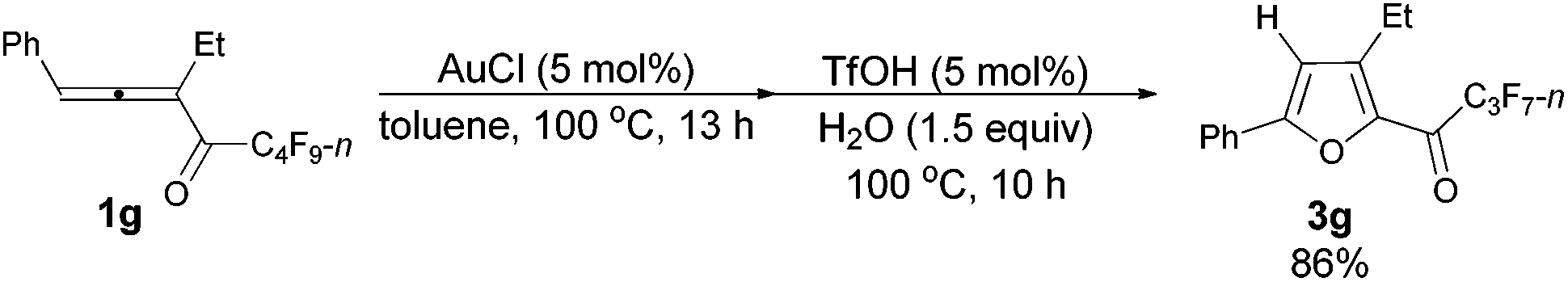 CH), 2.96 (q, J = 7.4 Hz, 2 H, CH2), 1.29 (t, J = 7.5 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.3 (t, J = 26.3 Hz), 159.2, 148.3, 143.6, 130.3, 129.1, 128.5, 125.4, 109.8, 20.0, 13.1; 19F NMR (282 MHz, CDCl3) δ −81.0 (t, J = 8.9 Hz, 3 F), −118.1 (q, J = 9.0 Hz, 2 F), −126.3 (s, 2 F).
CH), 2.96 (q, J = 7.4 Hz, 2 H, CH2), 1.29 (t, J = 7.5 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.3 (t, J = 26.3 Hz), 159.2, 148.3, 143.6, 130.3, 129.1, 128.5, 125.4, 109.8, 20.0, 13.1; 19F NMR (282 MHz, CDCl3) δ −81.0 (t, J = 8.9 Hz, 3 F), −118.1 (q, J = 9.0 Hz, 2 F), −126.3 (s, 2 F).
The reaction of 1-phenylbuta-1,2-dien-3-yl perfluorobutyl ketone 1h (0.3763 g, 1.00 mmol) with AuCl (97%, 0.0111 g, 0.046 mmol) in toluene (5 mL) at 100 °C for 3.5 h afforded crude product 2h. Then TfOH (99%, 4.5 μL, d = 1.695 g mL−1, 0.0076 g, 0.050 mmol) and H2O (27 μL, d = 1.0 g mL−1, 0.0270 g, 1.50 mmol) were added and reacted at 100 °C for another 13 h to afford 3h14 (0.3039 g, 86%) (chromatography eluent: petroleum ether/ethyl acetate = 500/1) as an oil: 1H NMR (300 MHz, CDCl3) δ 7.82–7.70 (m, 2 H, ArH), 7.53–7.38 (m, 3 H, ArH), 6.80 (s, 1 H,
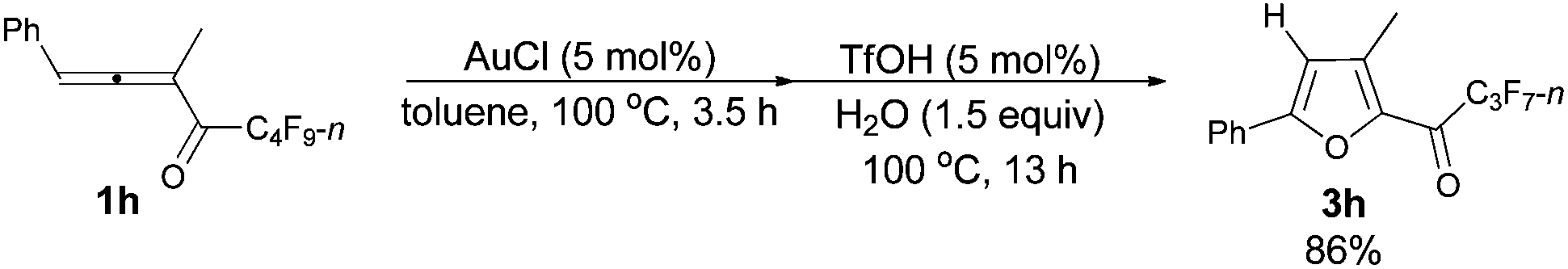 CH), 2.52 (s, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.5 (t, J = 26.2 Hz), 158.9, 144.2, 141.9, 130.3, 129.1, 128.4, 125.4, 111.6, 12.6; 19F NMR (282 MHz, CDCl3) δ −80.5 (t, J = 9.2 Hz, 3 F), −117.5 to −117.9 (m, 2 F), −125.8 to −126.0 (m, 2 F).
CH), 2.52 (s, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.5 (t, J = 26.2 Hz), 158.9, 144.2, 141.9, 130.3, 129.1, 128.4, 125.4, 111.6, 12.6; 19F NMR (282 MHz, CDCl3) δ −80.5 (t, J = 9.2 Hz, 3 F), −117.5 to −117.9 (m, 2 F), −125.8 to −126.0 (m, 2 F).
The reaction of 1-(4′-methylphenyl)hexa-1,2-dien-3-yl perfluorobutyl ketone 1i (0.2101 g, 0.50 mmol) with AuCl (97%, 0.0058 g, 0.024 mmol) in toluene (5 mL) at 100 °C for 11 h afforded crude product 2i. Then TfOH (99%, 2.3 μL, d = 1.695 g mL−1, 0.0039 g, 0.026 mmol) and H2O (13.5 μL, d = 1.00 g mL−1, 0.0135 g, 0.75 mmol) were added and reacted at 100 °C for another 5 h to afford 3i14 (0.1756 g, 88%) (chromatography eluent: petroleum ether/ethyl acetate = 500/1) as a solid: m.p. 33.5–34.0 °C (recrystallization solvent: n-hexane/ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 7.67 (d, J = 7.8 Hz, 2 H, ArH), 7.27 (d, J = 7.8 Hz, 2 H, ArH), 6.78 (s, 1 H,
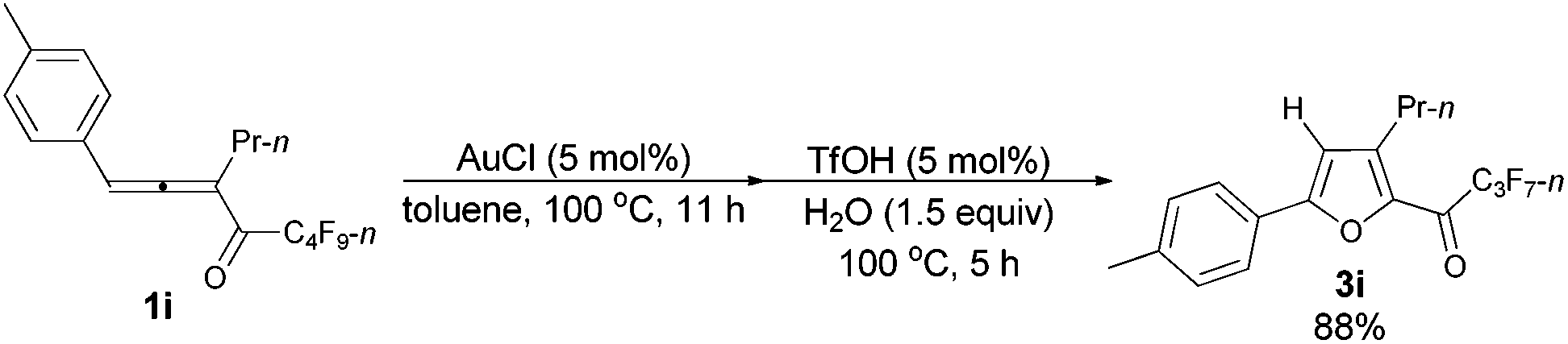 CH), 2.90 (t, J = 7.5 Hz, 2 H, CH2), 2.41 (s, 3 H, CH3), 1.77–1.63 (m, 2 H, CH2), 1.01 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.1 (t, J = 25.9 Hz), 159.4, 147.0, 143.7, 140.7, 129.8, 125.8, 125.5, 109.8, 28.4, 22.3, 21.5, 13.8; 19F NMR (282 MHz, CDCl3) δ −81.0 (t, J = 8.3 Hz, 3 F), −118.0 (q, J = 8.7 Hz, 2 F), −126.4 (s, 2 F).
CH), 2.90 (t, J = 7.5 Hz, 2 H, CH2), 2.41 (s, 3 H, CH3), 1.77–1.63 (m, 2 H, CH2), 1.01 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.1 (t, J = 25.9 Hz), 159.4, 147.0, 143.7, 140.7, 129.8, 125.8, 125.5, 109.8, 28.4, 22.3, 21.5, 13.8; 19F NMR (282 MHz, CDCl3) δ −81.0 (t, J = 8.3 Hz, 3 F), −118.0 (q, J = 8.7 Hz, 2 F), −126.4 (s, 2 F).
The reaction of 1-(4′-fluorophenyl)hexa-1,2-dien-3-yl perfluorobutyl ketone 1j (0.2112 g, 0.50 mmol) with AuCl (97%, 0.0061 g, 0.026 mmol) in toluene (5 mL) at 100 °C for 12 h afforded crude product 2j. Then TfOH (99%, 2.3 μL, d = 1.695 g mL−1, 0.0039 g, 0.026 mmol) and H2O (13.5 μL, d = 1.0 g mL−1, 0.0135 g, 0.75 mmol) were added and reacted at 100 °C for another 10 h to afford 3j14 (0.1648 g, 82%) (chromatography eluent: petroleum ether/ethyl acetate = 500/1) as an oil: 1H NMR (300 MHz, CDCl3) δ 7.77 (dd, J1 = 8.3 Hz, J2 = 5.6 Hz, 2 H, ArH), 7.17 (t, J = 8.4 Hz, 2 H, ArH), 6.78 (s, 1 H,
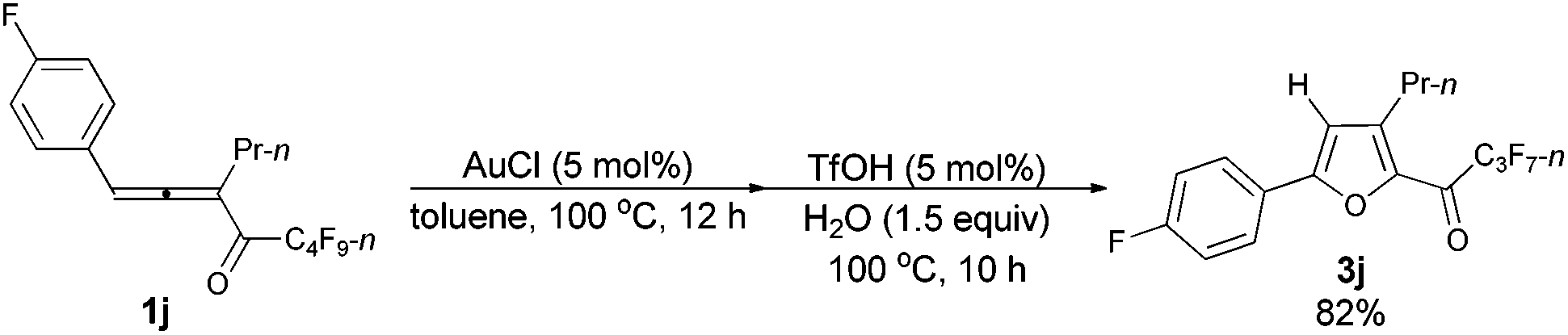 CH), 2.90 (t, J = 7.7 Hz, 2 H, CH2), 1.78–1.62 (m, 2 H, CH2), 1.02 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.3 (t, J = 25.7 Hz), 163.8 (d, J = 250.4 Hz), 158.0, 146.9, 143.9, 127.5 (d, J = 7.7 Hz), 124.9 (d, J = 3.8 Hz), 116.4 (t, J = 22.2 Hz), 110.1, 28.4, 22.3, 13.8; 19F NMR (282 MHz, CDCl3) δ 81.0 (t, J = 8.9 Hz, 3 F), −109.3 to −109.6 (m, 1 F), −118.0 (q, J = 8.9 Hz, 2 F), −126.4 (s, 2 F).
CH), 2.90 (t, J = 7.7 Hz, 2 H, CH2), 1.78–1.62 (m, 2 H, CH2), 1.02 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.3 (t, J = 25.7 Hz), 163.8 (d, J = 250.4 Hz), 158.0, 146.9, 143.9, 127.5 (d, J = 7.7 Hz), 124.9 (d, J = 3.8 Hz), 116.4 (t, J = 22.2 Hz), 110.1, 28.4, 22.3, 13.8; 19F NMR (282 MHz, CDCl3) δ 81.0 (t, J = 8.9 Hz, 3 F), −109.3 to −109.6 (m, 1 F), −118.0 (q, J = 8.9 Hz, 2 F), −126.4 (s, 2 F).
The reaction of 1-(2′-thienyl)hexa-1,2-dien-3-yl perfluorobutyl ketone 1k (0.0812 g, 0.20 mmol) with AuCl (97%, 0.0025 g, 0.010 mmol) in toluene (2 mL) at 100 °C for 13 h afforded crude product 2k. Then TfOH (99%, 0.9 μL, d = 1.695 g mL−1, 0.0015 g, 0.010 mmol) and H2O (5.4 μL, d = 1.0 g mL−1, 0.0054 g, 0.30 mmol) were added and reacted at 100 °C for another 10 h to afford 3k14 (0.0331 g, 43%) (chromatography eluent: petroleum ether/ethyl acetate = 500/1) as an oil: 1H NMR (300 MHz, CDCl3) δ 7.52 (d, J = 3.6 Hz, 1 H, ArH), 7.45 (d, J = 4.8 Hz, 1 H, ArH), 7.13 (d, J = 4.4 Hz, 1 H, ArH), 6.67 (s, 1 H,
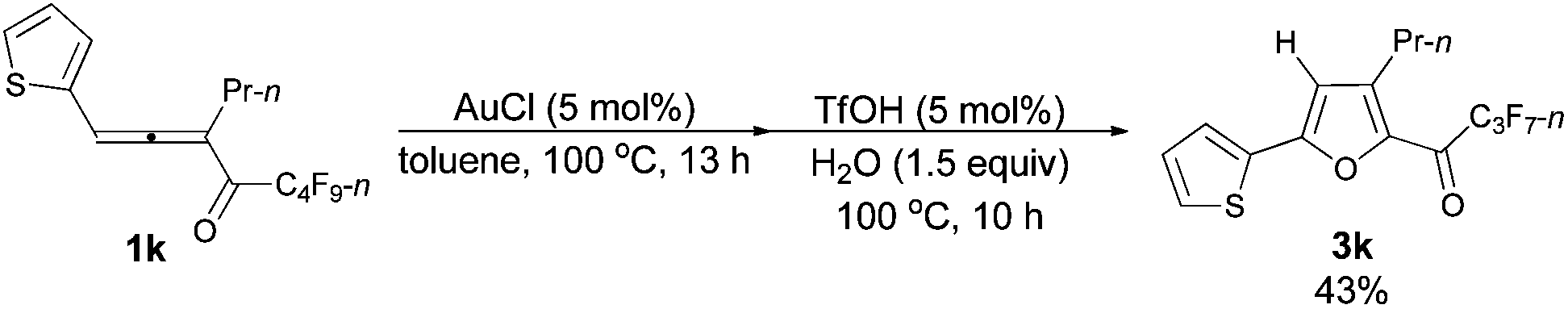 CH), 2.89 (t, J = 7.5 Hz, 2 H, CH2), 1.79–1.60 (m, 2 H, CH2), 1.01 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.0 (t, J = 26.4 Hz), 154.4, 147.0, 143.3, 131.4, 128.38, 128.36, 127.0, 110.0, 28.4, 22.2, 13.8; 19F NMR (282 MHz, CDCl3) δ −81.0 (t, J = 9.0 Hz, 3 F), −118.0 (q, J = 8.8 Hz, 2 F), −126.3 to −126.4 (m, 2 F).
CH), 2.89 (t, J = 7.5 Hz, 2 H, CH2), 1.79–1.60 (m, 2 H, CH2), 1.01 (t, J = 7.4 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3) δ 170.0 (t, J = 26.4 Hz), 154.4, 147.0, 143.3, 131.4, 128.38, 128.36, 127.0, 110.0, 28.4, 22.2, 13.8; 19F NMR (282 MHz, CDCl3) δ −81.0 (t, J = 9.0 Hz, 3 F), −118.0 (q, J = 8.8 Hz, 2 F), −126.3 to −126.4 (m, 2 F).
Acknowledgements
Financial support from the National Natural Science Foundation of China (21572202) is greatly appreciated. Shengming Ma is a Qiu Shi Adjunct Professor at Zhejiang University. The results presented in entry 2 of Table 2 (WWT), entry 5 of Table 4 and entry 1 of Table 8 (ZJS) have been kindly reproduced by Mr Wangteng Wu and Dr Jiasheng Zhang.Notes and references
- (a) H.-K. Lee, K.-F. Chan, C.-W. Hui, H.-K. Yim, X.-W. Wu and H. N. C. Wong, Pure Appl. Chem., 2005, 77, 139 CAS; (b) H. N. C. Wong, P. Yu and C.-Y. Yick, Pure Appl. Chem., 1999, 71, 1041 CrossRef CAS; (c) B. H. Lipshutz, Chem. Rev., 1986, 86, 795 CrossRef CAS.
- For most important examples, see: (a) F. Bellina and R. Rossi, Tetrahedron, 2006, 62, 7213 CrossRef CAS; (b) A. Fürstner and H. Weintritt, J. Am. Chem. Soc., 1998, 120, 2817 CrossRef; (c) A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582 CrossRef PubMed; (d) A. Fürstner, K. Reinecke, H. Prinz and H. Waldmann, ChemBioChem, 2004, 5, 1575 CrossRef PubMed; (e) A. Fürstner and E. J. Grabowski, ChemBioChem, 2001, 2, 706 CrossRef; (f) G. R. Pettit, J. McNulty, D. L. Herald, D. L. Doubek, J.-C. Chapuis, J. M. Schmidt, L. P. Tackett and M. R. Boyd, J. Nat. Prod., 1997, 60, 180 CrossRef CAS PubMed.
- (a) Y. Miyata, T. Nishinaga and K. Komatsu, J. Org. Chem., 2005, 70, 1147 CrossRef CAS PubMed; (b) M. Heylen, K. van den Broeck, C. Boutton, M. van Beylen, A. Persoons and C. Samyn, Eur. Polym. J., 1998, 34, 1453 CrossRef CAS; (c) F. Ferrero, L. Napoli, C. Tonin and A. Varesano, J. Appl. Polym. Sci., 2006, 102, 4121 CrossRef CAS; (d) S. Venkatraman, R. Kumar, J. Sankar, T. K. Chandrashekar, K. Sendhil, C. Vijayan, A. Kelling and M. O. Senge, Chem. – Eur. J., 2004, 10, 1423 CrossRef CAS PubMed; (e) A. Facchetti, A. Abbotto, L. Beverina, M. E. van der Boom, P. Dutta, G. Evmenenko, G. A. Pagani and T. J. Marks, Chem. Mater., 2003, 15, 1064 CrossRef CAS; (f) L.-Z. Zhang, C.-W. Chen, C.-F. Lee, C.-C. Wu and T.-Y. Luh, Chem. Commun., 2002, 2336 RSC; (g) P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207 CrossRef.
- (a) D. S. Ennis and T. L. Gilchrist, Tetrahedron, 1990, 46, 2623 CrossRef CAS; (b) T. R. Bailey, Synthesis, 1991, 242 CrossRef CAS; (c) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS; (d) P. Weyerstahl, A. Schenk and H. Marschall, Liebigs Ann., 1995, 1849 CrossRef CAS; (e) J. Fournier, S. Arseniyadis and J. Cossy, Angew. Chem., Int. Ed., 2012, 51, 7562 CrossRef CAS PubMed; (f) C. A. Schoenbaum, D. K. Schwartz and J. W. Medlin, Acc. Chem. Res., 2014, 47, 1438 CrossRef CAS PubMed; (g) W. Wu and H. Jiang, Acc. Chem. Res., 2014, 47, 2483 CrossRef CAS PubMed.
- For recent reviews, see: (a) N. T. Patil and Y. Yamamoto, ARKIVOC, 2007, x, 121 Search PubMed; (b) S. F. Kirsch, Org. Biomol. Chem., 2006, 4, 2076 RSC; (c) R.C. D. Brown, Angew. Chem., Int. Ed., 2005, 44, 850 CrossRef CAS PubMed; (d) B. A. Keay, Chem. Soc. Rev., 1999, 28, 209 RSC; (e) X. L. Hou, H. Y. Cheung, T. Y. Hon, P. L. Kwan, T. H. Lo, S. Y. Tong and H. N. C. Wong, Tetrahedron, 1998, 54, 1955 CrossRef CAS; (f) S. Schröter, C. Stock and T. Bach, Tetrahedron, 2005, 61, 2245 CrossRef; (g) C. Schmuck and D. Rupprecht, Synthesis, 2007, 3095 CrossRef CAS; (h) Y. Xiao and J. Zhang, Angew. Chem., Int. Ed., 2008, 47, 1903 CrossRef CAS PubMed; (i) P. Lenden, D. A. Entwistle and M. C. Willis, Angew. Chem., Int. Ed., 2011, 50, 10657 CrossRef CAS PubMed; (j) C. He, S. Guo, J. Ke, J. Hao, H. Xu, H. Chen and A. Lei, J. Am. Chem. Soc., 2012, 134, 5766 CrossRef CAS PubMed; (k) B. Lu, J. Wu and N. Yoshikai, J. Am. Chem. Soc., 2014, 136, 11598 CrossRef CAS PubMed.
- For transition metal-catalyzed cyclization of allenones, see ref. 6–10: (a) J. A. Marshall and E. D. Robinson, J. Org. Chem., 1990, 55, 3450 CrossRef CAS; (b) J. A. Marshall and X.-J. Wang, J. Org. Chem., 1991, 56, 960 CrossRef CAS; (c) J. A. Marshall and G. S. Bartley, J. Org. Chem., 1994, 59, 7169 CrossRef CAS; (d) J. A. Marshall and C. A. Sehon, J. Org. Chem., 1995, 60, 5966 CrossRef CAS.
- (a) A. S. K. Hashmi, Angew. Chem., Int. Ed. Engl., 1995, 34, 1581 CrossRef; (b) A. S. K. Hashmi, T. L. Ruppert, T. Knöfel and J. W. Bats, J. Org. Chem., 1997, 62, 7295 CrossRef CAS PubMed; (c) B. Alcaide, P. Almendros and T. Martínez del Campo, Eur. J. Org. Chem., 2007, 2844 CrossRef CAS.
- (a) A. S. K. Hashmi, L. Schwarz, J.-H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285 CrossRef CAS; (b) C.-Y. Zhou, P. W. H. Chan and C.-M. Che, Org. Lett., 2006, 8, 325 CrossRef CAS PubMed; (c) B. Alcaide, P. Almendros, J. M. Alonso and I. Fernández, J. Org. Chem., 2013, 78, 6688 CrossRef CAS PubMed.
- (a) A. V. Kel'in and V. Gevorgyan, J. Org. Chem., 2002, 67, 95 CrossRef; (b) J. T. Kim, A. V. Kel'in and V. Gevorgyan, Angew. Chem., Int. Ed., 2003, 42, 98 CrossRef CAS; (c) A. W. Sromek, A. V. Kel'in and V. Gevorgyan, Angew. Chem., Int. Ed., 2004, 43, 2280 CrossRef CAS PubMed; (d) A. W. Sromek, M. Rubina and V. Gevorgyan, J. Am. Chem. Soc., 2005, 127, 10500 CrossRef CAS PubMed; (e) T. Schwier, A. W. Sromek, D. M. L. Yap, D. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 9868 CrossRef CAS PubMed; (f) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 5195 CrossRef CAS PubMed; (g) A. S. Dudnik, A. W. Sromek, M. Rubina, J. T. Kim, A. V. Kel'in and V. Gevorgyan, J. Am. Chem. Soc., 2008, 130, 1440 CrossRef CAS PubMed; (h) A. S. Dudnik, Y. Xia, Y. Li and V. Gevorgyan, J. Am. Chem. Soc., 2010, 132, 7645 CrossRef CAS PubMed; (i) Y. Xia, A. S. Dudnik, V. Gevorgyan and Y. Li, J. Am. Chem. Soc., 2008, 130, 6940 CrossRef CAS PubMed.
- (a) S. Ma and J. Zhang, Chem. Commun., 2000, 117 RSC; (b) S. Ma, J. Zhang and L. Lu, Chem. – Eur. J., 2003, 9, 2447 CrossRef CAS PubMed; (c) S. Ma and L. Li, Org. Lett., 2000, 2, 941 CrossRef CAS PubMed; (d) Y. Xia, Y. Xia, R. Ge, Z. Liu, Q. Xiao, Y. Zhang and J. B. Wang, Angew. Chem., Int. Ed., 2014, 53, 3917 CrossRef CAS PubMed.
- (a) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (d) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2004 Search PubMed.
- For reviews referring to Brønsted acid-catalyzed cyclization of allenes, see: (a) A. S. K. Hashmi, Catal. Today, 2007, 122, 211 CrossRef CAS; (b) T. Cañeque, F. M. Truscott, R. Rodriguez, G. Maestri and M. Malacria, Chem. Soc. Rev., 2014, 43, 2916 RSC.
- G. He, C. Xue, C. Fu and S. Ma, Synlett, 2010, 281 CAS.
- C. Xue, X. Huang, S. Wu, J. Zhou, J. Dai, C. Fu and S. Ma, Chem. Commun., 2015, 51, 17112 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR, 13C NMR, 19F NMR and NOE spectra of all the compounds prepared. See DOI: 10.1039/c6qo00001k |
| This journal is © the Partner Organisations 2016 |