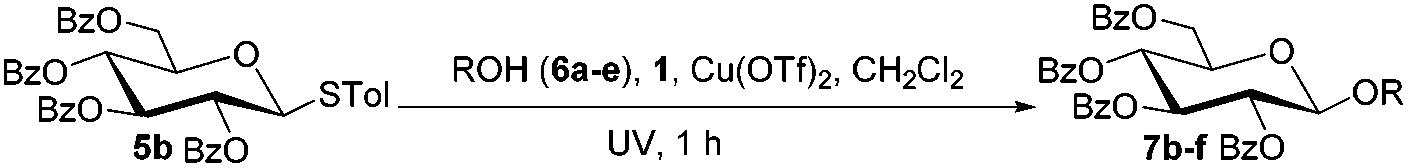DOI:
10.1039/C6QO00021E
(Research Article)
Org. Chem. Front., 2016,
3, 737-743
Light-driven highly efficient glycosylation reactions†
Received
15th January 2016
, Accepted 3rd April 2016
First published on 5th April 2016
Abstract
Glycosylation is unarguably the most important reaction in the field of glycochemistry. Photo-initiated reactions hold extraordinary potential in organic synthesis. Herein we report a novel light-driven strategy for the activation of thioglycosides via the merger of a CF3 radical pathway followed by the subsequent glycosylation with glycosyl acceptors. This protocol could efficiently activate thioglycosides, thereby greatly enhancing the substrate scope of reactions. In particular, the glycosylation reactions, which are completely inert under the existing photo-induced glycosylation conditions, proceeded smoothly in high yields. Many common protective groups were well tolerated under the coupling conditions. Both UV and visible light or even sunlight were used as the source of light for the reactions. The high efficiency of this light-driven glycosylation protocol was further highlighted by the rapid one-pot sequential assembly of oligosaccharides.
Introduction
Oligosaccharides and glycoconjugates play important roles in numerous physiological and pathological processes.1 To gain a better understanding of these biological events, pure and structurally well-defined materials are highly desirable. However, the availability of these saccharides is limited, due to their low content, inherent structural diversity, and microheterogeneity in nature. Therefore, chemical synthesis is a major approach to prepare oligosaccharides and glycoconjugates. Remarkable advances have been achieved in methods,2–6 strategies,7–9 and even an automated synthesizer10,11 for the chemical preparation of oligosaccharides. Extensive experimental and theoretical efforts have also been devoted to reveal the mechanistic details of glycosylation reactions.12–14 In recent years, reactions triggered by light offer a great variety of useful transformations.15–18 In particular, to move toward green and sustainable chemistry, significant progress has been made in visible-light mediated oxidation19 and reduction,20 C–H activation,21 as well as C–C and C–heteroatom bond-formation processes.22–24 Carbohydrate is a particular class of biomolecule linked by C–O bonds. However, only a handful of photo-induced glycosylations have been reported.25–31 Since the activation of glycosyl donors largely depends on the redox potential of a photosensitizer or photocatalyst, the existing methods require a careful adjustment of anomeric leaving groups to ensure a successful light-induced activation, and exhibit an extremely limited substrate scope/generality. Meanwhile, the present methods also suffer from severe shortages such as low yields, the incompatibility of functional groups with glycosylation conditions, the use of a large amount of glycosyl acceptors, and long reaction time. Thus, the progress of light-mediated glycosylations is far behind.
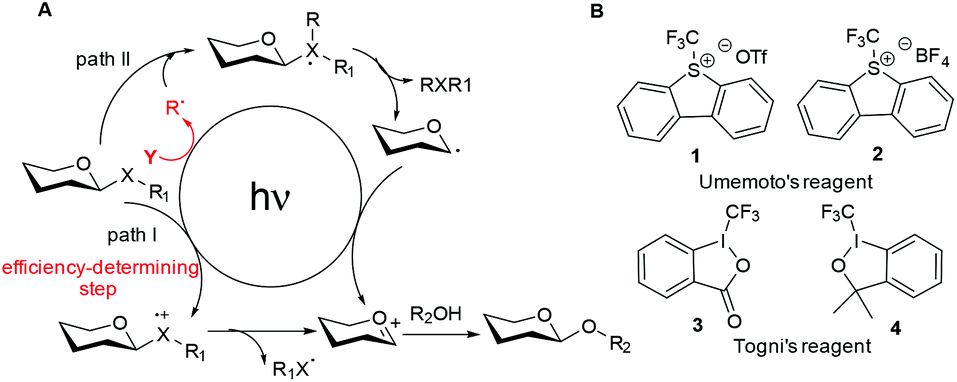
We have developed a glycosylation method via direct activation of thioglycosides upon UV irradiation.32 However, this method still lags behind the existing non-photoinduced methods due to some shortages such as the coupling efficiency and reaction time. To tackle this problem, we therefore hypothesized that the merger of an additional radical pathway with light-mediated glycosylation would lead to an alternative and potentially complementary mechanistic manifold, thereby activating a glycosyl donor regardless of their redox potential (Fig. 1A, path II). This strategy requires a radical precursor Y to readily generate an electrophilic radical R under illumination. For this purpose, several prerequisites must be satisfied for Y: (1) it must be inert under normal conditions, whereas reactive under illumination; (2) the radical must be generated in a controllable manner, yet be reactive for rapid activation of the donor. We noticed that light had been employed to generate the highly electrophilic trifluoromethyl radical by the use of some commercially available CF3 sources under mild conditions.33,34 Moreover, the trifluoromethyl radical could react with thiols to form trifluoromethyl sulfides via a radical mechanism.35,36 Thus, we envisaged that trifluoromethylating reagents37 such as Umemoto's reagents38 and Togni's reagents39 (Fig. 1B) might be ideal radical precursors for light-induced thioglycoside activation and subsequent glycosylation. Herein, we report a novel photochemical protocol for the activation of thioglycosides and highly efficient glycosylation by Umemoto's reagent.
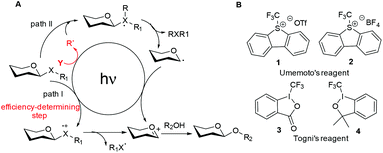 |
| | Fig. 1 (A) The traditional (path I) and designed (path II) mechanistic manifolds for light-induced glycosylation reactions. (B) Structures of Umemoto's reagents and Togni's reagents. | |
Results and discussion
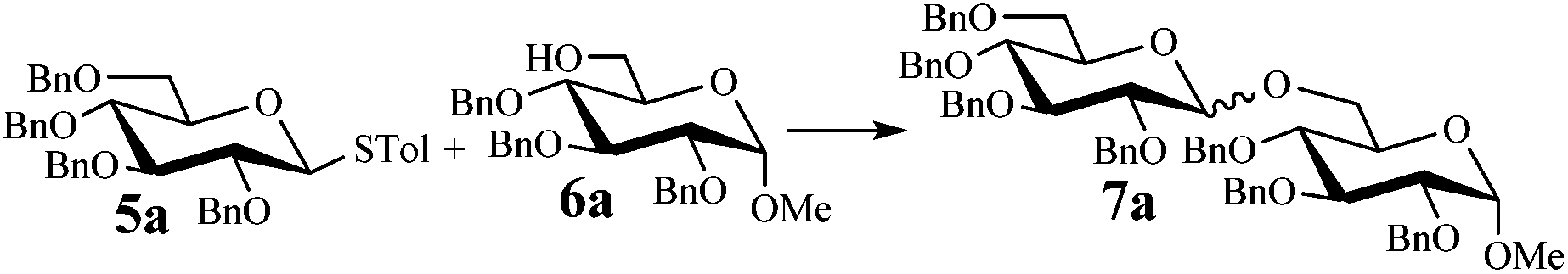
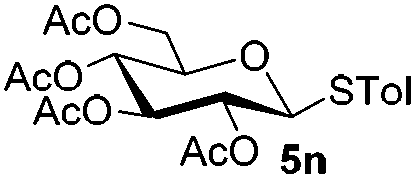
We began our investigations by examining the reaction of donor 5a with acceptor 6a under the irradiation of UV light. In the absence of photoirradiation, no desired product was detected by the use of either Umemoto's reagents or Togni's reagents (Table 1, entry 1; Table S1,† entries 1–16). In the presence of both photoirradiation and trifluoromethylating reagents 1–4, only a small amount (not more than 9% yield) of the desired product 7a was obtained in different solvents (Table 1, entries 2–5; Table S1,† entries 17–28). Additives including bases, photosensitizers, and metal salts, were able to improve the coupling yield a little (Table S1,† entries 29–37, from trace to 34%). The additive Cu(OTf)2 gave the best result (Table 1, entry 6, 39%). Notably, decreasing the reaction temperature boosted the yield to 72% (Table 1, entries 7 and 8). Further screening of the equivalent of Cu(OTf)2 (Table 1, entries 8–10) and Umemoto's reagent 1 (Table 1, entries 12–15) as well as the ratio of 5a/6a (Table 1, entries 10–12) established the standard conditions: donor 5a (1.3 equiv.), acceptor 6a (1.0 equiv.), 1 (1.5 equiv.), 4 Å MS, and CH2Cl2 in a quartz flask under the irradiation of UV light for one hour at −72 °C (Table 1, entry 16, 85%). Control experiments showed that though Cu(OTf)2 seemed to be not essential for the yield of 7a, it led to a higher yield and better reproducibility (Table 1, entries 16 and 17). After continuous experimentations we found that the yield of 7a was increased to 93% by using a pre-activation based glycosylation protocol (Table 1, entries 18 and 19; Table S1,† entries 38–43).40–42
Table 1 Optimization of the reaction conditions under the irradiation of UV lighta
|
|
| Entry |
5a/6a |
Activator (equiv.) |
Additive (equiv.) |
Yieldc (%) |
|
General conditions: donor, 1–4, acceptor (0.02 mmol, 1.0 equiv.), 4 Å MS (200 mg), CH2Cl2 (2.0 mL) in a quartz flask at −72 °C under the irradiation of UV for 1 h.
Yield was determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard, and the α/β ratios were 1.1/1 to 1.3/1.
Isolated yield.
The acceptor was added after the donor was pre-activated.
No UV irradiation, room temperature.
UV irradiation for 30 min, room temperature.
Room temperature.
0 °C.
MeCN was used as the solvent.
|
| 1e |
1.2/1 |
1 (1.8) |
— |
0b |
| 2f |
1.2/1 |
1 (1.8) |
— |
5b |
| 3f |
1.2/1 |
2 (1.8) |
— |
5b |
| 4i,f |
1.2/1 |
1 (1.8) |
— |
9b |
| 5i,f |
1.2/1 |
2 (1.8) |
— |
6b |
| 6g |
1.2/1 |
1 (1.8) |
Cu(OTf)2 (1.0) |
39 |
| 7h |
1.2/1 |
1 (1.8) |
Cu(OTf)2 (1.0) |
43 |
| 8 |
1.2/1 |
1 (1.8) |
Cu(OTf)2 (1.0) |
72 |
| 9 |
1.2/1 |
1 (1.8) |
Cu(OTf)2 (1.5) |
75 |
| 10 |
1.2/1 |
1 (1.8) |
Cu(OTf)2 (2.0) |
67 |
| 11 |
1.3/1 |
1 (2.0) |
Cu(OTf)2 (1.5) |
79 |
| 12 |
1.4/1 |
1 (2.0) |
Cu(OTf)2 (1.5) |
78 |
| 13 |
1.3/1 |
1 (1.0) |
Cu(OTf)2 (1.5) |
69 |
| 14 |
1.3/1 |
1 (1.3) |
Cu(OTf)2 (1.5) |
77 |
| 15 |
1.3/1 |
1 (1.4) |
Cu(OTf)2 (1.5) |
83 |
| 16 |
1.3/1 |
1 (1.5) |
Cu(OTf)2 (1.5) |
85 |
| 17 |
1.3/1 |
1 (1.5) |
— |
80 |
| 18d |
1.3/1 |
1 (1.5) |
Cu(OTf)2 (1.5) |
93 |
| 19d |
1.3/1 |
2 (1.5) |
Cu(OTf)2 (1.5) |
81 |
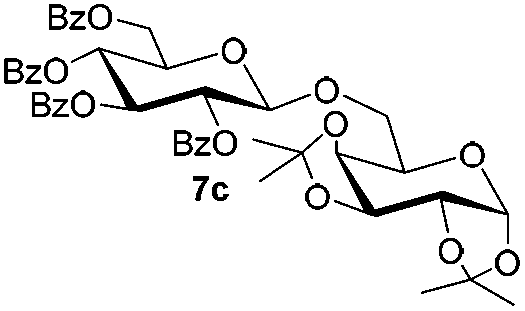
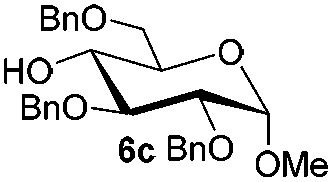
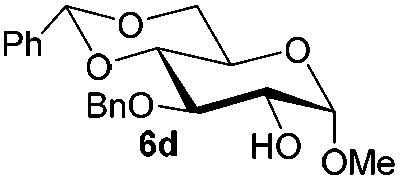
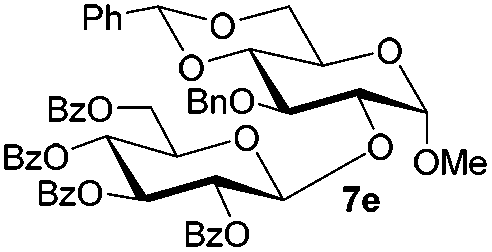
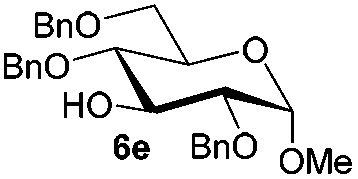
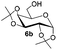
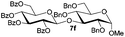
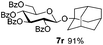
Under these optimized high-yielding reaction conditions, we next examined the glycosylation between the disarmed donor 5b and various acceptors (Table 2). The glycosyl donor 5b was completely inert under the traditionally photo-induced glycosylation conditions.25 Gratifyingly, the reaction of 5b with the glycosyl acceptors 6a–e having a free hydroxyl group at the C-6, C-4, C-2, and C-3 positions, respectively, proceeded in good to excellent yields under either the standard conditions or the pre-activation conditions. Yet the coupling yields were 6–17% higher when using the pre-activation protocol. This might be due to the formation of less byproducts of the reaction (such as the byproducts generated from acceptors). Impressively, the tough coupling reaction of the disarmed 5b with hindered 6c in carbohydrate chemistry also worked well (entry 3). Isopropylidene, benzoyl, and benzyl protective groups were well tolerated under these conditions.
Table 2 Glycosylations of acceptors 6a–e with donor 5b under the irradiation of UVa
|

|
| Entry |
Glycosyl acceptor |
Disaccharide |
Yieldc |
Yieldb,c |
|
General conditions: donor (0.026 mmol, 1.3 equiv.), 1 (0.03 mmol, 1.5 equiv.), acceptor (0.02 mmol, 1.0 equiv.), Cu(OTf)2 (0.03 mmol, 1.5 equiv.), 4 Å MS (200 mg), CH2Cl2 (2.0 mL) in a quartz flask at −72 °C under the irradiation of UV for 1 h.
The acceptor was added after the donor was pre-activated.
Isolated yield.
|
| 1 |
6a
|
|
82% |
99% |
| 2 |
|
|
92% |
98% |
| 3 |
|
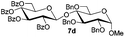
|
71% |
87% |
| 4 |
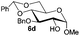
|
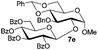
|
78% |
94% |
| 5 |
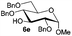
|
|
80% |
96% |
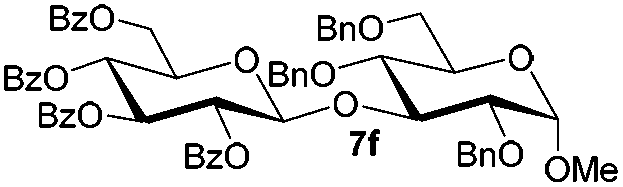
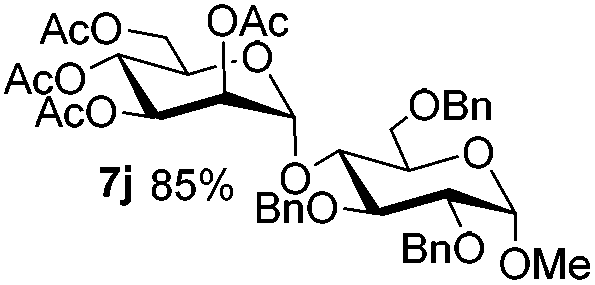
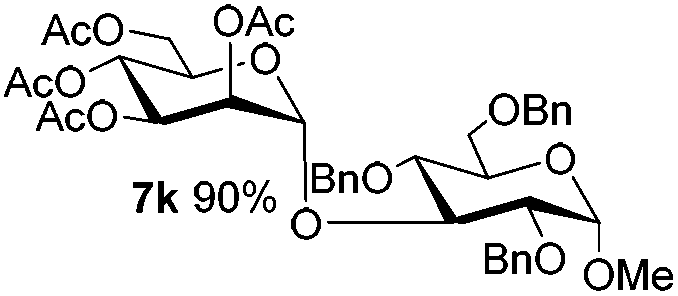
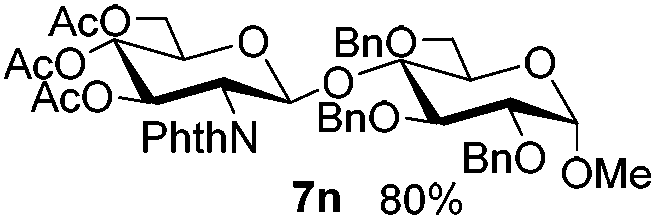
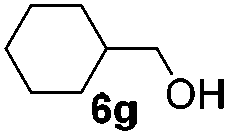
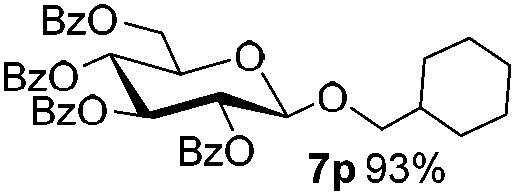
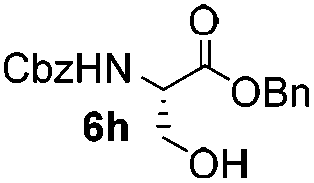
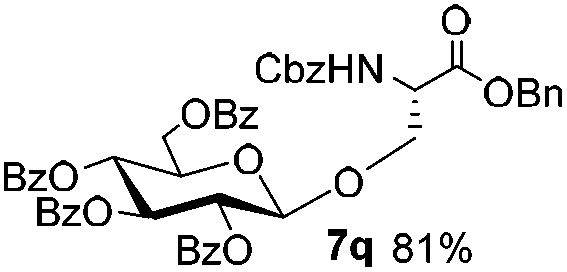
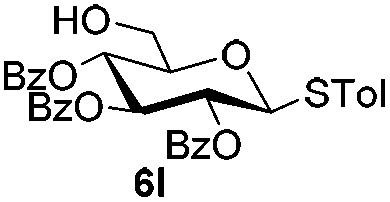
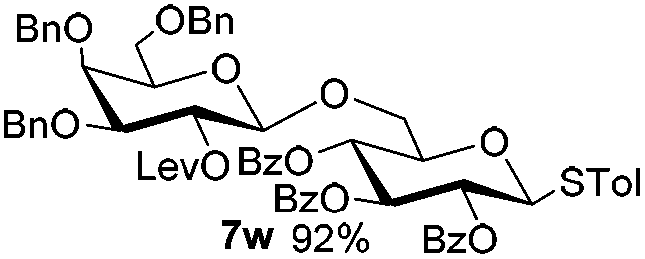
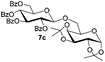
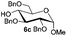
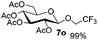
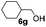
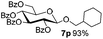
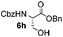
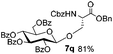
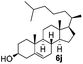
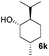
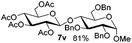
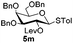
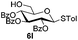
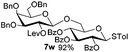
Encouraged by these results, the glycosylation between a wide range of glycosyl donors and acceptors was further evaluated by the use of the pre-activation protocol under UV illumination. The glycosyl acceptors, with a free hydroxyl group at different positions of glucose moiety, were efficiently glycosylated with disarmed galactosyl, mannosyl, rhamnosyl, or glucosaminyl donors in good to excellent yields (Table 3). The coupling reactions of the disarmed donors with non-sugar alcohols such as trifluoroethanol, cyclohexylmethanol, protected serine, adamantanol, cholesterol, and menthol, also proceeded smoothly in high yields (Table 4). Except for the SBox group, thioglycosides with an anomeric leaving group of 2-methylphenylthio, 2,6-dimethylphenylthio, ethylthio (SEt), or 2-pyridylthio (SPy), were able to be employed as glycosyl donors (Table 5). Acceptor 6l with a p-tolylthio leaving group was also glycosylated to afford disaccharide 7w in 92% isolated yield, which hinted the potential application of this protocol in preactivation-based one-pot oligosaccharide synthesis (Table 5, entry 6).
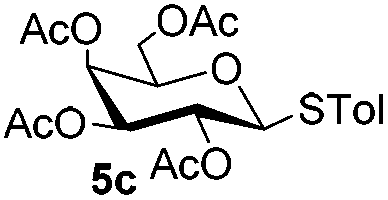
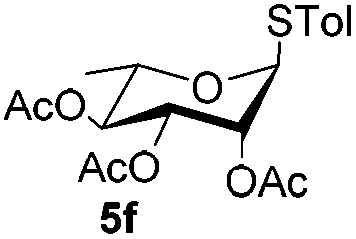
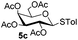
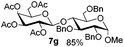
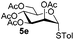
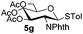
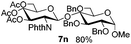
Table 3 Glycosyl coupling reactions of disarmed donors 5c–g under the irradiation of UVa
| Entry |
Donor |
Acceptor |
Disaccharide and yieldb |
|
General conditions: donor (0.026 mmol, 1.3 equiv.), 1 (0.03 mmol, 1.5 equiv.), acceptor (0.02 mmol, 1.0 equiv.), Cu(OTf)2 (0.03 mmol, 1.5 equiv.), 4 Å MS (200 mg), CH2Cl2 (2.0 mL) in a quartz flask at −72 °C under the irradiation of UV for 1 h by the use of the pre-activation protocol.
Isolated yield.
|
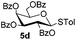
| 1 |
|
6c
|
|
| 2 |
5c
|
6e
|
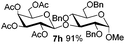
|
| 3 |
|
6a
|
|
| 4 |
|
6c
|
|
| 5 |
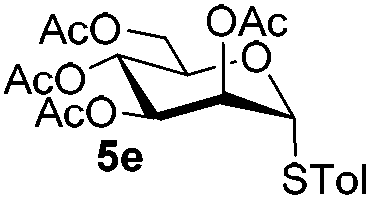
5e
|
6e
|
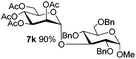
|
| 6 |
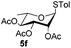
|
6c
|
|
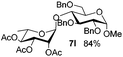
| 7 |
5f
|
6e
|
|
| 8 |
|
6c
|
|
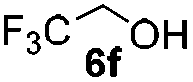
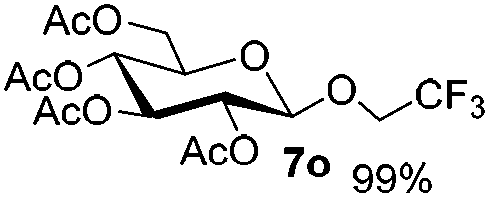
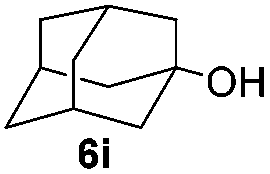
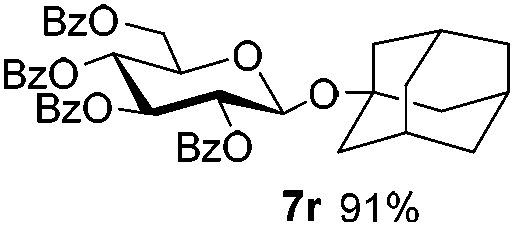
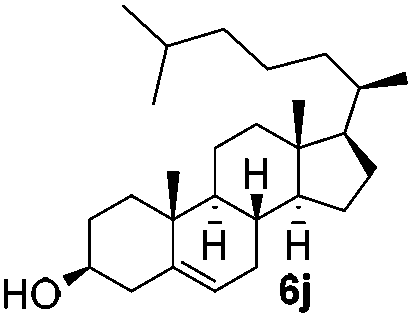
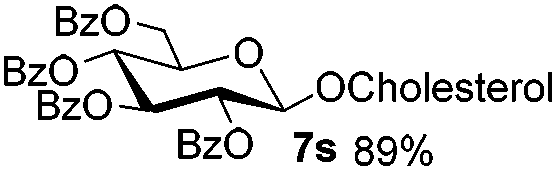
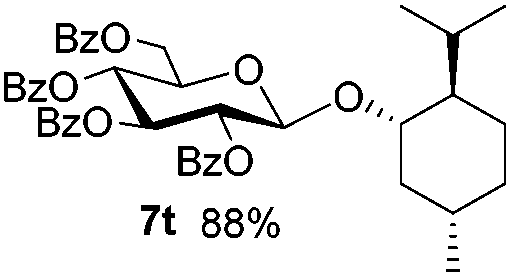
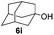
Table 4 Glycosylations of non-sugar acceptors 6f–k under the irradiation of UVa
| Entry |
Donor |
Acceptor |
Disaccharide and yieldb |
|
General conditions: donor (0.026 mmol, 1.3 equiv.), 1 (0.03 mmol, 1.5 equiv.), acceptor (0.02 mmol, 1.0 equiv.), Cu(OTf)2 (0.03 mmol, 1.5 equiv.), 4 Å MS (200 mg), CH2Cl2 (2.0 mL) in a quartz flask at −72 °C under the irradiation of UV for 1 h by the use of the pre-activation protocol.
Isolated yield.
|
| 1 |
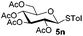
|
|
|
| 2 |
5b
|
|
|
| 3 |
5b
|
|
|
| 4 |
5b
|
|
|
| 5 |
5b
|
|
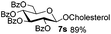
|
| 6 |
5b
|
|
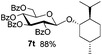
|
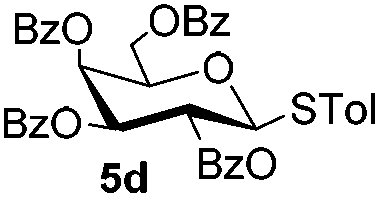
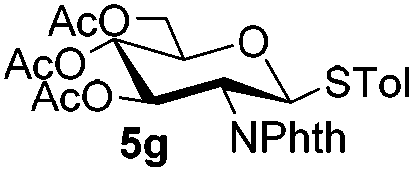
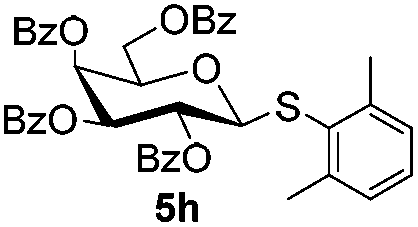
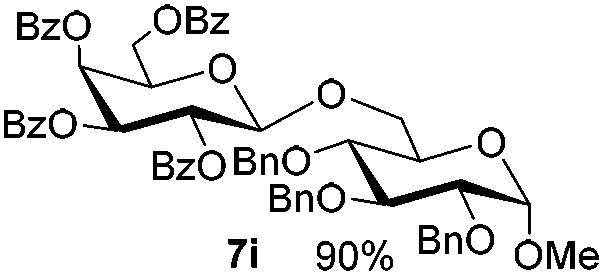
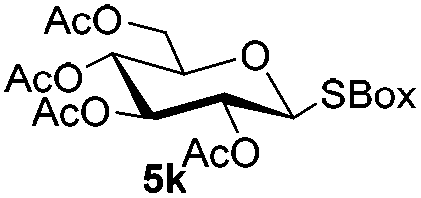
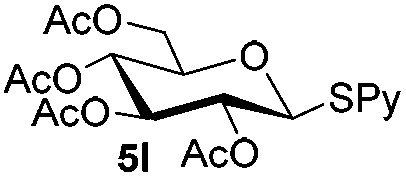
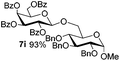
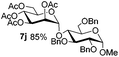
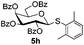
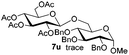
Table 5 Glycosyl coupling reactions of different thioglycosyl donors 5h–l under the irradiation of UVa
| Entry |
Donor |
Acceptor |
Disaccharide and yieldb |
|
General conditions: donor (0.026 mmol, 1.3 equiv.), 1 (0.03 mmol, 1.5 equiv.), acceptor (0.02 mmol, 1.0 equiv.), Cu(OTf)2 (0.03 mmol, 1.5 equiv.), 4 Å MS (200 mg), CH2Cl2 (2.0 mL) in a quartz flask at −72 °C under the irradiation of UV for 1 h by the use of the pre-activation protocol.
Isolated yield.
|
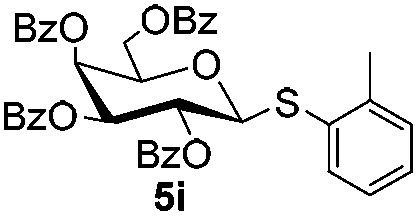
| 1 |
|
6a
|
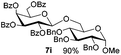
|
| 2 |
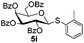
|
6a
|
7i 94% |
| 3 |
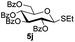
|
6a
|
|
| 4 |
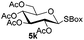
|
6a
|
|
| 5 |
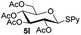
|
6c
|
|
| 6 |
|
|
|
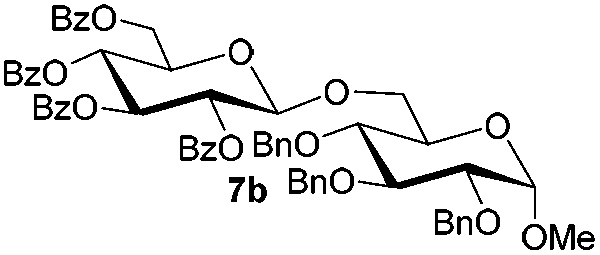
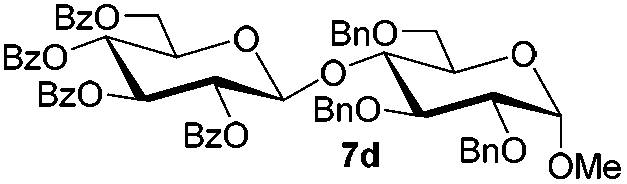
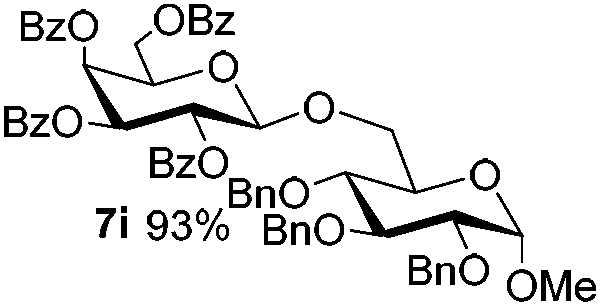
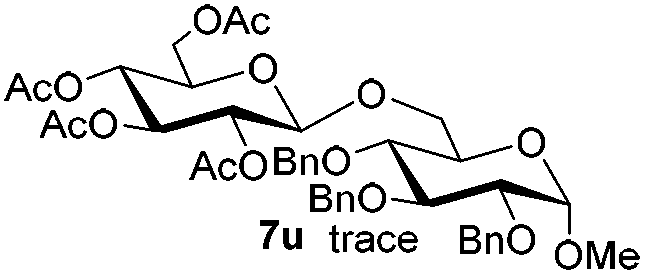
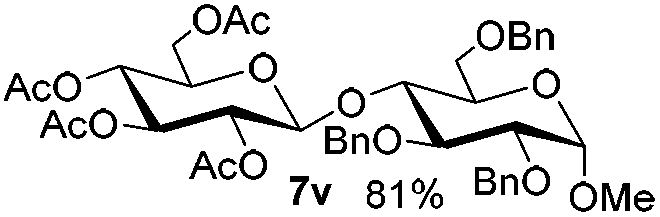
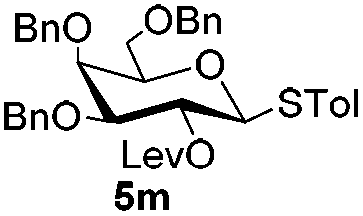
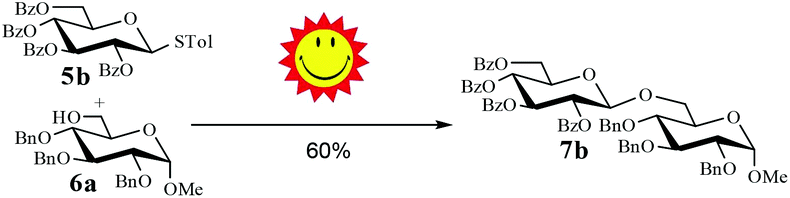
Having successfully examined the efficiency of the glycosylation reaction under the irradiation of UV light, we turned our attention to the development of a visible-light induced glycosylation by using thioglycosides as donors (Table S2†). We conducted our study on the coupling reaction of thioglycoside 5b and acceptor 6a by using different photo-catalysts upon irradiation by four 7W blue LEDs (λmax = 465 nm). It appeared that Ru(bpy)3(PF6)2 (0.05 equiv.) was the best photo-catalyst for this system. It was also found that the use of 2.6 equivalents of Umemoto's reagent 1 gave the best yield. Subsequently, the glycosylations of a wide range of thioglycoside donors and glycosyl acceptors were investigated under the optimized conditions. As shown in Table 6, all examples showed good coupling results, indicating that this protocol worked well in the construction of various disaccharides. The successful coupling of 5m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43 with the thioglycosyl acceptor 6l showed potential applications in reactivity-based one-pot oligosaccharide assembly (entry 17). Moreover, this protocol was highlighted by the smooth glycosylation reaction of 6a with 5b under the irradiation of sunlight (Fig. 2).
43 with the thioglycosyl acceptor 6l showed potential applications in reactivity-based one-pot oligosaccharide assembly (entry 17). Moreover, this protocol was highlighted by the smooth glycosylation reaction of 6a with 5b under the irradiation of sunlight (Fig. 2).
 |
| | Fig. 2 The glycosylation reaction of 6a with 5b under sunlight irradiation. Reagents and conditions: compound 5b (0.026 mmol, 1.3 equiv.), 1 (0.052 mmol, 2.6 equiv.), Ru(bpy)3(PF6)2 (0.001 mmol, 0.05 equiv.), 6a (0.02 mmol, 1.0 equiv.), Cu(OTf)2 (0.03 mmol, 1.5 equiv.), 4 Å MS (200 mg), CH2Cl2 (2.0 mL) in a glass flask at room temperature under sunlight irradiation for 6 h. | |
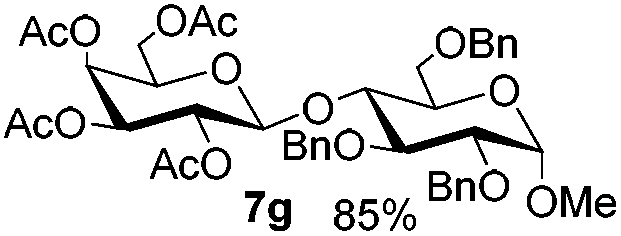
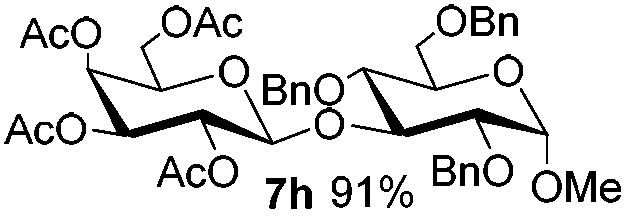
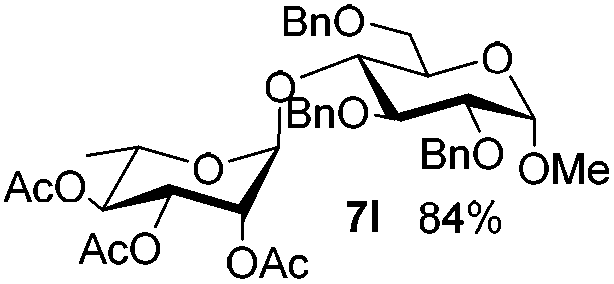
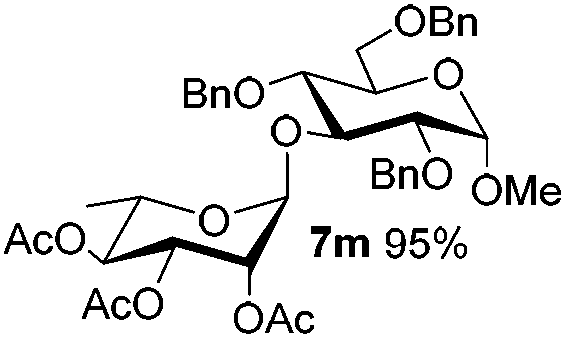
Table 6 Glycosylations under the irradiation of visible lighta
| Entry |
Donor |
Acceptor |
Disaccharide |
Yieldb (%) |
|
General conditions: donor (0.026 mmol, 1.3 equiv.), 1 (0.052 mmol, 2.6 equiv.), Ru(bpy)3(PF6)2 (0.001 mmol, 0.05 equiv.), acceptor (0.02 mmol, 1.0 equiv.), Cu(OTf)2 (0.03 mmol, 1.5 equiv.), 4 Å MS (200 mg), CH2Cl2 (2.0 mL) in a glass flask at room temperature under the irradiation of visible light for 4 h.
Isolated yield.
|
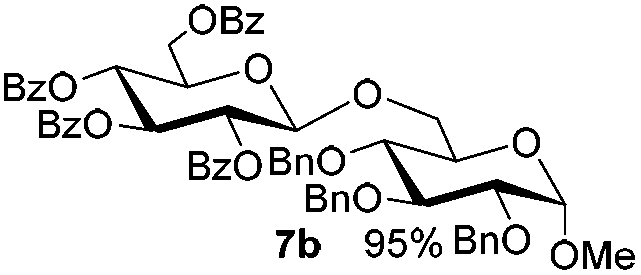
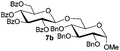
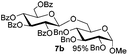
| 1 |
5b
|
6a
|
7b
|
94 |
| 2 |
5b
|
6b
|
7c
|
95 |
| 3 |
5b
|
6c
|
7d
|
75 |
| 4 |
5b
|
6d
|
7e
|
89 |
| 5 |
5b
|
6e
|
7f
|
90 |
| 6 |
5b
|
6g
|
7q
|
80 |
| 7 |
5b
|
6h
|
7r
|
65 |
| 8 |
5b
|
6i
|
7s
|
82 |
| 9 |
5b
|
6j
|
7t
|
62 |
| 10 |
5c
|
6e
|
7h
|
84 |
| 11 |
5d
|
6a
|
7i
|
88 |
| 12 |
5e
|
6e
|
7k
|
81 |
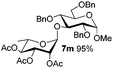
| 13 |
5f
|
6e
|
7m
|
80 |
| 14 |
5h
|
6a
|
7i
|
89 |
| 15 |
5i
|
6a
|
7i
|
84 |
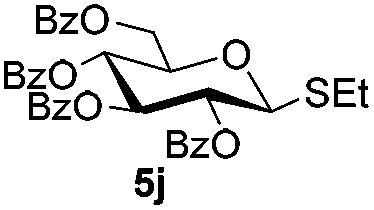
| 16 |
5j
|
6a
|
7b
|
85 |
| 17 |
5m
|
6l
|
7w
|
83 |
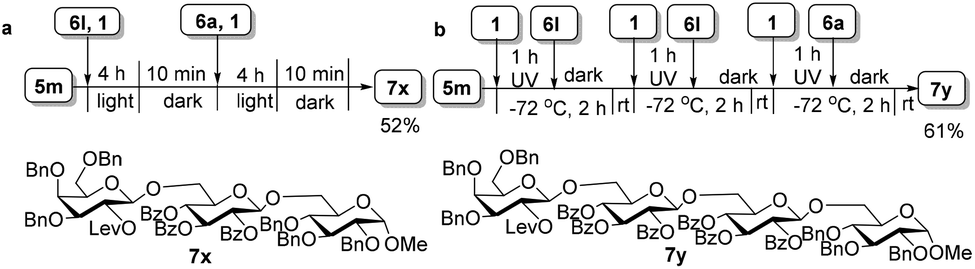
The high efficiency of the light-promoted glycosylations was further underlined by the one-pot assembly of trisaccharide 7x and tetrasaccharide 7y. As described in Fig. 3, the synthesis of trisaccharide 7xvia the reactivity-based one-pot strategy under visible-light irradiation was examined, providing trisaccharide 7x in 52% isolated yield (for more details, see ESI 3.5†). Under the irradiation of UV light, the preactivation-based one-pot oligosaccharide synthesis was also realized, and as exemplified by the synthesis of tetrasaccharide 7y, tetrasaccharide 7y was finally obtained in 61% isolated yield successfully without any intermediate isolation (for more details, see ESI 3.6†).
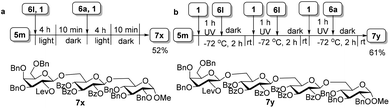 |
| | Fig. 3 One-pot synthesis of trisaccharide 7x and tetrasaccharide 7y. (a) Reactivity-based one-pot synthesis of trisaccharide 7x under the irradiation of visible light. (b) Preactivation-based one-pot synthesis of tetrasaccharide 7y under UV irradiation. Both yields are calculated based on the amount of 6a. | |
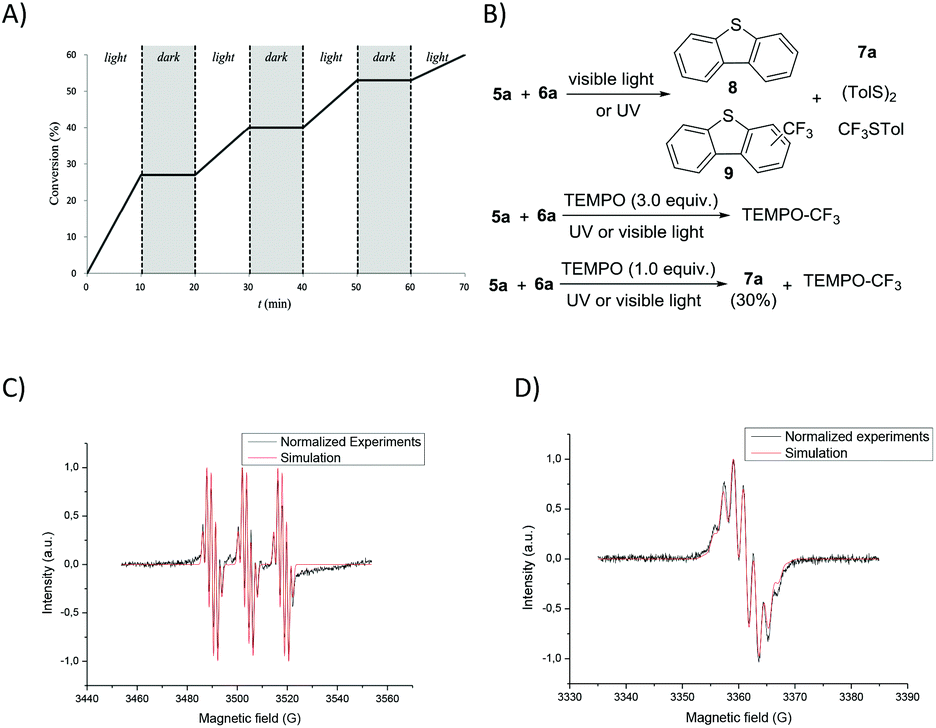
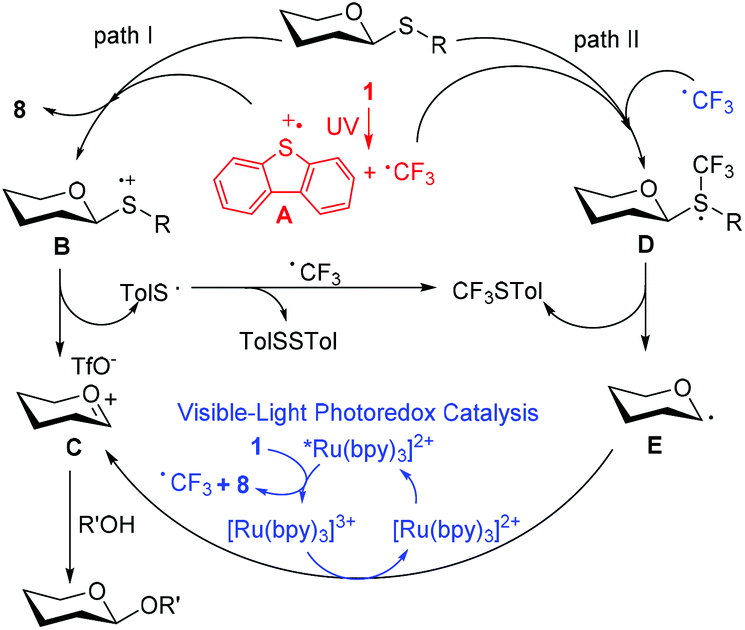
In order to gain mechanistic insight into the reaction, some preliminary mechanistic experiments were performed. On the removal of each individual component, light, Umemoto's reagent, or catalyst, the conversion of thioglycoside donors was suppressed completely. The light–dark interval reaction further illustrated the direct correlation between photolysis and the conversion of thioglycoside donors (Fig. 4A and ESI†). In the glycosylation reaction, dibenzothiophene (8), trifluoromethylated dibenzothiophenes (9), disulfide (TolSSTol), and p-tolyl(trifluoromethyl)sulfide were detected under the irradiation of UV light, whereas disulfide was absent under the irradiation of visible light. In the presence of a radical scavenger 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO, 3.0 equiv.), the conversion of the glycosyl donor was not detected even after prolonged exposure to light, and a TEMPO trapped CF3 adduct was detected by 19F NMR analysis.44 However, the glycosyl-TEMPO adduct was not detected in our experiments (Fig. 4B and ESI†). Furthermore, the ESR signals of the CF3 radical (aN = 14.2 G, aF (CF3) = 1.80 G, and aH = 1.19 G) were clearly observed by using N-tert-butyl-α-phenylnitrone (PBN) as a radical spin trapper under UV or visible light illumination at room temperature (Fig. 4C and the ESI†).45 Interestingly, the ESR signals of the dibenzothiophene radical cation (aH = 1.70 G), a well resolved seven-line spectrum, were observed when a solution of Umemoto's reagent was irradiated by UV light at 200 K (Fig. 4D and the ESI†).46 In the presence of thioglycoside donors, these signals disappeared under either visible or UV light illumination. These results showed that different radicals can be produced under different illuminations, that is, both CF3 radical and dibenzothiophene radical cation are produced under UV illumination, and only the CF3 radical is produced under visible light illumination. Replacement of 1 by either BrCCl3 or CBr4 led to no reaction, which might result from a different mechanism compared with the work of Ragains and Bowers (Table S2,† entries 17 and 18).25,47 The role of Cu(OTf)2 was also checked. It was found that Cu(OTf)2 played an important role in reducing the yield of 9 to increase the amount of CF3 radical in thioglycoside activation, instead of acting as an extra triflate source to stabilize the dioxalenium intermediate (Table S1, entry 44 and Scheme S1†). The Cu(OTf)2 played a far different role when compared with its function in our previous work.32
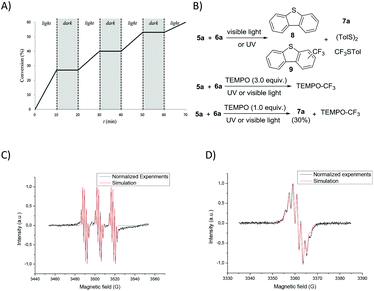 |
| | Fig. 4 Mechanistic investigations. (A) Light–dark interval experiments under the irradiation of visible light. (B) Control experiments in the presence of TEMPO. (C) EPR experiments for trapping the CF3 radical under the irradiation of visible light. The simulated parameters are similar to the literature report, with aN = 14.2 G, aF (CF3) = 1.80 G, and aH = 1.19 G. (D) Observation of the dibenzothiophene radical cation by EPR experiments at 200 K under the irradiation of UV light. The simulation was done with a radical coupled to eight equivalent hydrogen atoms. The hyperfine coupling aH = 1.70 G. | |
On the basis of the above observations and literature precedents,35,48,49 we proposed the possible mechanism (Fig. 5). In the presence of visible light, the reaction experienced a mechanism via path II. Firstly, Umemoto's reagent 1 was reduced by the excited state *[Ru(bpy)3]2+ to afford a CF3 radical, which reacted with the sulfur atom in thioglycoside donor to generate the radical intermediate D. The release of p-tolyl(trifluoromethyl)sulfide from D produced the glycosyl radical intermediate E, which was then oxidized by [Ru(bpy)3]3+ to give the glycosyl cation C. Finally, a glycosyl coupling reaction of the glycosyl cation C with an acceptor occurred. Under the irradiation of UV light, the homolytic generation of the CF3 radical and dibenzothiophene radical cation (A) was followed by a traditional glycosylation process (path I)50 and/or a CF3 radical-participated process (path II). The recombination of the CF3 radical and TolS radical or dimerization of the TolS radical provided p-tolyl(trifluoromethyl)sulfide or TolSSTol. The formation of a small amount of trifluoromethylated dibenzothiophenes (9) might result from the free-radical aromatic substitution reaction of dibenzothiophene with the CF3 radical. The observation that the UV-visible light absorbance of 1 is not affected by the presence of thioglycosides, may probably rule out the formation of an electron donor–acceptor complex between the thioglycoside and Umemoto's reagent.51
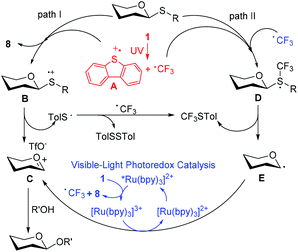 |
| | Fig. 5 A mechanistic proposal for the light-driven glycosylation. | |
Conclusions
In conclusion, an efficient and novel light-mediated glycosylation protocol using thioglycosides as glycosyl donors via the merger of a CF3 radical process has been developed. Our strategy, promoted by Umemoto's reagent 1 as the CF3 radical source under the irradiation of either UV or visible light, provides the glycosyl coupling products in high yields. Sunlight can also be used as the source of light. This strategy is able to efficiently activate thioglycosides, thereby exhibiting a wide substrate scope/generality. Noteworthily, it enables transformations that are completely inert under the traditional photo-induced glycosylation conditions, and is well tolerated with many protective groups. The high efficiency of this protocol is nicely highlighted by either the reactivity-based or the preactivation-based one-pot oligosaccharide synthesis. To the best of our knowledge, this is the most effective photo-driven glycosylation method. The results obtained by this protocol are comparable to and even in some cases better than those by the currently widely-used glycosylation methods. Moreover, the mechanistic studies demonstrated that this light-driven glycosylation underwent a radical process involving the CF3 radical and/or dibenzothiophene radical cation. Thus, this work not only sets up a new landmark of photo-glycochemistry, but also arouses a renewed interest in Umemoto's reagents and trifluoromethyl sulfonium salts as sources of radical cations.52 The disclosed protocol could find wide applications in oligosaccharide assembly and hold potential toward automated oligosaccharide synthesis.
Experimental
All reactions were performed under an argon atmosphere in an oven-dried quartz flask using standard Schlenk techniques, unless otherwise noted. Synthesis-grade solvents were used as purchased and the reaction mixtures were deoxygenated prior to illumination. Full experimental details and characterization data for all new compounds are included in the ESI.†
Acknowledgements
This work was financially supported by the grants (2012CB822100, 2013CB910700) from the Ministry of Science and Technology of China, the National Natural Science Foundation of China (Grant No. 21232002, 21572012), Beijing Natural Science Foundation (2142015), and Beijing Higher Education Young Elite Teacher Project (YETP0063). We thank Prof. Jingfen Lu and Dr Guoquan Liu for the assistance in EPR experiments and helpful discussions.
Notes and references
-
A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, 2009 Search PubMed.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046 CrossRef CAS PubMed.
-
A. V. Demchenko, Handbook of Chemical Glycosylation, Wiley–VC, 2008 Search PubMed.
- X.-M. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900 CrossRef CAS PubMed.
- B. Yu, J. S. Sun and X.-Y. Yang, Acc. Chem. Res., 2012, 45, 1227 CrossRef CAS PubMed.
- Y. Yang, X. Zhang and B. Yu, Nat. Prod. Rep., 2015, 32, 1331 RSC.
- Y. Wang, X.-S. Ye and L.-H. Zhang, Org. Biomol. Chem., 2007, 5, 2189 CAS.
- T. J. Boltje, T. Buskas and G. J. Boons, Nat. Chem., 2009, 1, 611 CrossRef CAS PubMed.
- C.-H. Hsu, S.-C. Hung, C.-Y. Wu and C.-H. Wong, Angew. Chem., Int. Ed., 2011, 50, 11872 CrossRef CAS PubMed.
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523 CrossRef CAS PubMed.
- P. H. Seeberger, Acc. Chem. Res., 2015, 48, 1450 CrossRef CAS PubMed.
- D. Crich, Acc. Chem. Res., 2010, 43, 1144 CrossRef CAS PubMed.
- M. Huang, G. E. Garrett, N. Birlirakis, L. Bohé, D. A. Pratt and D. Crich, Nat. Chem., 2012, 4, 663 CrossRef CAS PubMed.
- T. G. Frihed, M. Bols and C. M. Pedersen, Chem. Rev., 2015, 115, 4963 CrossRef CAS PubMed.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052 CrossRef CAS PubMed.
- T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000 CrossRef CAS PubMed.
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872 CrossRef CAS PubMed.
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725 CrossRef CAS PubMed.
- J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750 CrossRef CAS PubMed.
- W. J. Wever, M. A. Cinelli and A. A. Bowers, Org. Lett., 2013, 15, 30 CrossRef CAS PubMed.
- M. Nakanishi, D. Takahashi and K. Toshima, Org. Biomol. Chem., 2013, 11, 5079 CAS.
- G. W. Griffin, N. C. Bandara, M. A. Clarke, W.-S. Tsang, P. J. Garegg, S. Oscarson and B. A. Silwanis, Heterocycles, 1990, 30, 939 CrossRef CAS.
- S. Hashimoto, I. Kurimoto, Y. Fujii and R. Noyori, J. Am. Chem. Soc., 1985, 107, 1427 CrossRef CAS.
- I. Cumptsey and D. Crich, J. Carbohydr. Chem., 2011, 30, 469 CrossRef.
- R. Iwata, K. Uda, D. Takahashi and K. Toshima, Chem. Commun., 2014, 10695 RSC.
- T. Furuta, K. Takeuchi; and M. Iwamura, Chem. Commun., 1996, 157 RSC.
- R.-Z. Mao, F. Guo, D.-C. Xiong, Q. Li, J. Duan and X.-S. Ye, Org. Lett., 2015, 17, 5606 CrossRef CAS PubMed.
- A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed.
- T. Koike and M. Akita, Top. Catal., 2014, 57, 967 CrossRef CAS.
- N. J. Straathof, B. J. Tegelbeckers, V. Hessel, X. Wang and T. Noel, Chem. Sci., 2014, 5, 4768 RSC.
- T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156 CrossRef CAS.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2014, 115, 650 CrossRef PubMed.
- I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754 CrossRef CAS PubMed.
- P. Eisenberger, S. Gischig and A. Togni, Chem. – Eur. J., 2006, 12, 2579 CrossRef CAS PubMed.
- X. Huang, L. Huang, H. Wang and X.-S. Ye, Angew. Chem., Int. Ed., 2004, 43, 5221 CrossRef CAS PubMed.
- L. Yang, Q. Qin and X.-S. Ye, Asian J. Org. Chem., 2013, 2, 30 CrossRef CAS.
- D. Crich and S.-X. Sun, J. Am. Chem. Soc., 1998, 120, 435 CrossRef CAS.
- P. De Pouilly, A. Chenede, J.-M. Mallet and P. Sinay, Bull. Soc. Chim. Fr., 1993, 130, 256 CAS.
- X. Wang, Y. Ye, S. Zhang, J. Feng, Y. Xu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 16410 CrossRef CAS PubMed.
- C.-P. Zhang, H. Wang, A. Klein, C. Biewer, K. Stirnat, Y. Yamaguchi, L. Xu, V. Gomez-Benitez and D. A. Vicic, J. Am. Chem. Soc., 2013, 135, 8141 CrossRef CAS PubMed.
- L. Eberson, M. P. Hartshorn, O. Persson and F. Radner, Acta Chem. Scand., 1997, 51, 492 CrossRef CAS.
- M. Spell, X.-P. Wang, A. E. Wahba, E. Conner and J. Ragains, Carbohydr. Res., 2013, 369, 42 CrossRef CAS PubMed.
- T. Billard, N. Roques and B. R. Langlois, Tetrahedron Lett., 2000, 41, 3069 CrossRef CAS.
- T. Billard, N. Roques and B. R. Langlois, J. Org. Chem., 1999, 64, 3813 CrossRef CAS.
- T. Nokami, T. Itoh and K. K. T. Mong, Isr. J. Chem., 2015, 55, 297 CrossRef CAS.
- Y. Cheng, X. Yuan, J. Ma and S. Yu, Chem. – Eur. J., 2015, 21, 8355 CrossRef CAS PubMed.
- M. Schmittel and A. Burghart, Angew. Chem., Int. Ed. Engl., 1997, 36, 2550 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00021e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.