
A novel calix[4]arene-based bifunctional squaramide organocatalyst for enantioselective Michael addition of acetylacetone to nitroolefins†
Ummu
Vural
,
Mustafa
Durmaz
* and
Abdulkadir
Sirit
Department of Chemistry, Ahmet Kelesoglu Education Faculty, Necmettin Erbakan University, 42090 Konya, Turkey. E-mail: mdurmaz@konya.edu.tr
First published on 12th April 2016
Abstract
A new chiral calix[4]arene-based organocatalyst 3 bearing bis-squaramide moieties was designed and synthesized from p-tert-butylcalix[4]arene. This bifunctional squaramide organocatalyst was used in the enantioselective conjugate addition of acetylacetone to β-nitrostyrenes. The corresponding adducts were obtained in good to excellent yields with high enantioselectivities (up to 94% ee).
Introduction
The Michael addition of 1,3-dicarbonyl compounds to electron deficient nitroolefins is one of the most efficient carbon–carbon bond-forming reactions in organic chemistry since the resulting γ-nitrocarbonyl compounds are valuable intermediates which could be used for the preparation of a large number of pharmaceutical substances.1 Of the organocatalysts developed for this transformation, chiral bifunctional organocatalysts which utilize hydrogen bonding activation have attracted intense research efforts in the past decade.2 Particularly, the field of hydrogen bonding was dominated by chiral thioureas, ureas, diols, phosphoric acids and amidinium cations and great advances were achieved.3 Although the thiourea core has been considered as the gold mine of H-bond donor catalysts, the chiral squaramide organocatalysts developed by Rawal's group and later exploited by other groups have been determined as other effective dual H-bond donor catalysts in recent years.4 In addition to their dual proton donating capacity, rigidity, H-bond spacing, H-bond angle and pKa, their highly modular nature and ease of synthesis make them superior to other well-organized alternatives.5 Consequently, a wide variety of chiral squaramide-derived bifunctional organocatalysts have been efficiently applied to numerous organic transformations.6Calixarenes, the third generation of supramolecules, have been widely used for the construction of artificial host molecules, which have found numerous applications in various fields.7 The introduction of chiral substituents into the calixarene skeleton either by attaching chiral units at one of the calix rims or by synthesizing ‘inherently’ chiral derivatives could, in turn, lead to chirality of the artificial receptors. Although many chiral calixarenes containing chiral units at either the wide or the narrow rim have been prepared as chiral receptors and sensors,8 only a few calix[4]arene derivatives have been reported as efficient organocatalysts for asymmetric transformations.9
Recently, research groups of Jiang and Gaeta reported the first examples of calix[4]arene-based receptors in which two squaramide moieties were introduced at the endo (or lower) and exo (or upper) rims, and their recognition properties toward anionic guests, respectively .10 To further extend the application of calix[4]arene-based squaramides, herein we report the design and synthesis of a chiral organocatalyst based on a calix[4]arene scaffold carrying squaramide and tertiary amine as catalytic functionalities and its ability to serve as an organocatalyst in the asymmetric Michael addition of acetylacetone to nitroolefins.
Results and discussion
Synthesis
The synthesis of chiral calix[4]arene-based squaramide 3 is outlined in Scheme 1. We envisioned that calix[4]arene squarate 2 could be used as the starting material for the preparation of the target squaramide. First, the known precursor calix[4]arene diamine 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 was coupled with dimethyl squarate in MeOH as the solvent to give calix[4]arene squarate 2 in 67% yield. Subsequent condensation of 2 with (1S,2S)-N,N-dimethyl-1,2-diaminocyclohexane12 in DCM under mild conditions afforded the calixarene-squaramide 3 in 58% yield.
11 was coupled with dimethyl squarate in MeOH as the solvent to give calix[4]arene squarate 2 in 67% yield. Subsequent condensation of 2 with (1S,2S)-N,N-dimethyl-1,2-diaminocyclohexane12 in DCM under mild conditions afforded the calixarene-squaramide 3 in 58% yield.
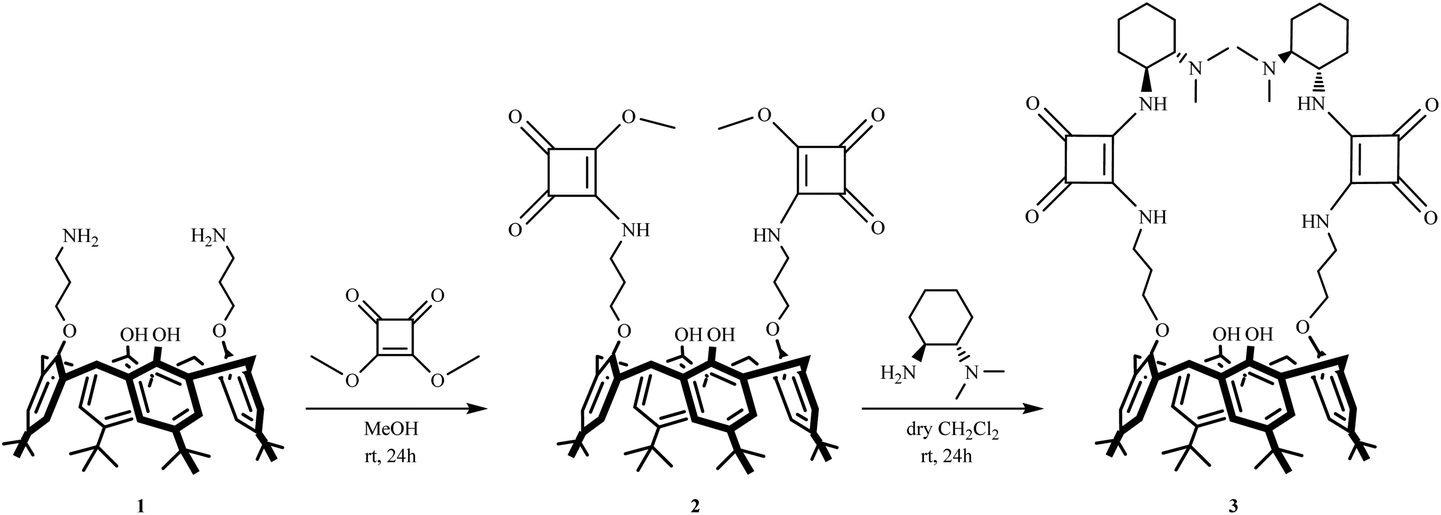 | ||
| Scheme 1 Synthesis of p-tert-butylcalix[4]arene squarate 2 and chiral squaramide 3 derivatives. | ||
The structures of newly synthesized calix[4]arene derivatives 2 and 3 were readily confirmed by spectral analysis. 1H NMR spectra of calix[4]arene squarate 2 showed a singlet for methoxy protons at 4.40 ppm and two sets of doublets due to the bridging methylene protons and two sets of singlets due to the tert-butyl groups.
In the lower field of the 1H NMR spectra of calix[4]arene squaramide 3, NH signals for the squaramide moiety were observed. In addition, the 1H and 13C NMR spectra also indicate that these macrocycles preferably adopt a cone conformation.
Enantioselective asymmetric Michael addition of 2,4-pentanedione to nitroalkenes
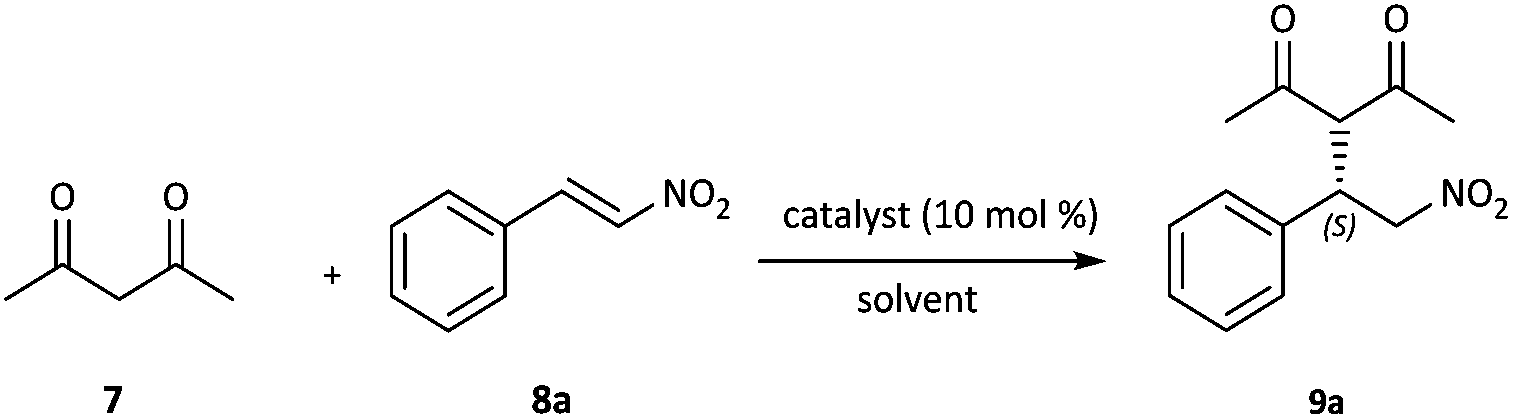
Squaramide-derived bifunctional organocatalysts bearing chirality inducers such as chiral diamines, amino alcohols, binaphthyls and cinchona alkaloids or multiple H-bond donors which might enhance enantiocontrol have been developed and successfully applied in the enantioselective Michael addition of acetylacetone to nitroolefins.13 Hence, the catalytic activity of 3 was initially evaluated in the conjugate addition of acetylacetone 7 to trans-β-nitrostyrene 8a and the results are summarized in Table 1. The Michael addition reaction proceeded smoothly in the presence of 10 mol% 3 in toluene at room temperature; after 4 h the product was obtained in good yield and ee (Table 1, entry 1). A brief survey of solvents revealed that both the yield and the enantioselectivity were highly dependent on the solvent. Aprotic solvents such as toluene, CH2Cl2, CHCl3, CCl4 and 1,2-dichloroethane resulted in higher product yields (Table 1, entries 1–5) relative to protic and more polar solvents such as CH3CN and MeOH (Table 1, entries 7 and 10). In addition, when the reaction was performed in brine, lower yield and enantioselectivity were obtained (Table 1, entry 11). This could be due to the competitive establishment of hydrogen bonding interactions between the protic solvents and substrates.14 The reaction could also be performed without a solvent. In this case, a slight decrease was observed in terms of chemical yield and selectivity (Table 1, entry 12). Thus, CH2Cl2, being both nonpolar and non-coordinating was found to be the best solvent. Running the reaction at 0 °C or in the presence of 5 mol% catalyst led to longer reaction times and some loss in yield and selectivity (Table 1, entries 13 and 14).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time [h] | Yieldb [%] | eec [%] |
| a The reactions were performed with 2 equiv. of 2,4-pentanedione and 1 equiv. of trans-β-nitrostyrene in 0.25 mL of the solvent in the presence of 10 mol% catalyst at room temperature for 4–24 h. b Isolated yields. c Determined by chiral HPLC analysis (Chiralpak AS-H), and the configuration was assigned by comparison of the retention time and specific rotation with literature data. d Reactions were performed at 0 °C. e 5 mol% catalyst was used. f 20 mol% catalyst was used. | |||||
| 1 | 3 | Toluene | 4 | 84 | 87 |
| 2 | 3 | CH2Cl2 | 4 | 91 | 91 |
| 3 | 3 | CHCl3 | 6 | 90 | 83 |
| 4 | 3 | CCl4 | 6 | 88 | 81 |
| 5 | 3 | ClCH2CH2Cl | 6 | 89 | 79 |
| 6 | 3 | EtOAc | 8 | 72 | 58 |
| 7 | 3 | CH3CN | 8 | 54 | 67 |
| 8 | 3 | THF | 12 | 68 | 74 |
| 9 | 3 | Et2O | 8 | 64 | 61 |
| 10 | 3 | MeOH | 24 | 25 | 27 |
| 11 | 3 | Brine | 4 | 45 | 70 |
| 12 | 3 | Neat | 6 | 79 | 82 |
| 13d | 3 | CH2Cl2 | 12 | 88 | 86 |
| 14e | 3 | CH2Cl2 | 16 | 86 | 84 |
| 15 | 6 | CH2Cl2 | 8 | 71 | 72 |
| 16f | 6 | CH2Cl2 | 8 | 74 | 77 |
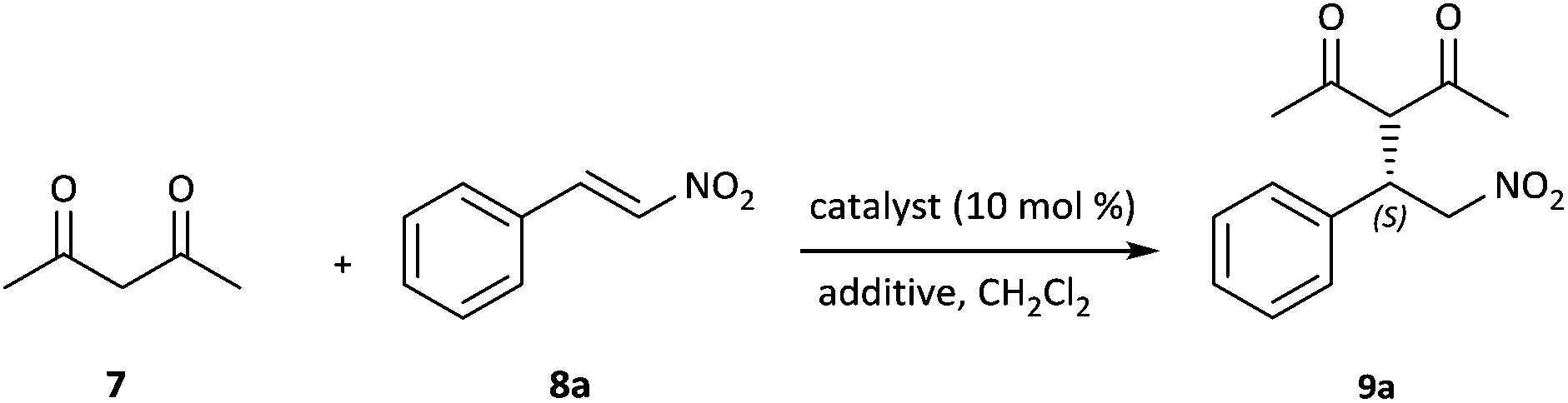
Typically, it is known that the presence of co-catalysts or additives has a significant effect on catalytic asymmetric reactions.15 Therefore, different additives were investigated to determine the optimal reaction conditions (Table 2, entries 1–9). From Table 2, we can see that the chemical yields and enantioselectivities varied greatly depending on the nature of the additive used. In each case, both yields and ee values were inferior.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Additive | Time [h] | Yieldb [%] | eec [%] |
| a The reactions were performed with 2 equiv. of 2,4-pentanedione and 1 equiv. of trans-β-nitrostyrene in 0.25 mL of CH2Cl2 in the presence of 10 mol% catalyst and 15 mol% additive at room temperature for 4–24 h. b Isolated yields. c Determined by chiral HPLC analysis (Chiralpak AS-H), and the configuration was assigned by comparison of the retention time and specific rotation with literature data. | |||||
| 1 | 3 | H2O | 4 | 88 | 61 |
| 2 | 3 | CH3COOH | 4 | 74 | 69 |
| 3 | 3 | PhCOOH | 6 | 65 | 72 |
| 4 | 3 | (S)-1,1′-Bi-2-naphthol | 6 | 67 | 74 |
| 5 | 3 | L-Proline | 12 | 70 | 58 |
| 6 | 3 | TFA | 8 | <5 | n.d. |
| 7 | 3 | p-TsOH | 24 | 59 | 63 |
| 8 | 3 | Pyridine | 24 | 73 | 59 |
| 9 | 3 | Et3N | 16 | 58 | 46 |
| 10 | 6 | p-tert-Butylphenol | 6 | 77 | 79 |
| 11 | 6 | Phenol | 6 | 85 | 83 |
In order to confirm the synergistic effect of the bifunctional organocatalyst 3 on the enantioselective Michael addition, the monomeric analogue 6 carrying both squaramide and tertiary amine subunits was also prepared starting from 3-(p-tert-butylphenoxy)propane amine 4 in two steps according to the procedures shown in Scheme 2 and used in the control experiments. In the first key control experiment, we examined the monomeric analog 6 as the catalyst, but lower yield and enantioselectivity were observed (Table 1, entry 15).
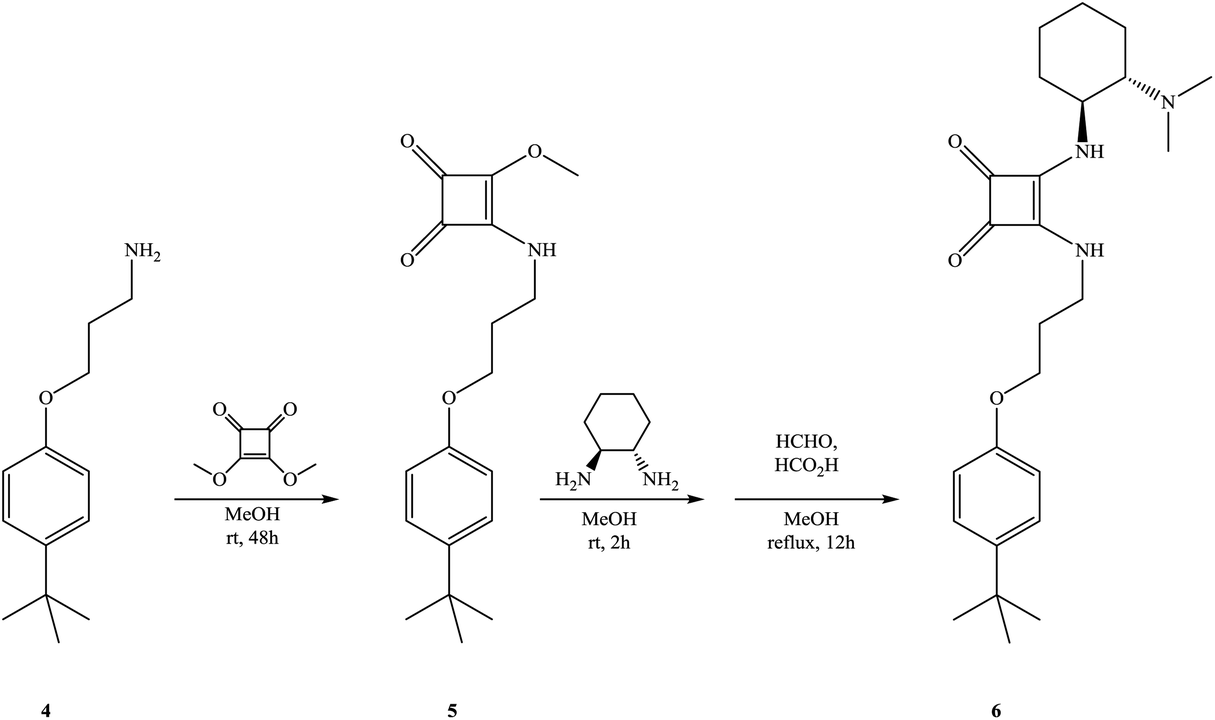 | ||
| Scheme 2 Synthesis of chiral p-tert-butylphenol analogue 6. | ||
To mimic the role of calixarene-squaramide 3, when 20 mol% of chiral analogue 6 was used, both the yield and enantioselectivity increased slightly (Table 1, entry 16). Control experiments using 6 as a catalyst and p-tert-butyl phenol or phenol as acidic additives were also performed (Table 2, entries 10 and 11) and the results clearly confirm the cooperative effect and special role of the calixarene skeleton on the enantioselective addition reaction.
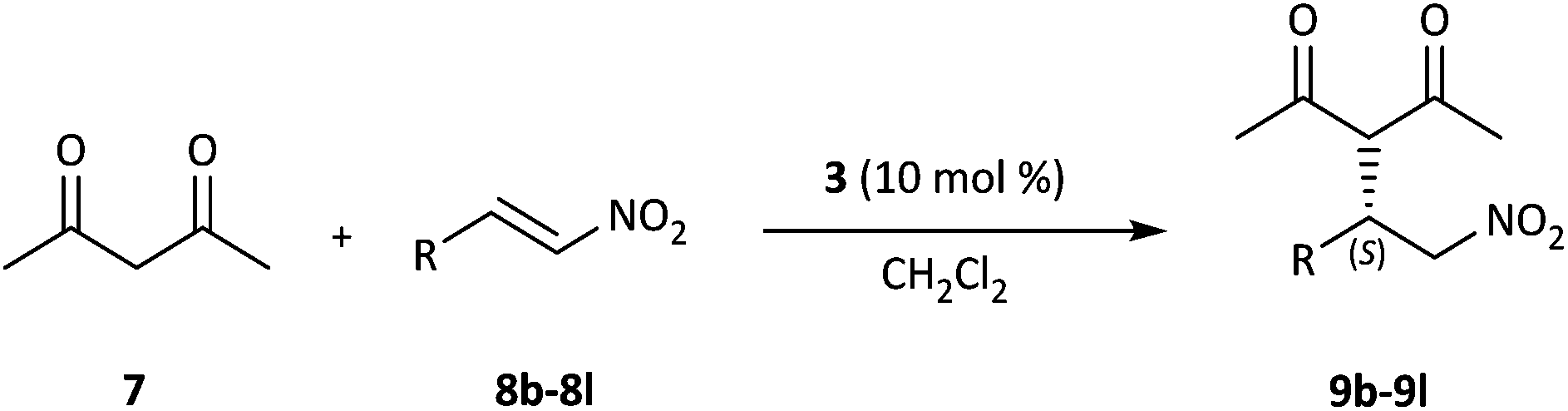
Under the optimized reaction conditions, we subsequently probed the substrate scope for the calixarene-squaramide 3-catalyzed enantioselective Michael addition reaction. A variety of substituted aromatic β-nitrostyrene derivatives 8b–8l, which bear electron-rich and electron-withdrawing groups as the electrophiles reacted smoothly with 2,4-pentanedione 7 to give the desired Michael adducts 9b–9l in 50–99% yields and with different enantioselectivities (Table 3, entries 1–11). Notably, the position and the electronic properties of the electron-withdrawing substituents (Br, F, Cl) are well tolerated by the conjugate addition reactions (Table 3, entries 5–11). But, the reactions of nitroalkenes 8c–8e bearing an electron-donating methoxy group on the benzene ring lowered both the yield and enantiomeric excess of the reaction (Table 3, entries 2–4).
|
|
||||
|---|---|---|---|---|
| Entry | R | Product | Yieldb [%] | eec [%] |
| a The reactions were performed with 2 equiv. of 2,4-pentanedione 7 and 1 equiv. of 8b–8l in 0.25 mL of CH2Cl2 in the presence of 10 mol% 3 at room temperature. b Isolated yields. c Determined by chiral HPLC analysis, and the configuration was assigned by comparison of the retention time and specific rotation with literature data. | ||||
| 1 | 4-Me-C6H4 (8b) | 9b | 76 | 74 (S) |
| 2 | 2-MeO-C6H4 (8c) | 9c | 50 | 68 (S) |
| 3 | 3-MeO-C6H4 (8d) | 9d | 50 | 70 (S) |
| 4 | 4-MeO-C6H4 (8e) | 9e | 58 | 72 (S) |
| 5 | 2-Br-C6H4 (8f) | 9f | 80 | 87 (S) |
| 6 | 3-Br-C6H4 (8g) | 9g | 84 | 85 (S) |
| 7 | 4-Br-C6H4 (8h) | 9h | 99 | 80 (S) |
| 8 | 2-F-C6H4 (8i) | 9i | 60 | 72 (S) |
| 9 | 4-F-C6H4 (8j) | 9j | 98 | 81 (S) |
| 10 | 4-Cl-C6H4 (8k) | 9k | 98 | 94 (S) |
| 11 | 2,4-Cl-C6H3 (8l) | 9l | 87 | 55 (S) |
On the basis of the results observed with other bifunctional organocatalysts and the observations obtained in this work, we propose a dual activation model for the Michael addition. First, the backbone of the calixarene catalyst was assumed to create equal reaction sites surrounded by two squaramides,16 in which the squaramide functionality interacts through hydrogen bondings with the nitro group of the nitroalkene, meanwhile the phenolic hydroxyl groups of the catalyst and the nitro group may form another hydrogen bond, which increase the electrophilicity of nitrostyrene and preorganise the reaction substrates. On the other hand, the tertiary amine deprotonates an acidic proton of acetylacetone and forms a ternary complex. However, the precise mechanism still needs further mechanistic study.
Conclusions
In summary, a new class of chiral calix[4]arene with bis-squaramide moieties has been developed and its ability as a bifunctional organocatalyst to catalyze the enantioselective conjugate addition of acetylacetone to various nitroolefins at room temperature is further evaluated. Catalyst 3 exhibited high performance in the asymmetric addition reaction and the corresponding products can be obtained in moderate to excellent yields with good enantioselectivities. Further studies to clarify the mechanism and catalytic performance of these types of catalysts in other asymmetric reactions are currently underway.Experimental
General
Melting points were determined on an Electrothermal 9100 apparatus in a sealed capillary. 1H and 13C NMR spectra were recorded in CDCl3 at room temperature on a Varian 400 MHz spectrometer. Chemical shifts were reported in ppm. Data were reported as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, br = broad), coupling constants (Hz). Fourier transform infrared (IR) spectra were recorded on a Thermo Scientific Nicolet iS5 spectrometer equipped with an ATR unit and are reported in wavenumbers (cm−1). HPLC measurements were carried out on Agilent 1100 equipment connected with Chiralpak Daicel AD-H, OD-H and AS-H columns. Optical rotations were measured on an Atago AP-100 digital polarimeter using a 1 dm cell. Elemental analyses were performed using a Leco CHNS-932 analyzer. Analytical TLC was performed using Merck prepared plates (silica gel 60 F254 on aluminum). Flash chromatography separations were performed on a Merck Silica Gel 60 (230–400 Mesh). All starting materials and reagents used were of standard analytical grade from Fluka, Merck, Aldrich, Acros or TCI and used without further purification. Dichloromethane was dried (CaCl2), distilled from CaH2 and stored over molecular sieves. Other commercial grade solvents were distilled, and then stored over molecular sieves. The drying agent employed was anhydrous MgSO4.Synthesis
General procedure for the enantioselective Michael addition
2,4-Pentanedione 7 (0.2 mmol) was added to a mixture of catalyst 3 (12 mg, 0.01 mmol, 10 mol%) and the corresponding nitroalkene 8 (0.1 mmol) in 0.25 mL CH2Cl2. After the reaction was completed (monitored by TLC), the resulting mixture was concentrated and the residue was subjected to column chromatography on silica gel (eluent, hexane/ethyl acetate) to afford the pure products 9a–9l. Compounds 9a–9l are known and the analytical and spectroscopic data are in accordance with those reported.4a,17 The enantiomeric excess of the products was determined by chiral HPLC analysis using chiral columns. Racemic samples of the Michael adducts were prepared using racemic catalysts.:15, flow rate: 1.0 mL min−1, wavelength = 210 nm: tR (major) = 15.1 min, tR (minor) = 24.1 min].
:10, flow rate: 1.0 mL min−1, wavelength = 210 nm: tR (major) = 10.3 min, tR (minor) = 16.5 min].
:3, flow rate: 0.5 mL min−1, wavelength = 210 nm: tR (major) = 37.9 min, tR (minor) = 40.9 min].
:15, flow rate: 1.0 mL min−1, wavelength = 210 nm: tR (major) = 9.4 min, tR (minor) = 11.7 min].
:10, flow rate: 1.0 mL min−1, wavelength = 210 nm: tR (major) = 15.2 min, tR (minor) = 23.4 min].
:3, flow rate: 0.5 mL min−1, wavelength = 220 nm: tR (major) = 21.8 min, tR (minor) = 27.3 min].
:15, flow rate: 1.0 mL min−1, wavelength = 210 nm: tR (major) = 17.7 min, tR (minor) = 33.4 min].
:15, flow rate: 1.0 mL min−1, wavelength = 210 nm: tR (major) = 16.8 min, tR (minor) = 29.9 min].
:10, flow rate: 1.0 mL min−1, wavelength = 220 nm: tR (major) = 10.2 min, tR (minor) = 11.5 min].
:10, flow rate: 1.0 mL min−1, wavelength = 210 nm: tR (major) = 13.1 min, tR (minor) = 25.1 min].
:15, flow rate: 1.0 mL min−1, wavelength = 220 nm: tR (major) = 14.6 min, tR (minor) = 29.1 min].
:10, flow rate: 1.0 mL min−1, wavelength = 210 nm: tR (major) = 9.7 min, tR (minor) = 12.0 min].
Acknowledgements
This work was supported by the Research Foundation of Necmettin Erbakan University (BAP-151210006) and the Scientific and Technological Research Council of Turkey (TUBITAK).Notes and references
- (a) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701 CrossRef CAS; (b) S. R. Harutyunyan, T. D. Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824 CrossRef CAS PubMed; (c) Y. Zhang and W. Wang, Catal. Sci. Technol., 2012, 2, 42 RSC.
- (a) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS PubMed; (b) F. Giacalone, M. Gruttadauria, P. Agrigento and R. Noto, Chem. Soc. Rev., 2012, 41, 2406 RSC; (c) X. Fang and C. J. Wang, Chem. Commun., 2015, 51, 1185 RSC; (d) C. Najera and M. Yus, Tetrahedron Lett., 2015, 56, 2623 CrossRef CAS.
- (a) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed; (b) O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051 RSC; (c) X. Yu and W. Wang, Chem. – Asian J., 2008, 3, 516 CrossRef CAS PubMed; (d) Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187 RSC.
- (a) J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS PubMed; (b) H. Konishi, T. Y. Lam, J. P. Malerich and V. H. Rawal, Org. Lett., 2010, 12, 2028 CrossRef CAS PubMed; (c) K. S. Yang, A. E. Nibbs, Y. E. Turkmen and V. H. Rawal, J. Am. Chem. Soc., 2013, 135, 16050 CrossRef CAS PubMed.
- For recent reviews on squaramides, see: (a) P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Adv. Synth. Catal., 2015, 357, 253 CrossRef CAS; (b) J. Aleman, A. Parra, H. Jiang and K. A. Jorgensen, Chem. – Eur. J., 2011, 17, 6890 CrossRef CAS PubMed; (c) R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330 RSC.
- (a) Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153 CrossRef CAS PubMed; (b) C. Min, X. Han, Z. Liao, X. Wu, H.-B. Zhou and C. Dong, Adv. Synth. Catal., 2011, 353, 2715 CrossRef CAS; (c) M. Isik, M. Y. Unver and C. Tanyeli, J. Org. Chem., 2015, 80, 828 CrossRef CAS PubMed; (d) G. Kardos and T. Soos, Eur. J. Org. Chem., 2013, 4490 CrossRef CAS.
- (a) C. D. Gutsche, Calixarenes: An Introduction, The Royal Society of Chemistry, Thomas Graham House, Cambridge, UK, 2nd edn, 2008 Search PubMed; (b) S. B. Nimse and T. Kim, Chem. Soc. Rev., 2013, 42, 366 RSC; (c) M. Yilmaz and S. Erdemir, Turk. J. Chem., 2013, 37, 558 CAS; (d) I. Oueslati, Curr. Org. Chem., 2015, 19, 2309 CrossRef CAS; (e) L. Mutihac, J. H. Lee, J. S. Kim and J. Vicens, Chem. Soc. Rev., 2011, 40, 2777 RSC; (f) M. Song, Z. Sun, C. Han, D. Tian, H. Li and J. S. Kim, Chem. – Asian J., 2014, 9, 2344 CrossRef CAS PubMed.
- (a) A. Sirit and M. Yilmaz, Turk. J. Chem., 2009, 33, 159 CAS; (b) S.-Y. Li, Y.-W. Xu, J.-M. Liu and C.-Y. Su, Int. J. Mol. Sci., 2011, 12, 429 CrossRef CAS PubMed; (c) S.-Y. Li, Q.-Y. Zheng, C.-F. Chen and Z.-T. Huang, Tetrahedron: Asymmetry, 2005, 16, 2816 CrossRef CAS; (d) J. Luo, Q.-Y. Zheng, C.-F. Chen and Z.-T. Huang, Tetrahedron, 2005, 61, 8517 CrossRef CAS.
- (a) Z.-X. Xu, G.-K. Li, C.-F. Chen and Z.-T. Huang, Tetrahedron, 2008, 64, 8668 CrossRef CAS; (b) R. Miao, Z.-X. Xu, Z.-T. Huang and C.-F. Chen, Sci. China, Ser. B: Chem., 2009, 52, 505 CrossRef CAS; (c) Z.-Y. Li, J.-W. Chen, Y. Liu, W. Xia and L. Wang, Curr. Org. Chem., 2011, 15, 39 CrossRef CAS; (d) Z.-Y. Li, J.-W. Chen, L. Wang and Y. Pan, Synlett, 2009, 2356 CAS; (e) Z.-Y. Li, C.-X. Lu, G. Huang, J.-J. Ma, H. Sun, L. Wang and Y. Pan, Lett. Org. Chem., 2010, 7, 461 CrossRef CAS; (f) E. Akceylan, A. Uyanik, S. Eymur, O. Sahin and M. Yilmaz, Appl. Catal., 2015, 499, 205 CrossRef CAS; (g) M. Durmaz, A. Tataroglu, H. Yilmaz and A. Sirit, Tetrahedron: Asymmetry, 2016, 27, 148 CrossRef CAS; (h) M. Durmaz and A. Sirit, Supramol. Chem., 2013, 25, 292 CrossRef CAS; (i) M. Durmaz and A. Sirit, Tetrahedron: Asymmetry, 2013, 24, 1443 CrossRef CAS.
- (a) C. Jin, M. Zhang, C. Deng, Y. Guan, J. Gong, D. Zhu, Y. Pan, J. Jiang and L. Wang, Tetrahedron Lett., 2013, 54, 796 CrossRef CAS; (b) C. Gaeta, C. Talotta, P. D. Sala, L. Margarucci, A. Casapullo and P. Neri, J. Org. Chem., 2014, 79, 3704 CrossRef CAS PubMed.
- Y. Lin, A. Leydier, E. Metay, A. Favre-Reguillon, D. Bouchu, S. Pellet-Rostaing and M. Lemaire, J. Inclusion. Phenom. Macrocyclic Chem., 2008, 61, 187 CrossRef CAS.
- (a) M. Kaik and J. Gawronski, Tetrahedron: Asymmetry, 2003, 14, 1559 CrossRef CAS; (b) A. Berkessel, S. Mukherjee, T. N. Müller, F. Cleemann, K. Roland, M. Brandenburg, J.-M. Neudörfl and J. Lee, Org. Biomol. Chem., 2006, 4, 4319 RSC.
- (a) Z. Dong, X. Jin, P. Wang, C. Min, J. Zhang, Z. Chen, H.-B. Zhou and C. Donga, ARKIVOC, 2011, 367 CAS; (b) Z. Dong, G. Qiu, H.-B. Zhou and C. Dong, Tetrahedron: Asymmetry, 2012, 23, 1550 CrossRef CAS; (c) H. Y. Bae, S. Some, J. S. Oh, Y. S. Lee and C. E. Song, Chem. Commun., 2011, 9621 RSC; (d) X. Xu, T. Cheng, X. Liu, J. Xu, R. Jin and G. Liu, ACS Catal., 2014, 4, 2137 CrossRef CAS; (e) B. Liu, X. Han, Z. Dong, H. Lv, H.-B. Zhou and C. Dong, Tetrahedron: Asymmetry, 2013, 24, 1276 CrossRef CAS.
- (a) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS PubMed; (b) J. M. Andres, R. Manzano and R. Pedrosa, Chem. – Eur. J., 2008, 14, 5116 CrossRef CAS PubMed; (c) Z.-W. Ma, Y.-X. Liu, L.-J. Huo, X. Gao and J.-C. Tao, Tetrahedron: Asymmetry, 2012, 23, 443 CrossRef CAS.
- (a) S. Hanessian and V. Pham, Org. Lett., 2000, 2, 2975 CrossRef CAS PubMed; (b) X. Shi, W. He, H. Li, X. Zhang and S. Zhang, Tetrahedron Lett., 2011, 52, 3204 CrossRef CAS; (c) Y. Liu, X. Wang and W. He, Org. Biomol. Chem., 2014, 12, 3163 RSC; (d) L. Hong, W. Sun, D. Yang, G. Li and R. Wang, Chem. Rev., 2016, 116, 4006 CrossRef CAS PubMed.
- K. Murai, S. Fukushima, S. Hayashi, Y. Takahara and H. Fujioka, Org. Lett., 2010, 12, 964 CrossRef CAS PubMed.
- (a) P. Gao, C. Wang, Y. Wu, Z. Zhou and C. Tang, Eur. J. Org. Chem., 2008, 4563 CrossRef CAS; (b) K. S. Rao, R. Trivedi and M. L. Kantam, Synlett, 2015, 221 CAS; (c) C. J. Wang, Z. H. Zhang, X. Q. Dong and X. J. Wu, Chem. Commun., 2008, 1431 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all new compounds and compounds 9a–9l. HPLC diagrams of compounds 9a–9l. See DOI: 10.1039/c6qo00135a |
| This journal is © the Partner Organisations 2016 |