
A versatile access to vicinal diamine motifs by highly anti-selective asymmetric vinylogous Mannich reactions: an efficient total synthesis of (+)-absouline†
Jian-Liang
Ye
*,
Hang
Chen‡
,
Yu-Feng
Zhang‡
and
Pei-Qiang
Huang
*
Department of Chemistry and Fujian Provincial Key Laboratory of Chemical Biology, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, P. R. China. E-mail: pqhuang@xmu.edu.cn; yejl@xmu.edu.cn
First published on 18th March 2016
Abstract
We report the asymmetric vinylogous Mannich reactions (VMRs) of N-Boc-2-tert-(butyldimethylsilyloxy)pyrrole (TBSOP) with N-tert-butanesulfinylimines. The reaction is highly anti-diastereoselective and shows good generality for the direct construction of a variety of vicinal anti-diamine motifs that are found in a number of biologically active alkaloids and medicinal agents. A VMR adduct was elaborated in six steps into the 1-aminopyrrolizidine alkaloid (+)-absouline, which constitutes the second and also the most efficient total synthesis of the natural enantiomer of the title alkaloid.
Introduction
anti-Vicinal diamines in general,1 and 1-aminopyrrolizidines2–6 in particular are structural motifs found in many biologically active alkaloids and medicinal agents.1–6 For example, 1-aminopyrrolizidine alkaloids (+)-absouline (E-1), (+)-isoabsouline (Z-1) (Fig. 1) and their N-oxides are found in two Caledonian plants Hugonia oreogena and Hugonia penicillanthemum,2a which were reported to exhibit modest antiviral activity.2a,b (+)-Laburnamine (2),3 which differs only at the acyl group from (+)-absouline (1), is found in Cytisus laburnum seeds3a,b and in Laburnum anagyroides seeds,3c and showed middle to high affinity to the α3β4, α7 and α4β2 nicotinic receptor subtype.3c Loline (3) is a representative of a group of thirteen alkaloids produced by mutualistic fungal endophytes living on forage grasses, which play a range of biological roles.4 Unnatural SC52246 (4) is a potent and selective 5HT3 serotonin receptor antagonist.5 Unnatural influenza neuraminidase inhibitor A-315675 (5) is a potent inhibitor of influenza A and B viral replication developed by the Abbott scientists.6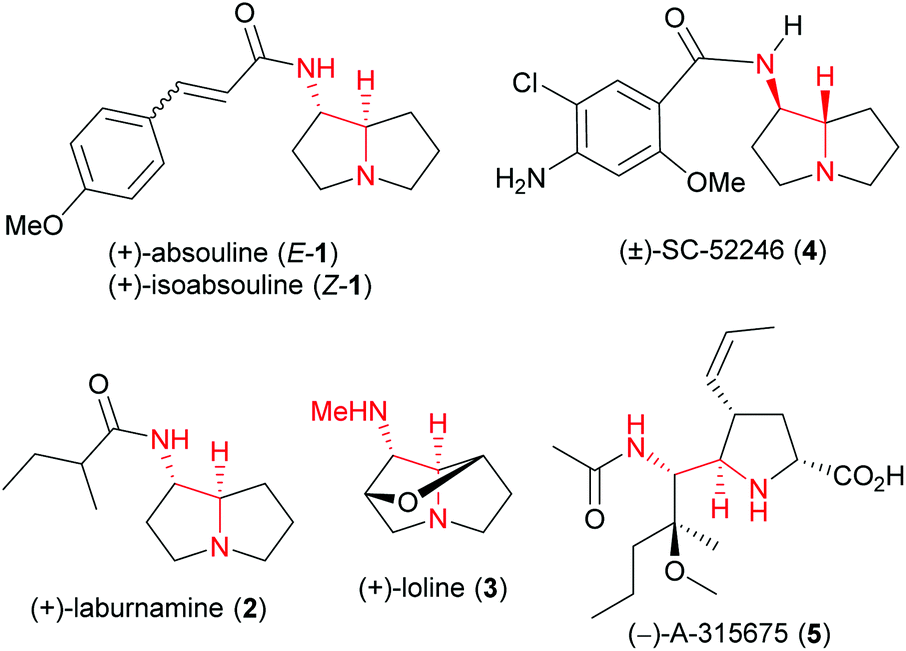 | ||
| Fig. 1 Representative natural products and medicinal agents bearing an anti-vicinal diamine motif. | ||
The unique structural feature and interesting bioactivities exhibited by 1-aminopyrrolizidine alkaloids make them attractive synthetic targets. However, although several syntheses of absouline7 (E-1), laburnamine8 (2), loline (3),9 and SC-522465 (4) have been reported, efficient enantioselective total syntheses are rare.7d–f,9e,g Specifically, for absouline (E-1), aza-conjugate addition-based highly efficient enantioselective total syntheses of (−)-absouline (E-1) have been achieved by Scheerer et al. (eight steps, 10% overall yield)7e and Davies et al. (eight steps, 20% overall yield),7f respectively. Surprisingly, the lengthy asymmetric synthesis of (+)-absouline (E-1), reported several years ago from these laboratories,7c remains the sole total synthesis of the natural enantiomer of this alkaloid. It is thus desirable to develop an alternative strategy for the efficient asymmetric total synthesis of (+)-absouline (E-1).
In recent years, we have been engaged in developing efficient methodologies for the asymmetric total syntheses of natural products9g,10 based on the vinylogous Mannich reaction (VMR)11 and the vinylogous Mukaiyama reaction (VMAR).11a,b Although the resulting heterocyclic amino alcohols can be converted into the corresponding vicinal diamines,9g a direct approach would be more efficient and more attractive. It was envisioned that the addition of a 2-silyloxypyrrole such as N-tert-butoxycarbonyl-2-(tert-butyldimethylsiloxy)pyrrole (TBSOP,127) to an imine such as 6 would afford the functionalized vicinal diamino adduct 8, which could be elaborated in a straightforward manner into 1-aminopyrrolizidine 9a or 1-aminoindolizidine 9b, and related alkaloids (Scheme 1).
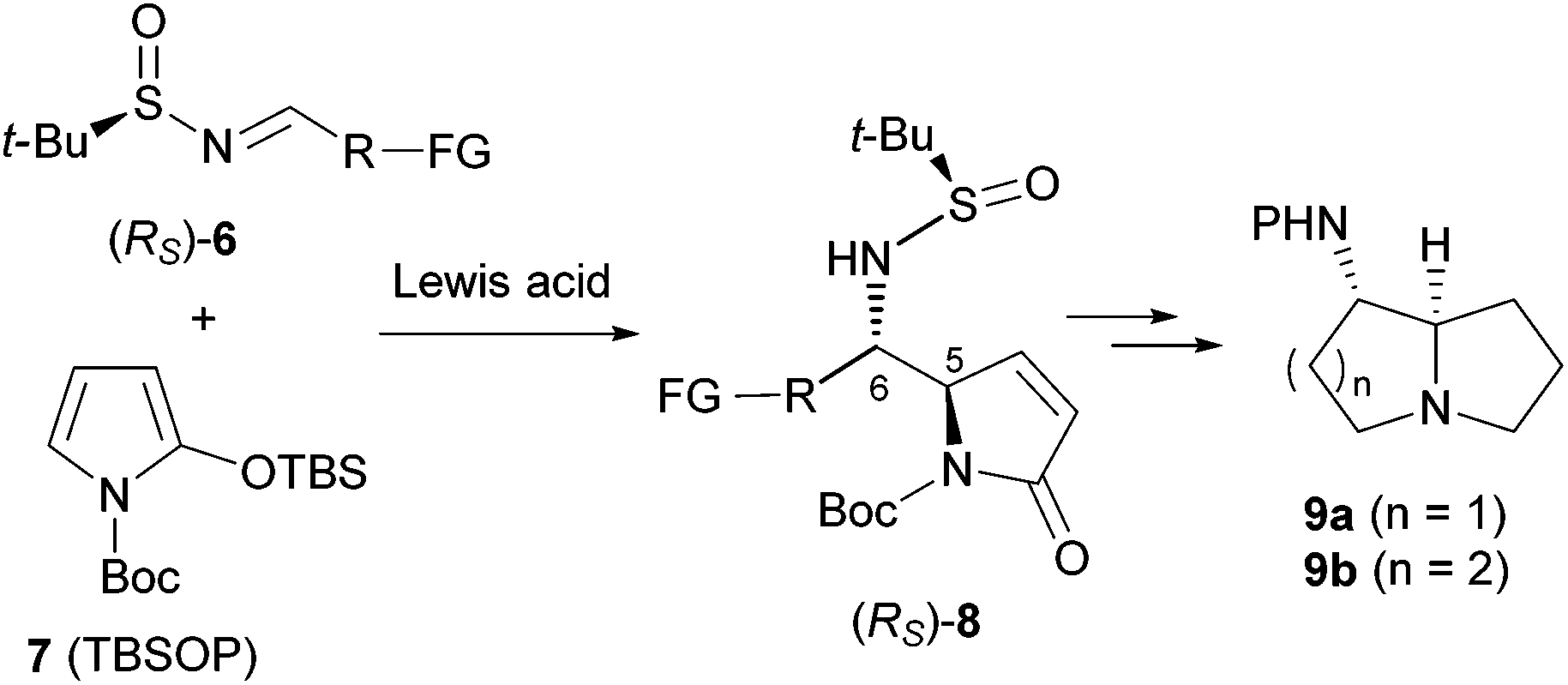 | ||
| Scheme 1 Vinylogous Mannich reaction-based approach to anti-vicinal diamine motifs. | ||
Thanks to the seminal work of Casiraghi/Rassu and Martin, the heterocyclic silyloxydiene-based VMRs and VMARs have become powerful methodologies for the syntheses of functionalized bioactive heterocycles and natural products. However, direct construction of the anti-vicinal diamine motif by asymmetric addition of a 2-silyloxypyrrole with an imine was less explored.6,13,14 The most significant advance in this field is undoubtedly the catalytic enantioselective VMR, developed independently by Casiraghi/Zanardi,14a,b and Hoveyda.14c Nevertheless, this methodology is still in its infancy. In fact, the reported methods are largely limited to aromatic14 and alkyne-substituted N-arylaldimines.14c When aliphatic N-arylimines were used as substrates, the cleavage of the N-aryl group of the resulting diamine products led to low yields.14b On the other hand, the chemistry developed during the synthesis of A-3156756 (5) is restricted to substrates 10 derived from a specific chiral non-racemic aldehyde (Scheme 2). Moreover, use of the Ellman N-tert-butanesulfinimine 10b led to 11b in a low yield (36%).6a In view of the widespread use of Ellman N-tert-butanesulfinimines (t-BS-imines 6) as versatile chiral and reliable amine templates,15 development of the Ellman sulfinimine-based asymmetric VMR would pave an avenue for the asymmetric synthesis of anti-vicinal diamine-containing bioactive molecules and alkaloids (Scheme 1). We report herein the asymmetric VMR of Ellman chiral sulfinimines (6) and TBSOP (7), and its application to the efficient asymmetric total synthesis of the naturally occurring (+)-(1S,7aR)-absouline (E-1).
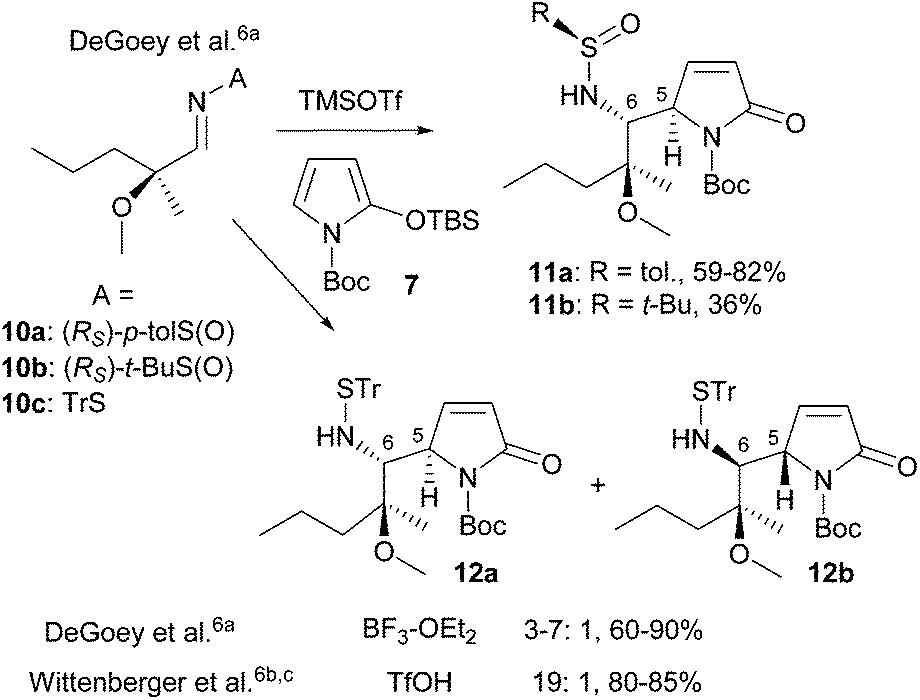 | ||
| Scheme 2 The VMRs investigated during the syntheses of A-315675. | ||
We first investigated the asymmetric VMRs of Ellman sulfinimines (6) with commercially available TBSOP (7). In view of the total synthesis of (+)-absouline (E-1), the known (RS)-t-BS-imine 6a,16a prepared in high yield from (RS)-N-tert-butanesulfinamide (13) and the known aldehyde 14a16b by Ellman's method,17 was selected as a model compound for investigating the asymmetric VMR. After screening the reaction conditions including Lewis acids (BF3·Et2O, TiCl4, Sm(OTf)3, Bi(OTf)3, Cu(OTf)2, TMSOTf), molar equivalents of both Lewis acids and TBSOP (7), optimal reaction conditions were determined, from which TMSOTf turned out to be the Lewis acid of choice (cf. ESI†). In the event, a mixture of TBSOP (7, 1.4 equiv.) and t-BS-imine 6a (1.0 equiv.) in CH2Cl2 was treated with TMSOTf (1.0 equiv.) at −78 °C for 7 h. In this manner, the desired addition product 8a was obtained in 96% yield as a single diastereomer (Scheme 3). The stereochemistry of 8a was determined to be RS,5R,6S (5,6-anti) by single-crystal X-ray diffraction crystallographic analysis (Scheme 3).18
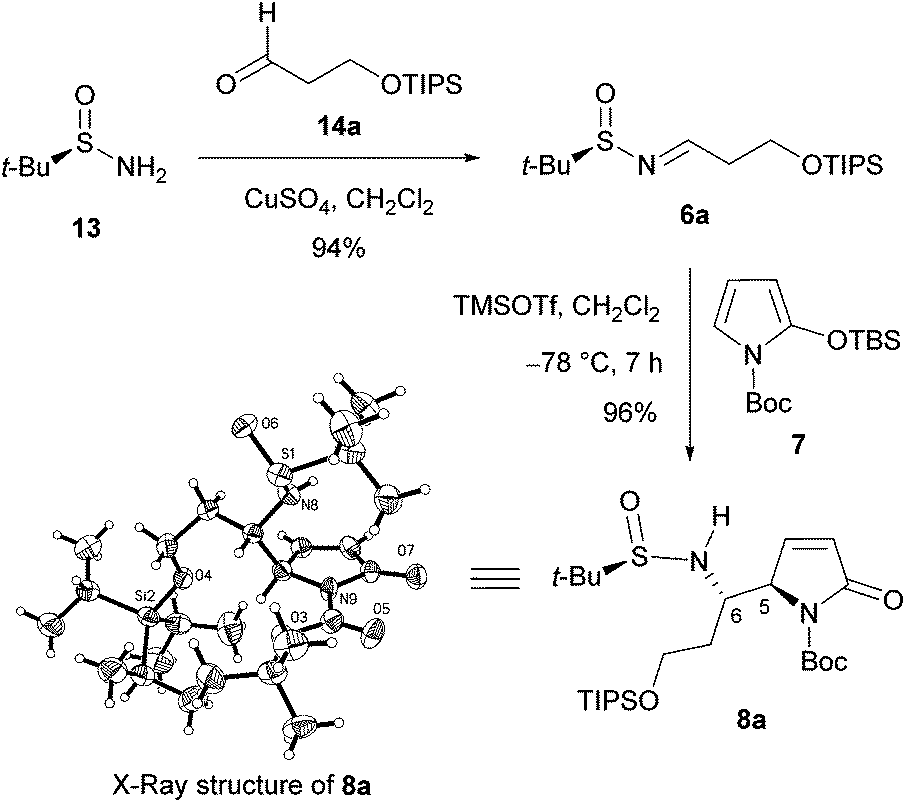 | ||
| Scheme 3 Synthesis and X-ray structure of 8a. | ||
To extend the scope of the asymmetric VMR, the reactions of TBSOP (7) with other (RS)-t-BS-imines 6b–o were examined, and the results are listed in Table 1. t-BS-imines 6b–o were prepared in high yields from (RS)-t-butanesulfinamide (13) and the corresponding aldehydes using either CuSO417 (for aliphatic aldehydes) or Ti(OEt)417 (for aromatic aldehydes) as a dehydrating agent (Table 1, col. 1). As in the case of 6a, the asymmetric VMRs of aliphatic t-BS-imines 6b–h produced the corresponding adducts 8b–h in excellent yields and anti-diastereoselectivities (86–98%, dr > 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, entries 2–8). The reactions of aromatic t-BS-imines 6i–o afforded the corresponding adducts 8i–o in 86–93% yields, and in 82:18 to >99:1 anti-diastereoselectivities (entries 9–15). A strong electron-donating group (MeO) at the para-position of the phenyl group decreased the anti-diastereoselectivity (86%, dr = 82:18, entry 12), while a strong electron-withdrawing group (NO2) seems to be beneficial for both the yield and anti-selectivity (93%, dr = 95:5, entry 14). The highest anti-diastereoselectivity (92%, >99:1) among the tested aromatic t-BS-imines was observed for the reaction of sterically hindered 1-naphthyl t-BS-imine 6o (entry 15).
:1, entries 2–8). The reactions of aromatic t-BS-imines 6i–o afforded the corresponding adducts 8i–o in 86–93% yields, and in 82:18 to >99:1 anti-diastereoselectivities (entries 9–15). A strong electron-donating group (MeO) at the para-position of the phenyl group decreased the anti-diastereoselectivity (86%, dr = 82:18, entry 12), while a strong electron-withdrawing group (NO2) seems to be beneficial for both the yield and anti-selectivity (93%, dr = 95:5, entry 14). The highest anti-diastereoselectivity (92%, >99:1) among the tested aromatic t-BS-imines was observed for the reaction of sterically hindered 1-naphthyl t-BS-imine 6o (entry 15).
|
|
|||
|---|---|---|---|
| Entry | Aldehyde | t-BS-imine 6 (% yield)d | Major diastereomer (anti/syn)c (% yield)d |
| a Prepared by Method A: CuSO4, CH2Cl2, rt, 12 h. b Prepared by Method B: Ti(OEt)4, CH2Cl2, rt, 12 h. c Ratio determined by 1H NMR analysis of crude mixture. d Isolated yield. | |||
| 1 | TIPSO(CH2)2CHO | (RS)-6a (94)a |
8a (>99:1) (96) |
| 2 | EtCHO | (RS)-6b (93)a |
8b (>99:1) (91) |
| 3 | n-PrCHO | (RS)-6c (90)a |
8c (>99:1) (98) |
| 4 | n-C5H11CHO | (RS)-6d (95)a |
8d (>99:1) (91) |
| 5 | Ph(CH2)2CHO | (RS)-6e (94)a |
8e (>99:1) (94) |
| 6 | 3-Cl-n-PrCHO | (RS)-6f (85)a |
8f (>99:1) (87) |
| 7 | i-PrCHO | (RS)-6g (94)a |
8g (>99:1) (86) |
| 8 | c-HexCHO | (RS)-6h (94)a |
8h (>99:1) (88) |
| 9 | PhCHO | (RS)-6i (95)b |
8i (93:7) (88) |
| 10 | p-i-PrC6H4CHO | (RS)-6j (94)b |
8j (92:8) (87) |
| 11 | 4-Ph-C6H4CHO | (RS)-6k (95)b |
8k (88:12) (88) |
| 12 | p-MeOC6H4CHO | (RS)-6l (94)b |
8l (82:18) (86) |
| 13 | p-ClC6H4CHO | (RS)-6m (91)b |
8m (88:12) (88) |
| 14 | p-O2NC6H4CHO | (RS)-6n (90)b |
8n (95:5) (93) |
| 15 | 1-NaphthylCHO | (RS)-6o (93)b |
8o (>99:1) (92) |
Single-crystal X-ray diffraction crystallographic analysis (Fig. 2) showed that the stereochemistry of aromatic 8m is the same as that of aliphatic 8a, namely, RS,5R,6S (5,6-anti).18 Comparison of the diagnostic resonances (cf. S3 in the ESI†) of the 1H NMR spectra of aliphatic 8b–h and aromatic 8i–o with those of 8a and 8m allowed concluding that they all possess the same 5,6-anti-stereochemistry as 8a and 8m.
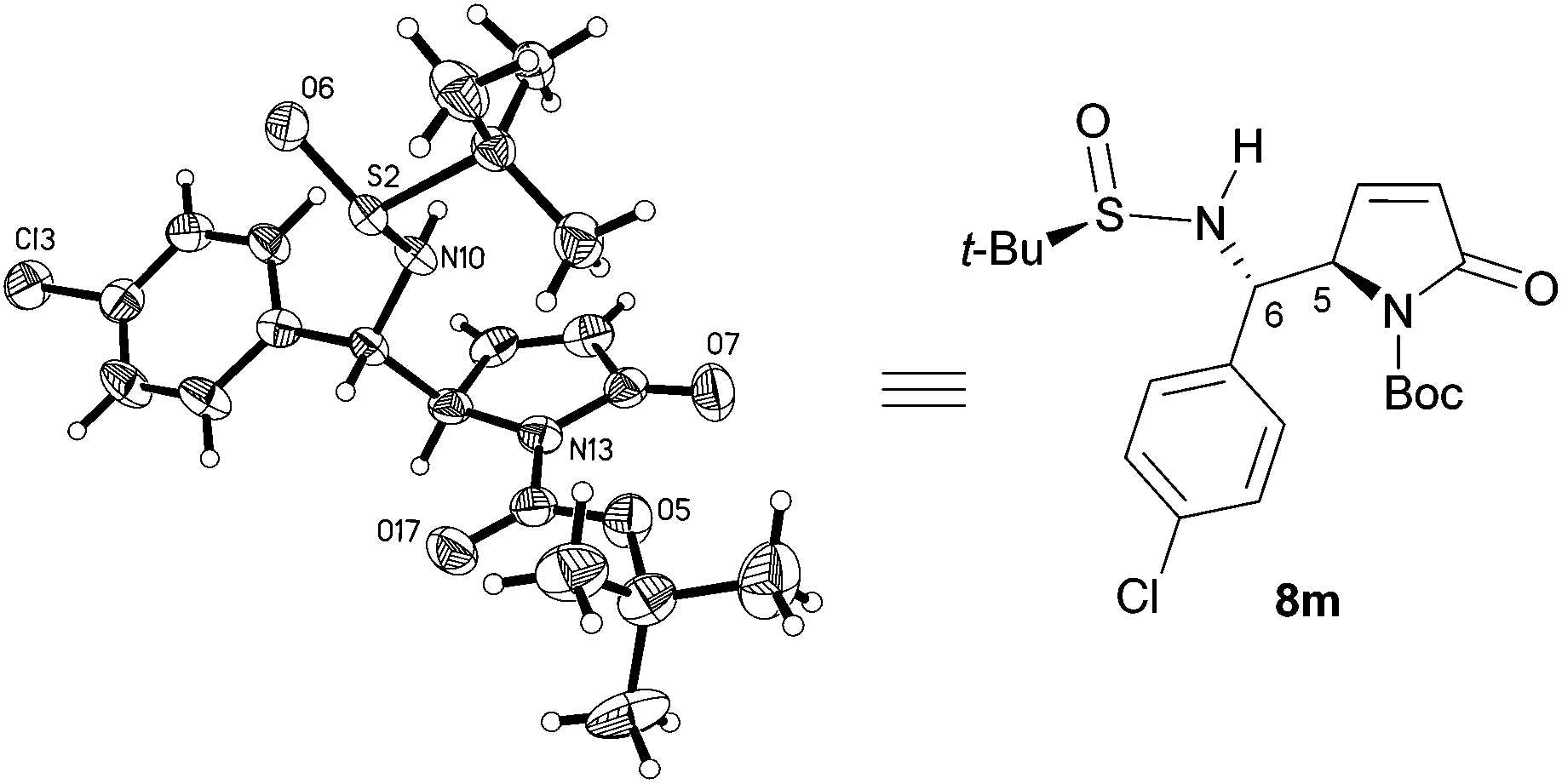 | ||
| Fig. 2 X-Ray structure of 8m. | ||
Interestingly, the stereochemical outcome including asymmetric induction at C5 of this anti-selective VMR (between TBSOP 7 and t-BS-imines 6) is in agreement not only with other VMRs of 2-silyloxypyrroles,6,14 but also with that observed for the VMR between 2-(tert-butyldimethylsiloxy)furan (TBSOF) and t-BS-imines 6.10a Thus, the same model10a (A in Fig. 3) could be used to account for the observed anti-selectivities of the VMRs, namely, according to the Cram–Davis’ open transition model,19,20 TBSOP (7) approaches t-BS-imines 6 by the si-face of the former and re-face of the latter, and the facial selectivity on the chiral sulfinimine is total. On the basis of this model, the lower diastereoselectivities and/or yields obtained from the branched t-BS-imines including isobutyl, cyclohexylmethyl and aromatic t-BS-imine could be attributed to the unfavorable steric interaction between those groups and the N-Boc group. The opposite effects of phenyl substituents OMe and Cl compared with that of NO2 might implicate a favorable secondary orbital interaction between the aromatic ring and the carbonyl group in Boc. The higher diastereoselectivity observed for the 1-naphthyl t-BS-imine 6o also supports this assumption.
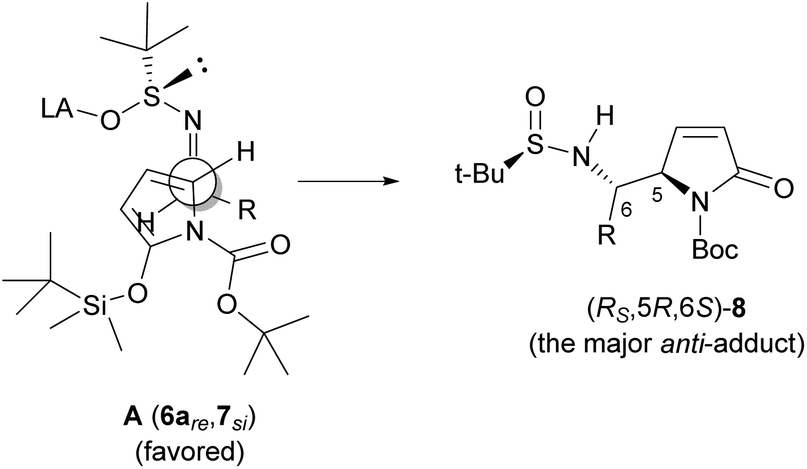 | ||
| Fig. 3 Plausible transition state for the anti-selective VMR leading to (RS,5R,6S)-8. | ||
After establishing a versatile and highly anti-diastereoselective approach to (RS,5R,6S)-8, we turned our attention to the total synthesis of (+)-absouline (E-1). Thus (RS,5R,6S)-8a, the required stereoisomer for the synthesis of (+)-absouline (E-1), was subjected to catalytic hydrogenation (10% Pd/C, H2 1 atm, 5 h, rt) of 8a, which produced 15 in 97% yield (Scheme 4). Chemoselective reduction of imide 15 was achieved by treating with BH3·SMe2 (4 equiv.) at −30 °C for 18.5 h, which produced N-Boc-pyrrolidine 16 in 85% yield. Treatment of the silyl ether 16 with 1.5 equiv. of TBAF at 0 °C for 1 h provided the alcohol 17 in 97% yield. Compound 17 was mesylated (MsCl, NEt3, CH2Cl2, 0 °C to rt, 3 h) to afford mesylate 18 in 90% yield. Successive treatment of 18 with TFA and K2CO3 in MeCN produced, in one-pot, the 1-aminopyrrolizidine derivative 19 in 90% yield. Finally, cleavage of the chiral auxiliary (conc. HCl, MeOH),17,21 followed by coupling of the resulting 1-aminopyrrolizidine with (E)-4-methoxycinnamic acid (DCC, DMAP) produced (+)-absouline (E-1) in 60% yield. The spectroscopy and optical rotation data of our synthetic (+)-absouline (E-1) matched well with those previously reported {[α]20D +51.3 (c 0.3, EtOH), [α]20D +40.0 (c 1.0, CHCl3); lit. natural (+)-absouline: [α]D +56 (c 1, EtOH);2a synthetic (+)-absouline: [α]20D +26 (c 1.05, CHCl3);7a [α]20D +46.0 (c 1.2, CHCl3);7c synthetic (−)-absouline: [α]20D −49 (c 0.2, EtOH)7f}.
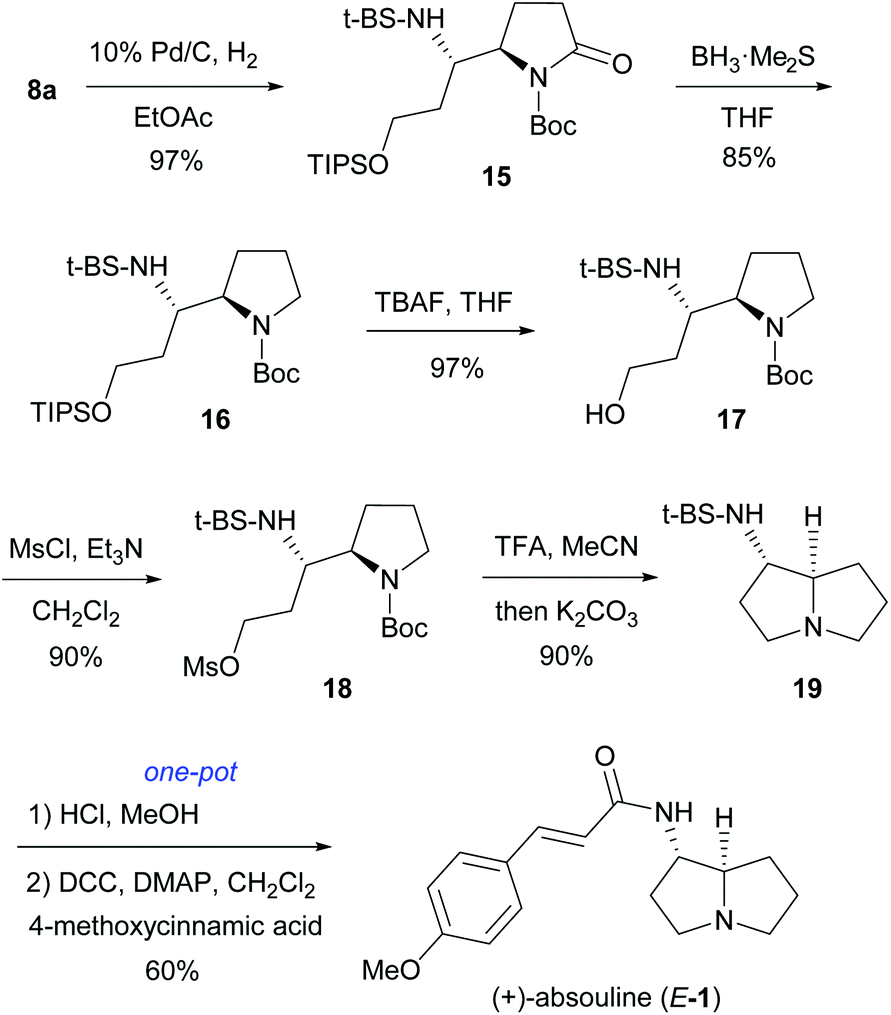 | ||
| Scheme 4 The synthesis of (+)-absouline (E-1). | ||
Conclusion
In summary, on the basis of the asymmetric vinylogous Mannich reactions (VMRs) between TBSOP and t-BS-imines, we have developed a direct, highly anti-stereoselective and versatile approach to anti-vicinal diamine structural motifs. The reaction is versatile for t-BS-imines bearing both linear and branched alkyl groups, as well as aromatic groups. Running under mild conditions, the reaction tolerates several functional groups, which allows further elaboration of adducts into bicyclic ring systems such as 1-aminopyrrolizidine. The power of this method has been demonstrated by a highly efficient total synthesis of the natural (+)-absouline (E-1). The method is applicable for the rapid construction of 1-aminoindolizidine ring system 9b and for the synthesis of other 1-aminopyrrolizidine-containing alkaloids such as (+)-isoabsouline (Z-1), (+)-laburnamine (2), and loline (3). Research along this line is in progress and the results will be reported in due course.Experimental section
General methods
1H NMR and 13C NMR spectra were recorded on a Bruker 400 or Bruker 500 (1H/400 or 500 MHz, 13C/100 or 125 MHz) spectrometer. Chemical shifts are expressed in parts per million (δ) relative to an internal standard of residual chloroform (7.26 ppm for 1H NMR and 77.0 ppm for 13C NMR) or DMSO-d6 (2.50 ppm for 1H NMR and 39.5 ppm for 13C NMR). ESI-mass spectra were recorded on a Bruker Dalton ESquire 3000 Plus LC-MS apparatus. Optical rotations were measured with a Perkin-Elmer 341 automatic polarimeter or an Anton Paar MCP 500 polarimeter. Melting points were uncorrected. Infrared spectra were recorded with a Nicolet Avatar 330 FT-IR spectrometer using the film KBr pellet technique. Silica gel (300–400 mesh) was used for flash column chromatography. THF was distilled over sodium benzophenone ketyl under Ar. Dichloromethane was distilled over calcium hydride under Ar.General procedure for the synthesis of compound (8)
To a cooled (−78 °C) solution of (RS)-t-BS-imine 622 (0.50 mmol) and commercially available tert-butyl-2-[(tert-butyldimethylsilyl)oxy]-1H-pyrrole-1-carboxylate (TBSOP) 7 (0.70 mmol) in CH2Cl2 (5 mL) was added dropwise TMSOTf (0.50 mmol) under an atmosphere of nitrogen. After being stirred for 7 h at the same temperature, the reaction was quenched with saturated aqueous NaHCO3 solution. The mixture was extracted with CH2Cl2 (5 mL × 3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to provide adduct 8. The diastereomeric ratio was determined by 1H NMR.:1, 1H NMR) as a white solid. Mp: 93–95 °C; [α]20D +116.4 (c 1.0, CHCl3); IR (film) νmax: 3345, 2943, 2886, 1776, 1743, 1718, 1368, 1317, 1161, 1102, 1054, 883, 772 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.00–1.10 (m, 18H, 6Me, superposed), 1.10–1.20 (m, 12H including a singlet at 1.12 (s, 9H)), 1.53 (s, 9H), 1.93–1.99 (m, 2H), 2.96 (d, J = 8.9 Hz, 1H), 3.89–4.01 (m, 2H), 4.28 (ddd, J = 3.2, 7.4, 16.1 Hz, 1H), 4.87 (t, J = 1.5 Hz, 1H), 6.21 (dd, J = 1.4, 6.2 Hz, 1H), 7.18 (dd, J = 2.0, 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 11.9 (3C), 18.0 (6C), 22.7 (3C), 28.1 (3C), 38.0, 55.3, 56.3, 60.6, 66.1, 83.2, 129.5, 145.8, 149.0, 169.1; HRMS calcd for C25H48N2O5SSiNa [M + Na]+: 539.2945; found: 539.2947.
:1, 1H NMR) as a white solid. Mp: 110–111 °C; [α]20D +183.0 (c 1.0, CHCl3); IR (film) νmax: 3268, 2923, 2852, 1566, 1462, 1453, 1366, 1185, 1150, 1036, 964, 890, 791, 736 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.13 (s, 9H), 1.17 (t, J = 7.6 Hz, 3H), 1.57 (s, 9H), 1.70–1.77 (m, 2H), 2.79 (d, J = 8.4 Hz, 1H), 4.01–4.04 (m, 1H), 4.68–4.69 (m, 1H), 6.22 (dd, J = 1.6, 6.2 Hz, 1H), 7.10 (dd, J = 2.0, 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 11.2, 22.8 (3C), 28.05, 28.14 (3C), 56.3, 58.0, 65.4, 83.3, 129.9, 144.9, 149.0, 168.8; HRMS calcd for C16H28N2O4SNa [M + Na]+: 367.1662; found: 367.1663.
:1, 1H NMR) as a white solid. Mp: 167–168 °C; [α]20D +249.2 (c 1.0, CHCl3); IR (film) νmax: 3296, 2959, 2929, 2872, 2359, 2340, 1774, 1741, 1712, 1461, 1368, 1342, 1317, 1293, 1255, 1160, 1104, 1052, 911, 847, 794, 770, 752 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.02 (t, J = 6.8 Hz, 3H), 1.13 (s, 9H), 1.57 (s, 9H), 1.60–1.70 (m, 3H), 1.86–1.95 (m, 1H), 2.75 (d, J = 8.5 Hz, 1H), 4.10–4.18 (m, 1H), 4.62–4.66 (m, 1H), 6.21 (dd, J = 1.6, 6.3 Hz, 1H), 7.10 (dd, J = 1.9, 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 13.7, 19.5, 22.7 (3C), 28.1 (3C), 36.9, 55.9, 56.2, 65.7, 83.3, 129.8, 145.0, 149.0, 168.9; HRMS calcd for C17H30N2O4SNa [M + Na]+: 381.1819; found: 381.1823.
:1, 1H NMR) as a white solid. Mp: 152–154 °C; [α]20D +210.2 (c 1.0, CHCl3); IR (film) νmax: 3226, 2956, 2928, 2861, 1775, 1742, 1714, 1458, 1369, 1319, 1293, 1256, 1161, 1106, 1053, 894, 846, 826, 795, 773, 752 cm−1; 1H NMR (400 MHz, CDCl3): δ 0.91 (t, J = 7.0 Hz, 3H), 1.13 (s, 9H), 1.30–1.40 (m, 4H), 1.47–1.70 (m, 4H), 1.58 (s, 9H), 2.67 (d, J = 8.8 Hz, 1H), 4.07–4.16 (m, 1H), 4.62–4.67 (m, 1H), 6.22 (dd, J = 1.4, 6.2 Hz, 1H), 7.09 (dd, J = 1.9, 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 13.9, 22.3, 22.6 (3C), 25.9, 28.1 (3C), 31.4, 34.9, 56.1, 56.2, 65.6, 83.2, 129.8, 145.0, 149.0, 168.8; HRMS calcd for C19H34N2O4SNa [M + Na]+: 409.2131; found: 409.2130.
:1, 1H NMR) as a white solid. Mp: 149–152 °C; [α]20D +125.6 (c 1.0, CHCl3); IR (film) νmax: 3291, 2979, 2941, 1774, 1742, 1723, 1603, 1477, 1453, 1369, 1317, 1294, 1196, 1157, 1132, 1076, 1051, 885, 752 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.15 (s, 9H), 1.43 (s, 9H), 1.85–1.96 (m, 1H), 1.97–2.08 (m, 1H), 2.82 (d, J = 8.6 Hz, 1H), 2.84–2.93 (m, 1H), 2.94–3.04 (m, 1H), 3.98–4.08 (m, 1H), 4.56 (dd, J = 2.0, 3.3 Hz, 1H), 6.21 (dd, J = 1.5, 6.2 Hz, 1H), 7.08 (dd, J = 2.0, 6.2 Hz, 1H), 7.20 (dd, J = 4.4, 8.8 Hz, 1H), 7.29–7.32 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 22.8 (3C), 28.0 (3C), 31.9, 36.5, 55.8, 56.4, 66.3, 83.3, 126.3, 128.6, 129.9, 140.6, 144.9, 148.8, 168.9; HRMS calcd for C22H32N2O4SNa [M + Na]+: 443.1975; found: 443.1977.
:1, 1H NMR) as a white solid. Mp: 140–142 °C; [α]20D +182.3 (c 1.0, CHCl3); IR (film) νmax: 3277, 2978, 2928, 2870, 1775, 1741, 1713, 1475, 1456, 1369, 1318, 1294, 1257, 1160, 1105, 1053, 955, 896, 845, 825, 795, 753, 643, 611 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.14 (s, 9H), 1.57 (s, 9H), 1.63–1.72 (m, 1H), 1.93–2.07 (m, 2H), 2.11–2.26 (m, 1H), 2.77 (d, J = 8.6 Hz, 1H), 3.61–3.71 (m, 2H), 4.09–4.17 (m, 1H), 4.62–4.65 (m, 1H), 6.24 (dd, J = 1.6, 6.2 Hz, 1H), 7.15 (dd, J = 2.0, 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 22.7 (3C), 28.2 (3C), 29.0, 31.5, 44.4, 55.7, 56.4, 66.0, 83.5, 130.1, 144.7, 149.2, 168.5; HRMS calcd for C17H29ClN2O4SNa [M + Na]+: 415.1429; found: 415.1426.
:1, 1H NMR) as a white solid. Mp: 158–159 °C; [α]20D +249.2 (c 1.0, CHCl3); IR (film) νmax: 3222, 2964, 2928, 2872, 1775, 1741, 1712, 1475, 1392, 1368, 1318, 1292, 1257, 1161, 1127, 1107, 1052, 891, 848, 816, 791, 752 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.14 (s, 9H), 1.19 (d, J = 6.4 Hz, 6H), 1.58 (s, 9H), 1.69–1.79 (m, 1H), 2.77 (d, J = 8.0 Hz, 1H), 3.78–3.84 (m, 1H), 4.81–4.84 (m, 1H), 6.20 (dd, J = 1.5, 6.2 Hz, 1H), 7.09 (dd, J = 1.8, 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 20.2, 21.0, 22.8 (3C), 28.2 (3C), 32.2, 56.3, 61.9, 64.6, 83.3, 129.8, 145.2, 148.9, 169.0; HRMS calcd for C17H30N2O4SNa [M + Na]+: 381.1819; found: 381.1814.
:1, 1H NMR) as a white solid. Mp: 158–159 °C; [α]20D +134.8 (c 1.0, CHCl3); IR (film) νmax: 3233, 2926, 2853, 1777, 1738, 1711, 1368, 1344, 1321, 1296, 1161, 1052, 729 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.14 (s, 9H), 1.20–1.50 (m, 6H), 1.57 (s, 9H), 1.70–2.12 (m, 5H), 2.75 (d, J = 8.0 Hz, 1H), 3.86–3.92 (m, 1H), 4.82–4.85 (m, 1H), 6.19 (dd, J = 1.5, 6.2 Hz, 1H), 7.08 (dd, J = 2.0, 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 22.8 (3C), 25.5, 25.8, 25.9, 28.1 (3C), 30.3, 30.7, 41.2, 56.2, 60.4, 64.2, 83.2, 129.6, 145.5, 148.8, 169.2; HRMS calcd for C20H34N2O4SNa [M + Na]+: 421.2132; found: 421.2136.
:7, 1H NMR) as a diastereomeric mixture, which was separated by repeated flash chromatography on silica gel (eluent: EtOAc/hexane = 3/1) to give (RS,5R,6S)-8i as a white solid. Mp: 149–150 °C; [α]20D +130.3 (c 1.0, CHCl3); IR (film) νmax: 3242, 2978, 2925, 2359, 2343, 1778, 1743, 1709, 1478, 1453, 1366, 1321, 1284, 1258, 1158, 1053, 894, 846, 815, 790, 772, 701 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.19 (s, 9H), 1.63 (s, 9H), 3.63 (d, J = 9.4 Hz, 1H), 4.89–4.92 (m, 1H), 5.40 (dd, J = 3.5, 9.4 Hz, 1H), 6.20 (d, J = 6.2 Hz, 1H), 6.85 (dd, J = 1.9, 6.2 Hz, 1H), 7.32–7.48 (m, 5H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 28.2 (3C), 56.7, 58.6, 67.4, 83.7, 126.3, 128.3, 129.1, 129.7, 138.3, 145.2, 149.2, 168.7; HRMS calcd for C20H28N2O4SNa [M + Na]+: 415.1662; found: 415.1658.
:8, 1H NMR) as a diastereomeric mixture, which was separated by repeated flash chromatography on silica gel (eluent: MeOH/CH2Cl2 = 1/80) to give (RS,5R,6S)-8j as a colorless oil. [α]20D +177.6 (c 1.0, CHCl3); IR (film) νmax: 3246, 2961, 2926, 2869, 1783, 1745, 1709, 1460, 1366, 1321, 1260, 1159, 1054, 896, 794; 1H NMR (400 MHz, CDCl3): δ 1.18 (s, 9H), 1.25 (d, J = 6.9 Hz, 6H), 1.63 (s, 9H), 2.92 (sept, J = 6.9 Hz, 1H), 3.53 (d, J = 9.3 Hz, 1H), 4.88 (ddd, J = 1.8, 2.0, 3.6 Hz, 1H), 5.36 (dd, J = 3.5, 9.3 Hz, 1H), 6.20 (dd, J = 1.6, 6.2 Hz, 1H), 6.88 (dd, J = 2.0, 6.2 Hz, 1H), 7.24–7.30 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 22.83, 22.87, 28.2 (3C), 33.8, 55.4, 56.6, 58.5, 67.4, 83.5, 126.3 (2C), 127.1 (2C), 129.6, 135.6, 145.4, 149.2, 168.7; HRMS calcd for C23H34N2O4SNa [M + Na]+: 457.2132; found: 457.2136.
:12, 1H NMR) as a diastereomeric mixture, which was separated by repeated flash chromatography on silica gel (eluent: MeOH/CH2Cl2 = 1/80) to give compound (RS,5R,6S)-8k (180 mg) and (RS,5S,6S)-8k (25 mg).
(RS,5R,6S)-8k: pale yellow solid. Mp: 131–133 °C; [α]20D +184.1 (c 1.0, CHCl3); IR (film) νmax: 3242, 2976, 2926, 2866, 1781, 1743, 1709, 1601, 1485, 1468, 1366, 1322, 1283, 1258, 1196, 1180, 1132, 1076, 896, 750; 1H NMR (400 MHz, CDCl3): δ 1.20 (s, 9H), 1.64 (s, 9H), 3.67 (d, J = 9.5 Hz, 1H), 4.95 (ddd, J = 1.8, 2.0, 3.6 Hz, 1H), 5.44 (dd, J = 3.4, 9.5 Hz, 1H), 6.22 (dd, J = 1.6, 6.2 Hz, 1H), 6.91 (dd, J = 2.0, 6.2 Hz, 1H), 7.36 (t, J = 7.4 Hz, 1H), 7.36 (t, J = 7.4 Hz, 1H), 7.41–7.48 (m, 4H), 7.57 (d, J = 7.8 Hz, 2H), 7.63 (d, J = 7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 28.2 (3C), 56.6, 58.5, 67.4, 83.7, 126.8 (2C), 127.1 (2C), 127.6, 127.7 (2C), 128.9 (2C), 129.8, 137.3, 140.3, 141.3, 145.2, 149.3, 168.6; HRMS calcd for C26H32N2O4SNa [M + Na]+: 491.1975; found: 491.1978.
(RS,5S,6S)-8k: pale yellow oil. [α]20D −252.9 (c 1.0, CHCl3); IR (film) νmax: 3243, 2977, 2926, 2867, 1776, 1745, 1713, 1602, 1488, 1456, 1367, 1319, 1285, 1259, 1158, 1076, 1052, 844, 824, 752; 1H NMR (400 MHz, CDCl3): δ 1.32 (s, 9H), 1.67 (s, 9H), 3.78 (d, J = 2.7 Hz, 1H), 5.14 (ddd, J = 1.8, 2.1, 5.1 Hz, 1H), 5.31 (dd, J = 2.7, 5.1 Hz, 1H), 5.98 (dd, J = 1.6, 6.2 Hz, 1H), 7.16 (dt, J = 1.8, 8.2 Hz, 1H), 7.29 (dd, J = 2.0, 6.2 Hz, 1H), 7.34 (t, J = 7.2 Hz, 1H), 7.39–7.45 (m, 2H), 7.46–7.55 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 28.2 (3C), 56.2, 58.4, 64.9, 83.8, 127.1 (2C), 127.2 (2C), 127.6, 128.0 (2C), 128.7, 128.8 (2C), 134.6, 140.2, 141.8, 146.5, 149.3, 168.6; HRMS calcd for C26H32N2O4SNa [M + Na]+: 491.1975; found: 491.1978.
:18, 1H NMR) as a diastereomeric mixture, which was separated by repeated flash chromatography on silica gel (eluent: MeOH/CH2Cl2 = 1/80) to give compound (RS,5R,6S)-8l (149 mg) and compound (RS,5S,6S)-8l (33 mg).
(RS,5R,6S)-8l: colorless oil. [α]20D +103.5 (c 1.0, CHCl3); IR (film) νmax: 3242, 2977, 2929, 1777, 1742, 1709, 1612, 1515, 1461, 1392, 1366, 1321, 1284, 1251, 1159, 1106, 1051, 1033, 936, 894, 818, 798, 753 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.18 (s, 9H), 1.62 (s, 9H), 3.56 (d, J = 9.2 Hz, 1H), 3.81 (s, 3H), 4.84–4.91 (m, 1H), 5.33 (dd, J = 3.2, 9.2 Hz, 1H), 6.20 (dd, J = 1.3, 6.2 Hz, 1H), 6.88 (dd, J = 1.9, 6.2 Hz, 1H), 6.93 (d, J = 8.3 Hz, 2H), 7.28 (d, J = 8.3 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 28.2 (3C), 55.3, 56.6, 58.1, 67.5, 83.6, 114.4 (2C), 127.5 (2C), 129.6, 130.3, 145.3, 149.3, 159.5, 168.7. HRMS calcd for C21H30N2O5SNa [M + Na]+: 445.1768; found: 445.1773.
(RS,5S,6S)-8l: colorless oil. [α]20D −279.3 (c 1.0, CHCl3); IR (film) νmax: 3234, 2976, 2928, 1775, 1744, 1712, 1612, 1515, 1458, 1393, 1338, 1319, 1283, 1252, 1180, 1158, 1106, 1051, 891, 841, 819, 757, 727 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.31 (s, 9H), 1.65 (s, 9H), 3.66 (d, J = 2.4 Hz, 1H), 3.76 (s, 3H), 5.07–5.12 (m, 1H), 5.23 (dd, J = 2.0, 5.0 Hz, 1H), 5.96 (dd, J = 1.5, 6.1 Hz, 1H), 6.78 (d, J = 8.6 Hz, 2H), 6.98 (d, J = 8.6 Hz, 2H), 7.24 (dd, J = 1.9, 6.1 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 28.2 (3C), 55.2, 56.1, 58.0, 64.8, 83.7, 113.9 (2C), 127.4, 128.6, 128.8 (2C), 146.6, 149.2, 159.8, 168.7. HRMS calcd for C21H30N2O5SNa [M + Na]+: 445.1768; found: 445.1773.
:12, 1H NMR) as a diastereomeric mixture, which was separated by repeated flash chromatography on silica gel (eluent: MeOH/CH2Cl2 = 1/80) to give (RS,5R,6S)-8m (165 mg) and (RS,5S,6S)-8m (23 mg).
(RS,5R,6S)-8m: white solid. Mp: 130–132 °C; [α]20D +197.8 (c 1.0, CHCl3); IR (film) νmax: 3270, 2977, 2928, 2866, 1779, 1744, 1710, 1493, 1461, 1367, 1322, 1281, 1259, 1196, 1158, 1132, 1076, 894, 810, 752; 1H NMR (400 MHz, CDCl3): δ 1.19 (s, 9H), 1.62 (s, 9H), 3.70 (d, J = 9.9 Hz, 1H), 4.90 (ddd, J = 1.8, 2.0, 3.6 Hz, 1H), 5.37 (dd, J = 3.6, 9.9 Hz, 1H), 6.22 (dd, J = 1.6, 6.2 Hz, 1H), 6.84 (dd, J = 2.0, 6.2 Hz, 1H), 7.33 (d, J = 8.5 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 28.2 (3C), 56.8, 58.2, 67.2, 83.8, 127.8 (2C), 129.2 (2C), 130.0, 134.3, 136.8, 144.7, 149.4, 168.2; HRMS calcd for C20H27ClN2O4SNa [M + Na]+: 449.1272; found: 449.1273.
(RS,5S,6S)-8m: colorless oil. [α]20D −247.7 (c 1.0, CHCl3); IR (film) νmax: 3243, 2961, 2924, 2854, 1772, 1747, 1717, 1558, 1457, 1366, 1318, 1260, 1157, 1076, 1051, 801; 1H NMR (400 MHz, CDCl3): δ 1.31 (s, 9H), 1.64 (s, 9H), 3.70 (d, J = 2.7 Hz, 1H), 5.11 (ddd, J = 1.8, 2.3, 5.2 Hz, 1H), 5.25 (dd, J = 2.7, 5.2 Hz, 1H), 5.99 (dd, J = 1.5, 6.2 Hz, 1H), 7.01 (d, J = 8.0 Hz, 2H), 7.22 (dd, J = 2.0, 6.2 Hz, 1H), 7.26 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 28.2 (3C), 56.3, 58.1, 64.6, 84.0, 128.8 (2C), 128.88 (2C), 128.94, 134.1, 134.9, 146.2, 149.3, 168.3. HRMS calcd for C20H27ClN2O4SNa [M + Na]+: 449.1272; found: 449.1273.
:5, 1H NMR) as a diastereomeric mixture, which was separated by repeated flash chromatography on silica gel (eluent: MeOH/CH2Cl2 = 1/80) to give compound (RS,5R,6S)-8n as a yellow solid. Mp: 100–102 °C; [α]20D +184.9 (c 1.0, CHCl3); IR (film) νmax: 3285, 2980, 2930, 2866, 1779, 1744, 1710, 1606, 1523, 1475, 1459, 1349, 1322, 1285, 1196, 1157, 1076, 893, 845, 754; 1H NMR (400 MHz, CDCl3): δ 1.19 (s, 9H), 1.62 (s, 9H), 4.02 (d, J = 10.3 Hz, 1H), 4.98 (ddd, J = 1.8, 1.9, 5.1 Hz, 1H), 5.47 (dd, J = 5.1, 10.4 Hz, 1H), 6.24 (dt, J = 1.6, 6.2 Hz, 1H), 6.84 (dd, J = 2.0, 6.2 Hz, 1H), 7.60 (d, J = 8.7 Hz, 2H), 8.25 (dd, J = 2.0, 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 28.2 (3C), 57.1, 58.4, 66.9, 84.1, 124.0 (2C), 127.7 (2C), 130.3, 144.3, 145.5, 147.8, 149.7, 167.8; HRMS calcd for C20H27N3O6SNa [M + Na]+: 460.1513; found: 460.1515.
:1, 1H NMR) as a diastereomeric mixture, which was separated by repeated flash chromatography on silica gel (eluent: MeOH/CH2Cl2 = 1/80) to give compound (RS,5R,6S)-8o as a pale yellow wax. [α]20D +55.8 (c 1.0, CHCl3); IR (film) νmax: 3284, 2979, 2929, 2866, 1780, 1743, 1705, 1601, 1514, 1474, 1457, 1358, 1322, 1258, 1195, 1156, 1076, 895, 796; 1H NMR (400 MHz, CDCl3): δ 1.22 (s, 9H), 1.72 (s, 9H), 3.80 (d, J = 9.5 Hz, 1H), 5.10 (ddd, J = 2.0, 2.1, 3.8 Hz, 1H), 6.18 (dd, J = 3.1, 10.7 Hz, 1H), 6.18 (dd, J = 1.6, 6.2 Hz, 1H), 6.70 (dd, J = 1.9, 6.2 Hz, 1H), 7.44 (d, J = 7.3 Hz, 1H), 7.47–7.58 (m, 2H), 7.64 (t, J = 7.5 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 8.49 (d, J = 8.5 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 28.4 (3C), 55.3, 56.8, 65.3, 83.9, 123.0, 123.9, 125.2, 126.2, 127.0, 129.0, 129.1, 129.6, 130.5, 133.9, 134.3, 145.6, 149.8, 168.4; HRMS calcd for C24H30N2O4SNa [M + Na]+: 465.1819; found: 465.1821.
:1) to give compound 15 (51 mg, yield: 97%) as a colorless oil. [α]20D +27.0 (c 1.0, CHCl3); IR (film) νmax: 3245, 2943, 2866, 1782, 1749, 1718, 1368, 1307, 1289, 1154, 1098, 1070, 1050, 882, 773 cm−1; 1H NMR (400 MHz, CDCl3): δ 0.95–1.15 (m, 21H), 1.17 (s, 9H), 1.55 (s, 9H), 1.79–1.88 (m, 2H), 1.89–2.11 (m, 2H), 2.40–2.57 (m, 2H), 3.33 (d, J = 7.2 Hz, 1H), 3.83–3.97 (m, 2H), 4.04 (ddd, J = 2.4, 7.2, 14.4 Hz, 1H), 4.27 (dt, J = 3.0, 9.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 11.5, 11.8, 17.1, 17.9 (6C), 22.6 (3C), 28.0 (3C), 32.1, 36.7, 55.7, 56.4, 60.7, 61.0, 83.1, 149.6, 174.3; HRMS calcd for C25H50N2O5SSiNa [M + Na]+: 541.3102; found: 541.3106.
:1) to give compound 16 (79 mg, yield: 85%) as a white wax. [α]20D +12.0 (c 1.0, CHCl3); IR (film) νmax: 3309, 2942, 2866, 1697, 1463, 1398, 1364, 1166, 1101, 882, 772, 682 cm−1; 1H NMR (400 MHz, CDCl3): δ 0.92–1.16 (m, 21H), 1.19 (s, 9H), 1.45 (s, 4.6H), 1.49 (s, 4.4H), 1.60–1.90 (m, 5H), 1.97 (br s, 1H), 3.07–3.23 (m, 1.5H), 3.45–3.71 (m, 1H), 3.76–4.04 (m, 4H), 4.10–4.25 (m, 0.5H), 4.12–4.31 (m, 1H); 1H NMR (400 MHz, DMSO-d6, temp = 80 °C): δ 1.00–1.15 (m, 21H), 1.17 (s, 9H), 1.42 (s, 9H), 1.60–1.85 (m, 5H), 1.90–2.00 (m, 1H), 3.05–3.14 (m, 1H), 3.35–3.43 (m, 1H), 3.65–3.90 (m, 4H), 4.50 (d, J = 8.4 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 11.9 (3C), 18.0 (6C), 22.8 (3C), 23.7, 24.1, 26.5, 27.5, 28.5 (3C), 36.2, 37.6, 47.2, 47.7, 55.6, 55.9, 56.1, 60.6, 60.9, 62.2, 79.3, 79.7, 154.4, 155.0; 13C NMR (100 MHz, DMSO-d6, temp = 80 °C): δ 11.2 (3C), 17.4 (6C), 22.3 (3C), 22.9, 25.2, 27.8 (3C), 36.5, 46.5, 54.5, 55.0, 60.3, 60.6, 77.7, 153.1; HRMS calcd for C25H52N2O4SSiNa [M + Na]+: 527.3309; found: 527.3309.
:1) to give compound 19 (44 mg, yield: 90%) as a yellow wax. [α]20D −25.5 (c 1.0, CHCl3); IR (film) νmax: 3438, 3200, 2961, 2514, 1676, 1458, 1416, 1366, 1200, 1177, 1129, 1054, 831, 799, 720 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.24 (s, 9H), 1.71–1.81 (m, 1H), 1.90–2.11 (m, 2H), 2.29–2.43 (m, 3H), 2.78–2.97 (m, 2H), 3.62–3.69 (m, 1H), 3.76–3.83 (m, 1H), 3.87–3.95 (m, 1H), 4.01–4.09 (m, 1H), 4.62 (d, J = 8.3 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 22.6 (3C), 25.2, 29.9, 32.9, 52.7, 54.9, 56.5, 60.5, 72.2; HRMS calcd for C11H23N2OS [M + H]+: 231.1526; found: 231.1526.
:1) to give (+)-absouline (E-1) (22 mg, yield: 60%) as a white powder. Mp: 164–166 °C [lit.: synthetic (−)-absouline: mp: 165 °C7d]; [α]20D +51.3 (c 0.3, EtOH), [α]20D +40.0 (c 1.0, CHCl3) {lit.: natural (+)-absouline: [α]D +56 (c 1, EtOH);2a synthetic (+)-absouline: [α]20D +26 (c 1.05, CHCl3);7a [α]20D +46.0 (c 1.2, CHCl3);7c synthetic (−)-absouline: [α]20D −34 (c 0.91, CHCl3);7a [α]20D −37 (c 1.8, CHCl3), [α]20D −51 (c 0.4, EtOH);7d [α]25D −28 (c 1.17, CHCl3), [α]25D −48 (c 0.8, EtOH);7e [α]20D −49 (c 0.2, EtOH)7f}; IR (film) νmax: 3270, 2959, 2868, 1656, 1603, 1575, 1545, 1512, 1304, 1288, 1256, 1223, 1173, 1031, 828 cm−1; 1H NMR (500 MHz, CDCl3): δ 1.65–1.95 (m, 4H), 1.97–2.08 (m, 1H), 2.23–2.30 (m, 1H), 2.60–2.68 (m, 2H), 2.85–3.04 (m, 1H), 3.21–3.32 (m, 2H), 3.82 (s, 3H), 4.21–4.27 (m, 1H), 6.18 (d, J = 6.0 Hz, 1H), 6.29 (d, J = 15.6 Hz, 1H), 6.88 (d, J = 8.8 Hz, 2H), 7.44 (d, J = 8.8 Hz, 2H), 7.58 (d, J = 15.6 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 25.4, 30.7, 33.0, 53.4, 55.2, 55.3, 55.5, 71.0, 114.2, 118.2, 127.6, 129.3, 140.7, 160.9, 166.1; HRMS calcd for C17H23N2O2 [M + H]+: 287.1754; found: 287.1758.
Acknowledgements
The authors are grateful for financial support from the NSF of China (21332007), the National Basic Research Program (973 Program) of China (Grant No. 2010CB833200), the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) of Ministry of Education.References
- For reviews on the asymmetric syntheses of vicinal diamines, see:
(a) D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580–2627 CrossRef CAS
; (b) H. Kim, S. M. So, J. Chin and B. M. Kim, Aldrichimica Acta, 2008, 41, 77–88 CAS
-
(a) K. Ikhiri, A. Ahond, C. Poupat, P. Potier, J. Pusset and T. Sevenet, J. Nat. Prod., 1987, 50, 626–630 CrossRef CAS
-
(a) F. Galinovsky, O. Vogl and H. Nesvadba, Sci. Pharm., 1953, 21, 256–258 CAS
- C. L. Schardl, R. B. Grossman, P. Nagabhyru, J. R. Faulkner and U. P. Mallik, Phytochemistry, 2007, 68, 980–996 CrossRef CAS PubMed
-
(a) D. L. Flynn, D. L. Zabrowski, D. P. Becker, R. Nosal, C. I. Villamil, G. W. Gullikson, C. Moummi and D. C. Yang, J. Med. Chem., 1992, 35, 1486–1489 CrossRef CAS PubMed
-
(a) D. A. DeGoey, H.-J. Chen, W. J. Flosi, D. J. Grampovnik, C. M. Yeung, L. L. Klein and D. J. Kempf, J. Org. Chem., 2002, 67, 5445–5453 CrossRef CAS PubMed
- For racemic syntheses of absouline, see:
(a) C. Christine, K. Ikhiri, A. Ahond, A. A. Mourabit, C. Poupat and P. Potier, Tetrahedron, 2000, 56, 1837–1850 CrossRef CAS
- For racemic synthesis of laburnamine, see: M. Norton Matos, C. A. M. Afonso and R. A. Batey, Tetrahedron, 2005, 61, 1221–1244 CrossRef CAS
- For racemic total syntheses, see:
(a) J. J. Tufariello, H. Meckler and K. Winzenberg, J. Org. Chem., 1986, 51, 3556–3557 CrossRef CAS
-
(a) S.-T. Ruan, J.-M. Luo, Y. Du and P.-Q. Huang, Org. Lett., 2011, 13, 4938–4941 CrossRef CAS PubMed
- For comprehensive recent reviews on the heterocyclic silyloxydiene-based VMRs and VMARs and synthetic applications, see:
(a) G. Casiraghi, L. Battistini, C. Curti, G. Rassu and F. Zanardi, Chem. Rev., 2011, 111, 3076–3154 CrossRef CAS PubMed
-
(a) G. Rassu, G. Casiraghi, P. Spanu, L. Pinna, G. G. Fava, M. B. Ferrari and G. Pelosi, Tetrahedron: Asymmetry, 1992, 3, 1035–1048 CrossRef CAS
-
(a) F. Zanardi, L. Battistini, G. Rassu, L. Pinna, M. Mor, N. Culeddu and G. Casiraghi, J. Org. Chem., 1998, 63, 1368–1369 CrossRef CAS
-
(a) C. Curti, L. Battistini, B. Ranieri, G. Pelosi, G. Rassu, G. Casiraghi and F. Zanardi, J. Org. Chem., 2011, 76, 2248–2252 CrossRef CAS PubMed
- For reviews, see:
(a) J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984–995 CrossRef CAS PubMed
-
(a) J. S. Villadsen, H. M. Stephansen, A. R. Maolanon, P. Harris and C. A. Olsen, J. Med. Chem., 2013, 56, 6512–6520 CrossRef CAS PubMed
- G. Liu, D. A. Cogan, T. D. Owens, T. P. Tang and J. A. Ellman, J. Org. Chem., 1999, 64, 1278–1284 CrossRef CAS
- CCDC 1046181 (8a) and 1445824 (8m) contain the supplementary crystallographic data for this paper.
- F. A. Davis and W. McCoull, J. Org. Chem., 1999, 64, 3396–3397 CrossRef CAS PubMed
- For theoretical studies on the origins of diastereoselectivity of the vinylogous Mannich reactions between 2-methoxyfuran and pyrrolinium ion, see: S. K. Bur and S. F. Martin, Org. Lett., 2000, 2, 3445–3447 CrossRef CAS PubMed
- N. Plobeck and D. Powell, Tetrahedron: Asymmetry, 2002, 13, 303–310 CrossRef CAS
- For the syntheses of (RS)-t-BS-imines 6 see: ESI.†.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of compounds 6j, 6k, 8a–8o, 15–19, and (+)-absouline (E-1); crystallographic structure files for 8a and 8m (CIF). CCDC 1046181 and 1445824. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00022c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |