
Cobalt-catalyzed synthesis of quinolines from the redox-neutral annulation of anilides and alkynes†
Qiangqiang
Yan
a,
Zhengkai
Chen
b,
Zhanxiang
Liu
a and
Yuhong
Zhang
*ac
aZJU-NHU United R&D Center, Department of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: yhzhang@zju.edu.cn
bDepartment of Chemistry, Zhejiang Sci-Tech University, Hangzhou 310018, China
cState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, China
First published on 17th March 2016
Abstract
A new method of cobalt-catalyzed annulation of anilides and internal alkynes for the efficient synthesis of quinoline scaffolds was developed. The combination of cobalt catalyst with the Lewis acid Zn(OTf)2 ensured the success of the annulation to give the quinoline heterocycles in high efficiency. The transformation is supposed to undergo ortho C–H activation and nucleophilic addition of C–Co species toward the amides. Attractive features of this system include the use of a low cost cobalt catalyst, the easy-handling operation, and readily available substrates.
Nucleophilic addition of organometallic reagents to polar unsaturated substrates constitutes an important class of carbon–carbon bond forming transformations. Traditionally, the active C–M species are generated from the carbon–halogen bonds or carbon–metal bonds, which often suffer from harsh reaction conditions and tedious prefunctionalization steps.1 To overcome these drawbacks, the access of active carbon–transition metal (C–TM) species from hydrocarbon materials has currently aroused much attention.2 In addition, significant efforts have been made in the area of transition-metal-catalyzed direct addition of C–H bonds into unsaturated bonds via the formation of active C–TM species by C–H cleavage.2,3 In particular, the annulation of aryl ketones or imines with alkynes via ortho C–H activation has been reported for the construction of valuable fused carbocycles by using the rhodium, ruthenium, and rhenium catalysts.4 While aryl amides are readily commercially available starting materials and fundamental building blocks in organic synthesis, the insertion of transition metal–carbon bonds via C–H cleavage as nucleophiles toward aryl amides remains a challenge due to their low electrophilicity.
Recently, considerable progress has been made on the addition of C–TM toward activated amides such as imides and carboxamides. Shi and coworkers reported the rhodium-catalyzed annulation of benzimides with alkynes to achieve the indenones (Scheme 1a).5 They found that the addition of the C–Rh bond formed from C–H cleavage to imide is much easier than that to amide. Kanai, Matsunaga, and coworkers explored the cobalt-catalyzed annulation of N-carbamoyl indoles and alkynes, in which the addition process of alkenyl-Cp*Co species to a carbamoyl moiety was involved to give the pyrroloindolones.6 They demonstrated the unique nucleophilic activity of the organocobalt species over the organorhodium species. Loh and coworkers disclosed the synthesis of pyrroloquinolinones by Rh-catalyzed annulation of N-carbamoyl indolines with alkynes.7 Very recently, Wang and Yu reported the ruthenium-catalyzed cycloaddition of enamides and alkynes for the assembly of pyridines (Scheme 1b).8 However, the anilides were inactive in this ruthenium catalytic system.
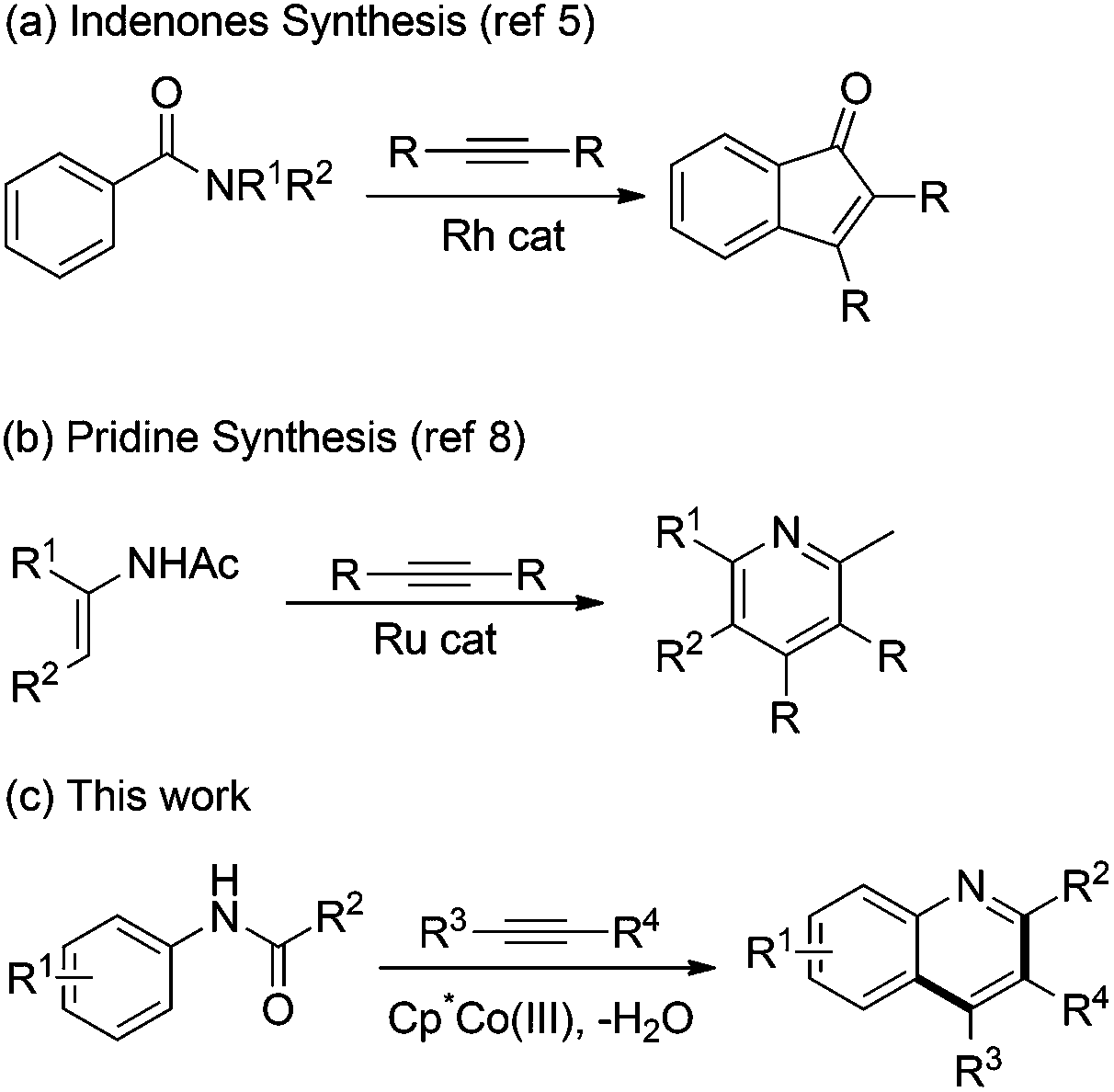 | ||
| Scheme 1 Addition of C–M bond to amide through C–H activation. | ||
To the best of our knowledge, the activation of unreactive arene C–H bonds requires the assistance of an adjacent directing group. Thus, there are two key challenges for the annulation of anilides with alkynes: (1) enhancing the electrophilicity of the directing group which mask the amino groups. (2) Increasing the nucleophilic activity of the C–TM species. Herein, we report the cobalt-catalyzed annulation of anilides with internal alkynes for the rapid synthesis of quinolines (Scheme 1c). It was found that the C–M bond of the alkenyl–CoIII species could be more polarized than that of the RhIII species and be more nucleophilic to react with a less electrophilic group.6 Furthermore, the knowledge3l,7 that Lewis acids improve the electrophilicity of amides permits a significant improvement of the annulation by using Zn(OTf)2, which may play a significant role in increasing the electrophilicity of ketones via coordination.9
Quinolines are important heterocycles and widely occur in natural products, biologically active molecules, and particular pharmaceuticals.10 Much attention has been focused on the efficient synthesis of quinolines over the past few decades11,12 and the protocol from the annulation of commercially available anilides and alkynes is highly in demand.13 We initially chose the acetanilide 1a and diphenylacetylene 2a as the modular substrate in the presence of the [Cp*Co(CO)I2]/AgSbF6 combination and catalytic KOAc in TFE at 90 °C to start our investigation. Disappointingly, no desired product was observed (Table 1, entry 1). To our delight, the replacement of KOAc with Li2CO3 gave the product 3a in 15% yield (entry 2). Considering the activation of the counter anion toward the cobalt catalyst, different silver salts were screened. We were pleased to find that catalytic AgNTf2 and AgOTf could remarkably improve the reaction efficiency (entries 3–5). The Lewis acid was regarded as a good activator to the carbonyl group. Among various Lewis acids tested, such as Cu(OTf)2, FeCl3, Sc(OTf)3 and Zn(OTf)2, Zn(OTf)2 exhibited the best reactivity to afford the quinoline product 3a in 71% yield (entries 6–9), presumably due to the increased electrophilicity of the carbonyl group of acetanilide assisted by Zn(OTf)2. Other inorganic or organic bases were also examined and the effect was inferior to that of Li2CO3 (entries 10–12). On the other hand, Li2CO3 may play a dual role in both promoting C–H activation and inhibiting the hydration of alkyne substrates to carbonyl compounds.14 Elevation of the reaction temperature to 120 °C led to a slight increase of the chemical reactivity and higher temperature failed to further improve the yield (entries 13 and 14). The solvent effect of the reaction was investigated by the employment of different solvents and TFE was the optimal choice (entries 15–18). When the reaction proceeded in the absence of [Cp*Co(CO)I2] or AgSbF6, no desired product was isolated, highlighting the necessity of [Cp*Co(CO)I2] or AgSbF6 for the reaction (entry 19).
|
|
||||
|---|---|---|---|---|
| Entry | Additive | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), [Cp*Co(CO)I2] (10 mol%), AgSbF6 (20 mol%), additive (20 mol%) and base (20 mol%) in solvent (1.5 mL) under air at 90 °C for 10 h. b Isolated yield. c At 120 °C. d At 130 °C. e Without [Cp*Co(CO)I2] or AgSbF6. | ||||
| 1 | — | KOAc | TFE | Trace |
| 2 | — | Li2CO3 | TFE | 15 |
| 3 | AgNTf2 | Li2CO3 | TFE | 56 |
| 4 | AgOAc | Li2CO3 | TFE | 25 |
| 5 | AgOTf | Li2CO3 | TFE | 52 |
| 6 | Cu(OTf)2 | Li2CO3 | TFE | 45 |
| 7 | FeCl3 | Li2CO3 | TFE | Trace |
| 8 | Sc(OTf)3 | Li2CO3 | TFE | 30 |
| 9 | Zn(OTf)2 | Li2CO3 | TFE | 71 |
| 10 | Zn(OTf)2 | CaO | TFE | 62 |
| 11 | Zn(OTf)2 | Na2CO3 | TFE | 47 |
| 12 | Zn(OTf)2 | Et3N | TFE | Trace |
| 13 | Zn(OTf)2 | Li2CO3 | TFE | 76c |
| 14 | Zn(OTf)2 | Li2CO3 | TFE | 76d |
| 15 | Zn(OTf)2 | Li2CO3 | DCE | 52c |
| 16 | Zn(OTf)2 | Li2CO3 | DMF | NRc |
| 17 | Zn(OTf)2 | Li2CO3 | 1,4-Dioxane | NRc |
| 18 | Zn(OTf)2 | Li2CO3 | Toluene | NRc |
| 19 | Zn(OTf)2 | Li2CO3 | TFE | NRc,e |
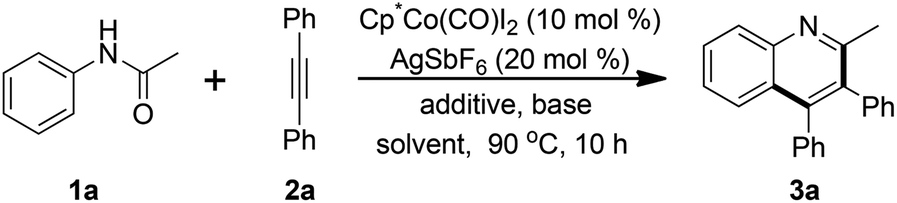
With the optimum reaction conditions in hand, the generality and limitation of the reactions was investigated (Table 2). A wide variety of acetanilides bearing electron-donating or electron-withdrawing groups at the para positions were subjected to the optimized conditions and to our satisfaction, moderate to good yields of the desired quinoline products were obtained (3a–3k). It was observed that the electron factors of acetanilide did not exert an obvious influence on the reaction. It was noteworthy that the acetanilide attached with a nitro group smoothly coupled with 2a to afford the corresponding quinoline product 3l in reasonable yield. The reaction proceeded smoothly with the use of several meta substituted or di-substituted acetanilides (3m–3o). With regard to the meta substituted substrates, the reactive site was located at the less-hindered position. The ortho substituted acetanilide could be compatible with the reaction and reduced reactivity was observed (3p), presumably due to the steric hindrance of the fluoro group. When (naphthalen-2-yl)acetamide was employed in the reaction system, the benzo[g]quinoline product 3q was furnished in 63% yield. Other types of amides, such as phenyl or ethyl amides, participated in the reaction successfully to afford the relevant quinolines (3r–3s) in acceptable yields.
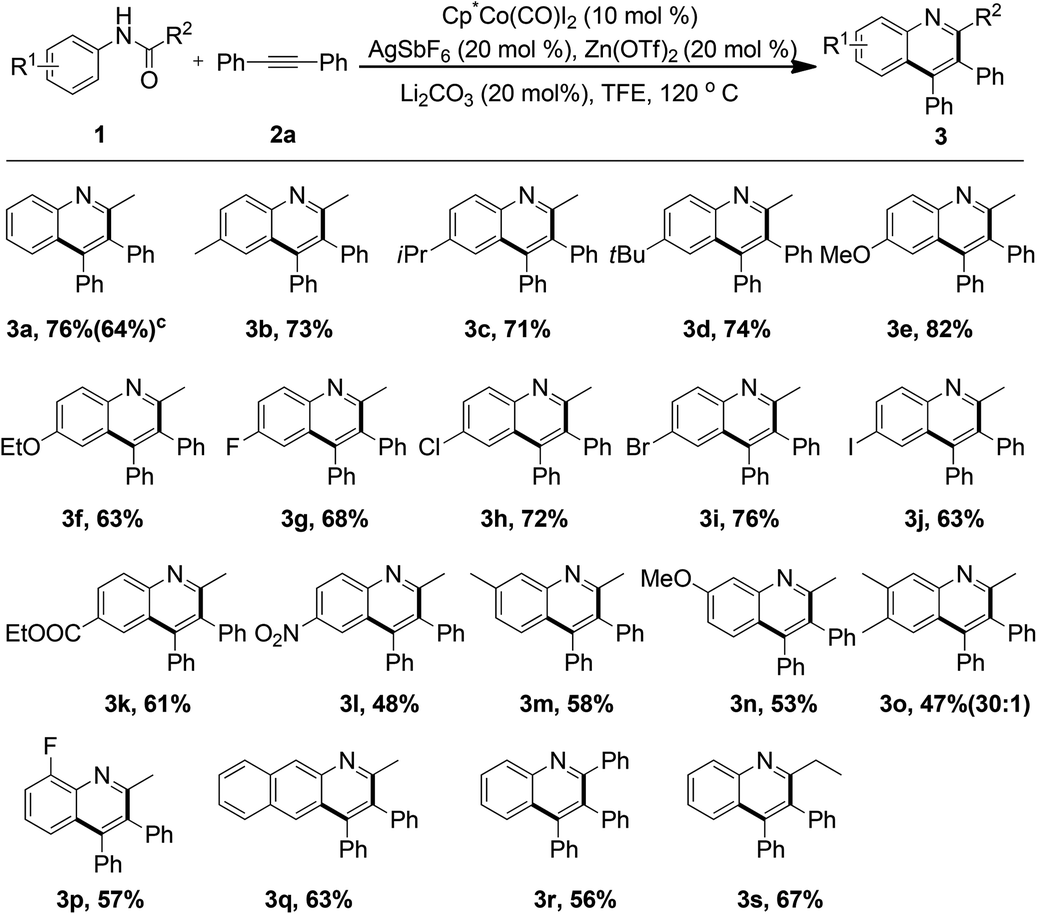
To further evaluate the versatility and compatibility of the present protocol, attention was next turned toward exploring the scope of different alkynes. Various symmetrically substituted diarylacetylenes were coupled with acetanilide 1a under the current reaction conditions to deliver the structurally diverse quinoline motifs in 52–73% yield (Table 3, 4a–4g). A number of electron-donating groups or halogen groups were well tolerated. Notably, a heterocyclic moiety, such as thiophene, could also be readily incorporated into the quinoline ring (4h). The applicability of the transformation was extended when a range of unsymmetrical alkynes were subjected to the reaction, and moderate yields of the products were observed (4i–4m, 61–66%). At this stage, the regioselectivity of the reaction was excellent (10–30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and another isomer was not isolated. More significantly, the alkyl-disubstituted alkyne could also participate smoothly in the reaction to lead to the corresponding product 4n, albeit in moderate yield. This observation further expanded the generality and practicability of the reaction.
:1) and another isomer was not isolated. More significantly, the alkyl-disubstituted alkyne could also participate smoothly in the reaction to lead to the corresponding product 4n, albeit in moderate yield. This observation further expanded the generality and practicability of the reaction.
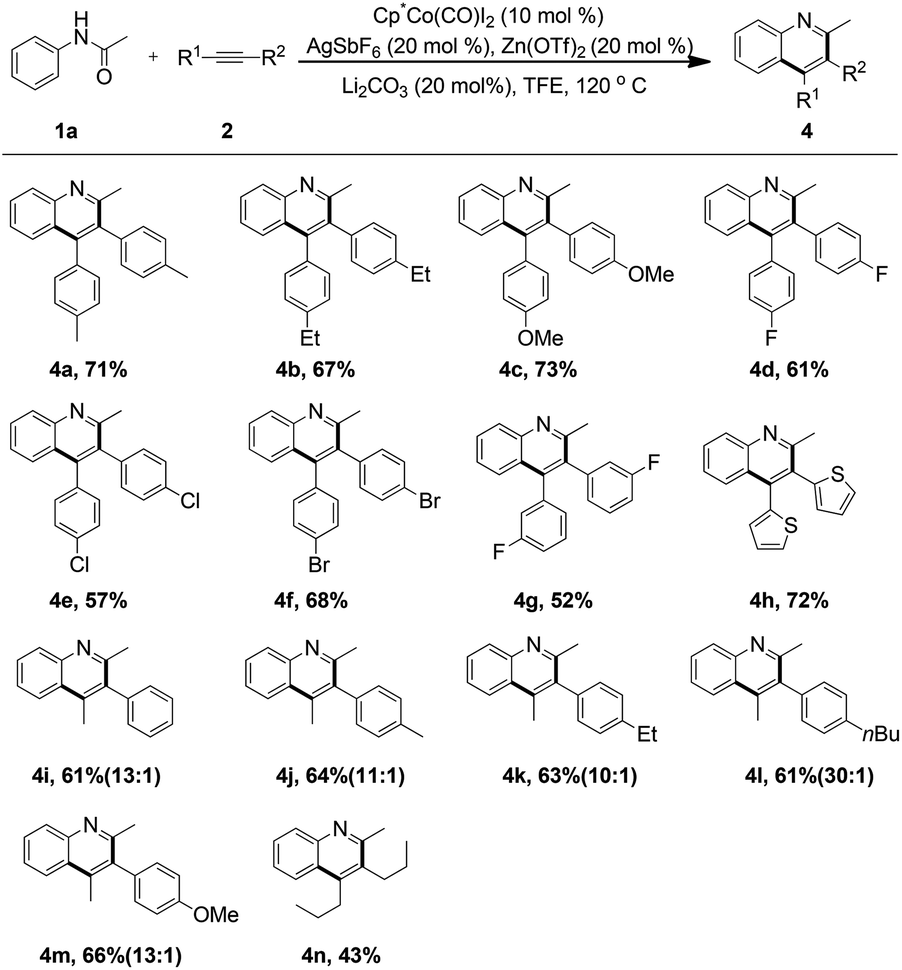
To gain further insights into the reaction mechanism, an intermolecular deuterium-labeling experiment was carried out in Scheme 2. The high kinetic isotope effect (KIE) value of 3.4 was observed from the competitive reactions of 1a and 1a-d5 with 2a (Scheme 2, eqn (1)). Moreover, a similar result was obtained from two parallel reactions (Scheme 3, eqn (2)). The preliminary results indicated that C–H activation was presumably involved in the rate-determining step.
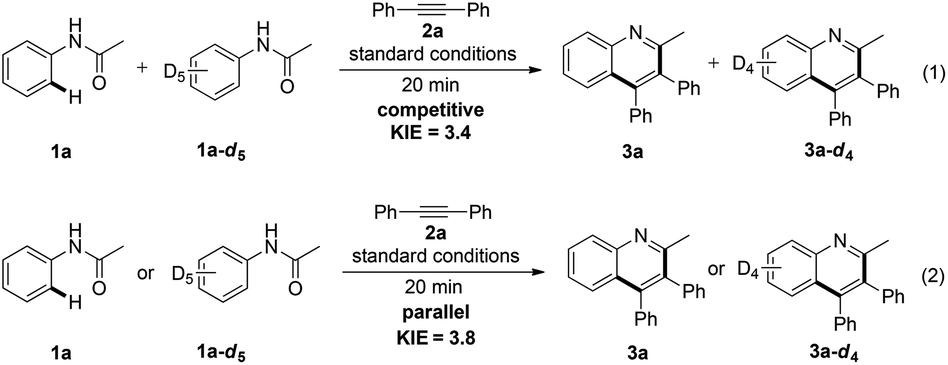 | ||
| Scheme 2 Deuterium labeling experiments. | ||
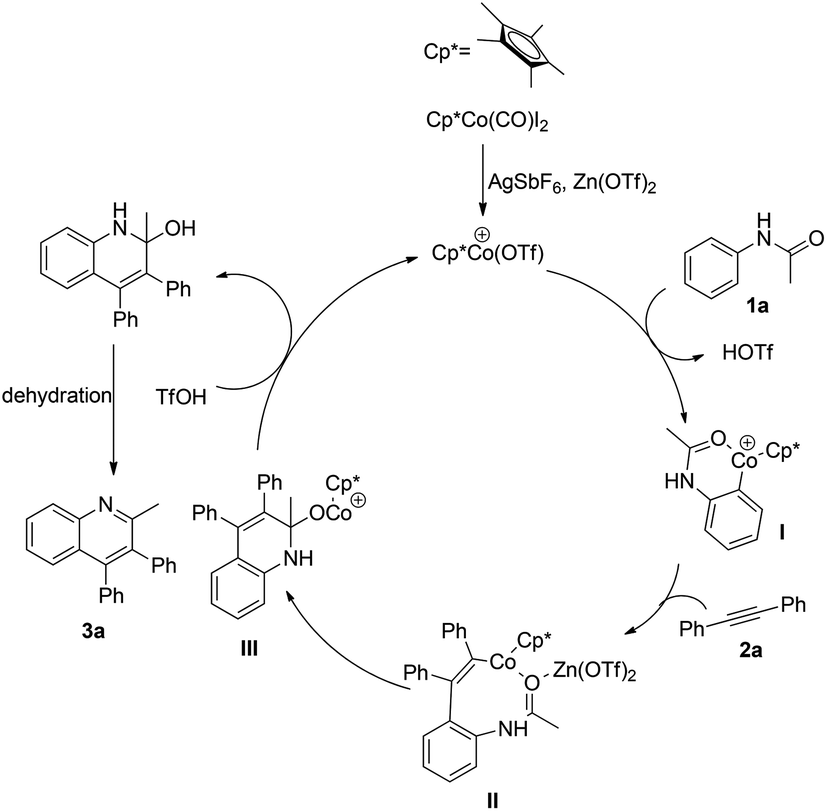 | ||
| Scheme 3 Plausible mechanism. | ||
On the basis of our understanding of the cobalt mediated catalytic cycle and the previous report,15 a plausible reaction mechanism is illustrated in Scheme 2. First, in the presence of AgSbF6 and Zn(OTf)2, an active cationic Co(III) complex is formed. Then cobalt catalyzed ortho C–H activation of acetanilide led to the six-membered cobaltacyclic intermediate I, followed by the migratory insertion of 2a into the C–Co bond to give the intermediate II. The analysis of MALDI-TOF-MS provided evidence for the possible formation of the intermediate II, which partially supports the proposed mechanism (see the ESI†). Subsequently, the C–Co bond of intermediate II could insert into the activated carbonyl group assisted by the Lewis acid Zn(OTf)2, furnishing the intermediate III, which could be protonated with the assistance of TfOH to produce a tertiary alcohol and regenerate the active Co(III) catalyst. Finally, the desired quinoline product 3a was delivered upon the dehydration of the tertiary alcohol.
In summary, we have developed a highly efficient approach for the direct synthesis of quinoline heterocycles with acetanilide and internal alkynes. The reaction pathway possibly involved the Co-catalyzed ortho C–H activation and nucleophilic addition of C–Co species toward the amides. The combined effect of the cobalt catalyst with Lewis acid Zn(OTf)2 ensured the success of the annulation. Notable features of the transformation included high efficiency, broad substrate scope and good functional group tolerance. Further investigations toward a more detailed mechanism and practical applications in the synthesis of useful complex molecules are underway in our laboratory.
Funding from the National Basic Research Program of China (no. 2011CB936003), the Natural Science Foundation of China (no. 21272205, 21472165) and the Program for Zhejiang Leading Team of S&T Innovation (2011R50007) is acknowledged.
Notes and references
- (a) G. S. Silverman and P. E. Rakita, Handbook of Grignard Reagents, Marcel Dekker, New York, 1996 Search PubMed; (b) H. G. Richey Jr. Grignard Reagents: New Development, Wiley, Chichester, U.K., 2000 Search PubMed; (c) S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069 CrossRef CAS PubMed; (d) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169 CrossRef CAS PubMed; (e) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS PubMed; (f) N. Miyaura, Synlett, 2009, 2039 CAS; (g) M. Yus, J. C. GonzalezGomez and F. Foubelo, Chem. Rev., 2011, 111, 7774 CrossRef CAS PubMed.
- For recent reviews, see: (a) G. Yan, X. Wu and M. Yang, Org. Biomol. Chem., 2013, 11, 5558 RSC; (b) X.-S. Zhang, K. Chen and Z.-J. Shi, Chem. Sci., 2014, 5, 2146 RSC; (c) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468 CrossRef CAS PubMed.
- (a) W. D. Jones, G. P. Foster and J. M. Putinas, J. Am. Chem. Soc., 1987, 109, 5047 CrossRef CAS; (b) Y. Fukumoto, K. Sawada, M. Hagihara, N. Chatani and S. Murai, Angew. Chem., Int. Ed., 2002, 41, 2779 CrossRef CAS; (c) C. Zhou and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 2302 CrossRef CAS PubMed; (d) Y. Kuninobu, Y. Nishina, C. Nakagawa and K. Takai, J. Am. Chem. Soc., 2006, 128, 12376 CrossRef CAS PubMed; (e) Y. Kuninobu, Y. Nishina, T. Takeuchi and K. Takai, Angew. Chem., Int. Ed., 2007, 46, 6518 CrossRef CAS PubMed; (f) Y. Kuninobu, Y. Fujii, T. Matsuki, Y. Nishin and K. Takai, Org. Lett., 2009, 11, 2711 CrossRef CAS PubMed; (g) K. Tsuchikama, Y. Hashimoto, K. Endo and T. Shibata, Adv. Synth. Catal., 2009, 351, 2850 CrossRef CAS; (h) B. Qian, S. Guo, J. Shao, Q. Zhu, L. Yang, C. Xia and H. Huang, J. Am. Chem. Soc., 2010, 132, 3650 CrossRef CAS PubMed; (i) Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676 CrossRef CAS PubMed; (j) H. Mizuno, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 1251 CrossRef CAS PubMed; (k) A. S. Tsai, M. E. Tauchert, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 1248 CrossRef CAS PubMed; (l) B. Zhou, Y. Hu and C. Wang, Angew. Chem., Int. Ed., 2015, 54, 13659 CrossRef CAS PubMed; (m) J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202 CrossRef CAS PubMed; (b) T. Fukutani, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Commun., 2009, 5141 RSC; (c) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050 CrossRef CAS PubMed; (d) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed; (e) K. Muralirajan, K. Parthasarathy and C.-H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 4169 CAS; (f) F. W. Patureau, T. Besset, N. Kuhl and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2154 CrossRef CAS PubMed; (g) R. K. Chinnagolla and M. Jeganmohan, Eur. J. Org. Chem., 2012, 417 CrossRef CAS.
- B.-J. Li, H.-Y. Wang, Q.-L. Zhu and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 3948 CrossRef CAS PubMed.
- H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424 CrossRef CAS PubMed.
- X.-F. Yang, X.-H. Hu and T.-P. Loh, Org. Lett., 2015, 17, 1481 CrossRef CAS PubMed.
- J. Wu, W. Xu, Z.-X. Yu and J. Wang, J. Am. Chem. Soc., 2015, 137, 9489 CrossRef CAS PubMed.
- (a) F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780 CrossRef CAS PubMed; (b) X. Zhang, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2014, 53, 10794 CrossRef CAS PubMed; (c) X. Zhang, Z. Qi, J. Gao and X. Li, Org. Biomol. Chem., 2014, 12, 9329 RSC; (d) Q. Wang, F. Xie and X. Li, J. Org. Chem., 2015, 80, 8361 CrossRef CAS PubMed; (e) D. Kang and S. Hong, Org. Lett., 2015, 17, 1938 CrossRef CAS PubMed; (f) Q. Wang, Y. Li, Z. Qi, F. Xie, Y. Lan and X. Li, ACS Catal., 2016, 6, 1971 CrossRef CAS.
- (a) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC; (b) M. Balasubramanian and J. G. Keay, in Comprehensive Heterocyclic Chemistry II, ed. A. E. McKillop, A. R. Katrizky, C. W. Rees and E. F. V. Scrivem, Elsevier, Oxford, 1996, vol. 5, p. 245 Search PubMed; (c) A. P. Gorka, A. deDios and P. D. Roepe, J. Med. Chem., 2013, 56, 5231 CrossRef CAS PubMed; (d) K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245 CrossRef CAS PubMed; (e) M. Rouffet, C. A. F. de Oliveira, Y. Udi, A. Agrawal, I. Sagi, J. A. McCammon and S. M. Cohen, J. Am. Chem. Soc., 2010, 132, 8232 CrossRef CAS PubMed; (f) S. Andrews, S. J. Burgess, D. Skaalrud, J. X. Kelly and D. H. Peyton, J. Med. Chem., 2010, 53, 916 CrossRef CAS PubMed.
- For selected reviews and examples on traditional methods, see: (a) J. Marco-Contelles, E. Prez-Mayoral, A. Samadi, M. C. Carreiras and E. Soriano, Chem. Rev., 2009, 109, 2652 CrossRef CAS PubMed; (b) J. Li, Name Reactions in Heterocyclic Chemistry, Wiley Interscience, Chichester, 2004, p. 375 Search PubMed; (c) Y. Wu, L. Liu, H. Li, D. Wang and Y. Chen, J. Org. Chem., 2006, 71, 6592 CrossRef CAS PubMed; (d) R. Zong, H. Zhou and R. P. Thummel, J. Org. Chem., 2008, 73, 4334 CrossRef CAS PubMed.
- For selected examples on the C–H activation pathway, see: (a) S. Kaur, M. Kumar and V. Bhalla, Chem. Commun., 2015, 51, 16327 RSC; (b) X. Liu, X. Li, H. Liu, Q. Guo, J. Lan, R. Wang and J. You, Org. Lett., 2015, 17, 2936 CrossRef CAS PubMed; (c) S. P. Shukla, R. K. Tiwari and A. K. Verma, J. Org. Chem., 2012, 77, 10382 CrossRef CAS PubMed; (d) G. Song, X. Gong and X. Li, J. Org. Chem., 2011, 76, 7583 CrossRef CAS PubMed; (e) X. Ji, H. Huang, Y. Li, H. Chen and H. Jiang, Angew. Chem., Int. Ed., 2012, 51, 7292 CrossRef CAS PubMed; (f) X. Yi and C. Xi, Org. Lett., 2015, 17, 5836 CrossRef CAS PubMed.
- During our preparation of this manuscript, the Co-catalyzed synthesis of quinoline has been reported: L. Kong, S. Yu, X. Zhou and X. Li, Org. Lett., 2016, 18, 588 CrossRef CAS PubMed.
- S. Liang, G. B. Hammond and B. Xu, Chem. Commun., 2015, 51, 903 RSC.
- (a) D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508 CrossRef CAS PubMed; (b) S. Prakash, K. Muralirajan and C.-H. Cheng, Angew. Chem., Int. Ed., 2016, 55, 1844 CrossRef CAS PubMed; (c) J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103 CrossRef CAS PubMed; (d) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for new compounds. See DOI: 10.1039/c6qo00059b |
| This journal is © the Partner Organisations 2016 |