
Tetramethoxy-bay-substituted perylene bisimides by copper-mediated cross-coupling†
Pawaret
Leowanawat
,
Agnieszka
Nowak-Król
and
Frank
Würthner
*
Universität Würzburg, Institut für Organische Chemie & Center for Nanosystems Chemistry, Am Hubland, 97074 Würzburg, Germany. E-mail: wuerthner@chemie.uni-wuerzburg.de
First published on 18th February 2016
Abstract
CuBr-mediated cross-coupling of 1,6,7,12-tetrabromoperylene-3,4,9,10-tetracarboxylic acid ester or 1,6,7,12-tetrabromoperylene-3,4:9,10-bis(dicarboximide) with sodium methoxide affords the respective tetramethoxy-perylene dye derivatives which are excellent green (λem ≈ 555 nm) and red (λem ≈ 635 nm) emitters, respectively, with high fluorescence quantum yields (60–70%).
Introduction
Perylene bisimides (PBIs) are among the most useful fluorescence dyes due to their unique combination of high fluorescence quantum yield (often close to unity) and outstanding thermal and (photo-)chemical stability.1 Compared to other esteemed fluorescence dyes such as for instance BODIPYs2 or squaraines,3 there is only one disadvantage for PBIs: due to the fixed length of the chromophore absorption and emission wavelengths cannot be adjusted over a wider spectral range. Thus, the absorption and emission maxima of strong PBI emitters remain limited to a spectral range from about 500 nm to about 580 nm. The most successful strategy to cover this spectral range is a substitution of the 1,6,7,12 bay positions by halogen or oxygen heteroatoms where the former affords a hypsochromic and the latter a bathochromic shift compared to the parent PBI dye (λabs = 523–527 nm; λem = 530–532 nm in dichloromethane).1 Functionalization with nitrogen donor atoms such as 1,7-dipyrrolidino proved to be a successful strategy to push the absorption (λabs ≈ 685 nm) and emission (λem ≈ 720 nm) bands towards longer wavelengths,4 but with a concomitant loss of the fluorescence quantum yield and photostability of the now far more electron rich π-scaffold owing to a different character of the highest occupied molecular orbital (HOMO). Likewise, other strategies such as a substitution of PBIs in the 2,5,8,11 positions5 or a longitudinal expansion to higher homologous rylene bisimides6 were introduced as alternative strategies but in general do not afford the outstandingly high fluorescence quantum yields appreciated for PBIs. One exception is the terrylene bisimides which indeed can afford excellent emitters at around λem = 670–710 nm with fluorescence quantum yields up to 90%.6Accordingly, the most successful strategy to tune the absorption and emission wavelengths of PBIs remains bay substitution.7 Here, 1,7-disubstitution from 1,7-dibromo-PBIs by all kinds of transition metal catalyzed cross-coupling reactions is synthetically more appealing whilst 1,6,7,12-tetrasubstitution is more challenging and usually involves 1,6,7,12-tetrachloro PBIs as starting materials. The latter can afford, however, more bathochromically absorbing and emitting dyes, e.g. 1,7-diphenoxy-PBIs absorb at λabs ≈ 550 nm while 1,6,7,12-tetraphenoxy-PBIs absorb at λabs ≈ 575 nm.1a However, the fourfold substitution proved to be far more challenging and only special nucleophiles like phenoxy and thiolates could be reacted successfully with tetrahalogenated PBIs, pointing at a radical-nucleophilic aromatic substitution pathway.8 Thus, whilst 1,7-dialkoxylated PBIs are easily accessible,9 their 1,6,7,12-tetraalkoxylated counterparts are still unknown compounds in this very popular field of research. Here we introduce an efficient synthesis of the first derivatives of this class of dyes and reveal their outstanding absorption and emission properties which extend the spectral range of PBI emitters beyond the 600 nm threshold.
Results and discussion
Synthesis
The starting material for our research was 1,6,7,12-tetrabromo-perylene bisanhydride 2 which was obtained according to the procedure of Zhu and co-workers by bromination of perylene tetracarboxylic bisanhydride 1 in oleum.10 This compound can be converted either to perylene bisimides, e.g. PBI 3 by imidization (Route A) or to the respective perylene tetracarboxylic esters (PTE), e.g. PTE 4 (Route B) (Scheme 1). Initially, we selected the short synthetic pathway via Route A to obtain tetramethoxy-bay-substituted PBI 7c by employing CuBr-mediated cross-coupling (Ullmann-type reaction) of PBI 3 and sodium methoxide under conditions reported by Aalten et al.11 for the synthesis of anisole derivatives. This reaction produced 7c in 20% yield along with the decomposition products. We noticed that the prolonged reaction time resulted in the consumption of the product and low recovered yield. This is consistent with the previously reported observation that electron-deficient PBI derivatives may undergo a nucleophilic attack of methoxide on one of the carbonyl groups, which initiates a rearrangement.12 To avoid this plausible process, we decided to explore an alternative route (Route B) involving a cross-coupling reaction between PTE 4 and NaOMe, with subsequent cyclization and imidization to a PBI 7 in the last step (Scheme 1). To this end, compound 4 was synthesized by adapting the reported procedure that employs DBU-promoted saponification of bisanhydride 2 and the consecutive alkylation of the intermediate with bromobutane.13 The following CuBr-mediated cross-coupling of PTE 4 with NaOMe afforded tetramethoxy-bay-substituted PTE 5 along with several side-products. Careful chromatography allowed us to isolate the desired PTE 5 in 33% yield. The structure of this key intermediate was confirmed by single crystal X-ray analysis (vide infra). Furthermore, MS and NMR data revealed that 5 was accompanied by compounds of incomplete methoxylation as well as dehalogenation. The main side-product formed in the reaction was 1,6,7-trimethoxy-substituted PBI 5′ isolated in 26% yield. The analogous reaction with the nucleophiles of lower nucleophilicity, e.g. butoxide gave rise exclusively to the dehalogenation products. These results clearly show how challenging the fourfold substitution of the bromine atoms in the bay areas is. In the next step tetraester 5 was converted into bisanhydride 6 in a quantitative yield by applying an excess of p-TsOH under conditions reported by Jager and co-workers.14 Subsequent imidization of compound 6 with the selected amines in propionic acid at 120 °C gave rise to the desired tetramethoxy-bay-substituted PBIs 7a–c in moderate to very high yields.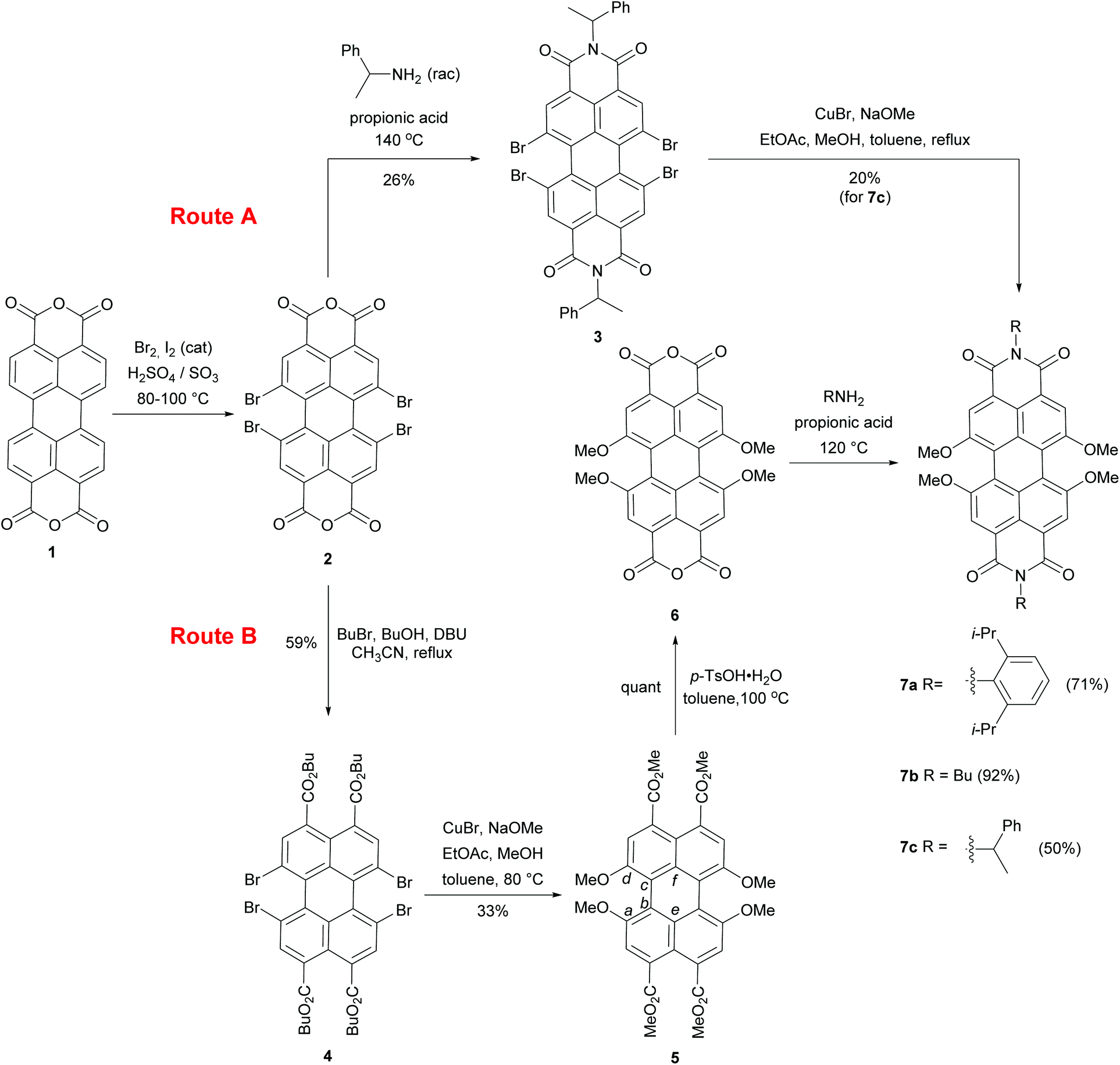 | ||
| Scheme 1 Synthesis of perylene tetraesters and perylene bisimides bearing four methoxy substituents in 1,6,7,12 bay positions. | ||
Solid state structural analysis
Single crystals of 5 suitable for X-ray structural analysis were grown by slow evaporation of a saturated toluene solution of 5. Compound 5 crystallizes in the monoclinic P21/c space group with molecules located at general positions. Fig. 1a and b show the molecular structure of 5. Due to the steric encumbrance effect of four methoxy groups at bay positions the perylene core is highly twisted. The distortion of the perylene π-scaffold was estimated following two different approaches. In the first approach, the perylene scaffold was considered as two separate naphthalene subunits and the rotational twist of the mean naphthalene planes with respect to each other was assigned as the twist angle of 31.4°. In the second method we measured dihedral angles associated with the positions of either four bay carbon atoms (a) Ca, Cb, Cc, and Cd (∠(Ca–Cb–Cc–Cd), for numbering see Scheme 1) or four inner carbons (b) Ce, Cb, Cc, and Cf (∠(Ce–Cb–Cc–Cf)) for each bay area of the PBI scaffold. The dihedral angles determined in this way are 37.4(3)°, 33.5(3)° for ∠(Ca–Cb–Cc–Cd) and 30.7(3)°, 31.9(3)° for ∠(Ce–Cb–Cc–Cf), i.e. slightly different for both sides of the perylene core, providing evidence of a non-centrosymmetrical structure. The unsymmetrical twisting has been noted before for other bay-substituted PBIs7 and is probably not originating from packing effects in the crystal lattice but an intrinsic feature of this molecular scaffold. All the C–C bond lengths within the two naphthalene moieties range between 1.368(3) and 1.433(3) Å and the longest bonds in the perylene backbone are accordingly those connecting the two naphthalene planes at distances of 1.475(3) and 1.465(3) Å, similar to other perylene derivatives.15 This feature makes the central six-membered ring more susceptible to distortions than the rest of the molecule.1a,16 When compared with the torsion angles of tetrachloro-substituted perylene, this value is somewhat smaller. For the perylene bearing four chlorine atoms dihedral angles between the two naphthalene planes of 38.6° and 36.7° were reported and attributed to the repulsion between the chlorine atoms at bay positions.17,18 In contrast to the latter example the torsion angle in tetraphenoxy PBI was markedly smaller (∼25°).19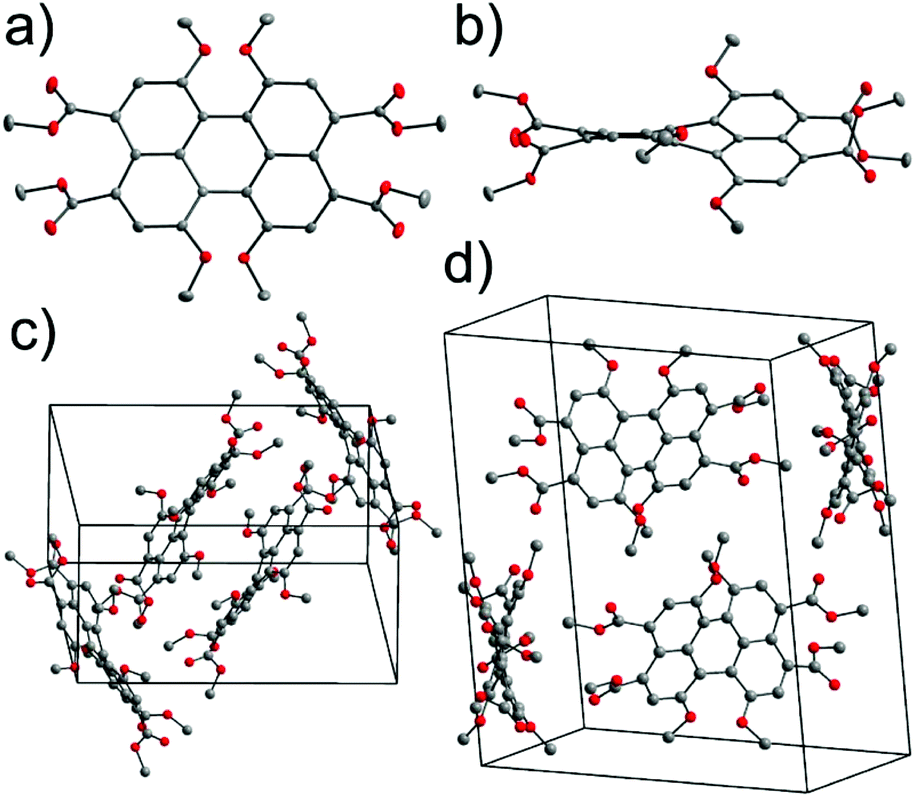 | ||
| Fig. 1 Molecular structure in front view (a), side view (b), and packing (c and d) of compound 5 in the single crystal as determined by single crystal X-ray analysis at 100 K. C, grey; O, red. Hydrogen atoms and solvent molecules were omitted for clarity. | ||
Another effect of sterical congestion is observed for the four carboxylic acid esters at the 3,4,9,10 peri positions. Thus, whilst the carbonyl groups are fully conjugated to the perylene core for the bisanhydrides and bisimides, for these tetraesters such conjugation is prohibited for sterical reasons. As we will show later this has important consequences for the absorption and emission properties of PTE 5. The packing arrangement of 5 is defined by two orientations of the PTE molecules with a mutual orientation of two molecules that is characterized by a dihedral angle of 60.2° between their mean planes and a CH–π interaction represented by a distance of 3.58 Å between the methyl group of one molecule and the centroid located in the center of the Cc–Cf bond (Fig. S1, ESI†) of an adjacent PTE. The distance between the mean planes of longitudinally slipped parallel PBI molecules is only 3.37 Å. However, the considerable transverse offset imparted by the contorted π-scaffolds prevents any effective overlap between the perylene π-planes (Fig. 1c and d).
The X-ray analysis data for 5 showed a substantial twist for PTE bearing four methoxy groups. This twist could also be confirmed by DFT calculations at the B3LYP/def2-SVP level for the simplified model compound of 7 (imide substituents were replaced by methyl groups), as well as for 5 for comparison. The torsion angles between the two naphthalene planes in compound 5 and the model PBI 7 calculated for vacuum are 25.75° and 28.76°, respectively, which shows that the twist in both molecules should be rather similar. These distortions find their consequences in the optical properties of the studied molecules.
Absorption and emission properties
The optical properties of the chromophores 3–7 were investigated in solution by steady-state absorption and emission spectroscopy.
Fig. 2a presents the absorption spectra of PBI 7a (blue line) in comparison with the PBI bearing phenoxy substituents (PBI 9, red line) at the bay positions, and parent PBI 8 (black line). The structures of reference PBIs 8 and 9 are shown in Chart 1. The spectrum of unsubstituted PBI 8 displays three sharp vibronic progressions for the S0–S1 transition with maxima between ca. 450–530 nm characterized by high molar absorption coefficients (ca. 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 for 0–0 transition). In pronounced contrast, for compound 7a and reference 9 broadening of the lineshapes and the loss of the vibronic fine structures were observed. It is noteworthy that these features are distinctive (with some exceptions) of most of the bay-substituted PBIs and are attributed to the core distortion out of planarity arising from a steric congestion in the bay area.15 A solution of PBI 7a in dichloromethane (DCM) exhibits purple-blue color. The spectrum of this compound reveals a broad absorption band at 610 nm, and a second vibronic progression at 566 with a shoulder around 520 nm corresponding to the S0–S1 transition. Along with the considerable broadening of these bands the drop of their maximal absorption intensities were observed. Accordingly, ε of the 0–0 transition decreased significantly to 60500 M−1 cm−1. On the other hand, the intensity of the band with a maximum at ca. 442 nm, assigned to the S0–S2 transition, increased. A similar signature can be observed in the spectrum of phenoxy-substituted PBI 9. Most interestingly, the maximum of the lowest absorption band of 7a is shifted by around 80 nm (2600 cm−1) towards lower energies as compared to the corresponding transition of PBI 8. The large bathochromic shift of this transition reflects the substantial electronic interactions between relatively strong electron-donating methoxy groups and an electron-deficient PBI core. Accordingly, replacement of OMe with OPh groups results in a less pronounced bathochromic shift vs.8 (of about 50 nm; 1600 cm−1), which coincides with the less electron-donating character of the OPh substituent. The spectra of the other two tetramethoxy-PBIs 7b and 7c closely resemble the spectrum of 7a in terms of lineshapes and positions (for details see Table 1 and Fig. S5 and S6, ESI†). In the case of tetraesters 4 and 5 the characteristic perylene fine structures are almost completely lost (Fig. 2b and S4, ESI†). The key intermediate – tetramethoxy-PTE 5 is orange in DCM solution with an absorption maximum at 505 nm. The significant hypsochromic shift vs. PBIs 7 is obviously due to the lack of conjugation to the electron-accepting carbonyl functions which are twisted out of the naphthalene planes (compare Fig. 1).
000 M−1 cm−1 for 0–0 transition). In pronounced contrast, for compound 7a and reference 9 broadening of the lineshapes and the loss of the vibronic fine structures were observed. It is noteworthy that these features are distinctive (with some exceptions) of most of the bay-substituted PBIs and are attributed to the core distortion out of planarity arising from a steric congestion in the bay area.15 A solution of PBI 7a in dichloromethane (DCM) exhibits purple-blue color. The spectrum of this compound reveals a broad absorption band at 610 nm, and a second vibronic progression at 566 with a shoulder around 520 nm corresponding to the S0–S1 transition. Along with the considerable broadening of these bands the drop of their maximal absorption intensities were observed. Accordingly, ε of the 0–0 transition decreased significantly to 60500 M−1 cm−1. On the other hand, the intensity of the band with a maximum at ca. 442 nm, assigned to the S0–S2 transition, increased. A similar signature can be observed in the spectrum of phenoxy-substituted PBI 9. Most interestingly, the maximum of the lowest absorption band of 7a is shifted by around 80 nm (2600 cm−1) towards lower energies as compared to the corresponding transition of PBI 8. The large bathochromic shift of this transition reflects the substantial electronic interactions between relatively strong electron-donating methoxy groups and an electron-deficient PBI core. Accordingly, replacement of OMe with OPh groups results in a less pronounced bathochromic shift vs.8 (of about 50 nm; 1600 cm−1), which coincides with the less electron-donating character of the OPh substituent. The spectra of the other two tetramethoxy-PBIs 7b and 7c closely resemble the spectrum of 7a in terms of lineshapes and positions (for details see Table 1 and Fig. S5 and S6, ESI†). In the case of tetraesters 4 and 5 the characteristic perylene fine structures are almost completely lost (Fig. 2b and S4, ESI†). The key intermediate – tetramethoxy-PTE 5 is orange in DCM solution with an absorption maximum at 505 nm. The significant hypsochromic shift vs. PBIs 7 is obviously due to the lack of conjugation to the electron-accepting carbonyl functions which are twisted out of the naphthalene planes (compare Fig. 1).
 | ||
| Fig. 2 (a) Absorption spectra of selected PBIs: 7a (blue line, 9.8 × 10−6 M), 8 (black line, 8.5 × 10−6 M), 9 (red line, 1.10 × 10−5 M) in CH2Cl2; (b) absorption and emission spectra of 5 (orange lines), 7a (blue lines), 8 (black lines), and 9 (red lines) in CH2Cl2 normalized to the 0–0 transition. Solid and dashed lines depict absorption and emission spectra, respectively. | ||
 | ||
| Chart 1 Structures of the reference compounds 8 and 9. | ||
| Cpd | λ abs [nm] | λ em [nm] | Stokes shift [cm−1] | Φ fl , |
|---|---|---|---|---|
| a Absorption maximum. b Fluorescence maximum. c Relative fluorescence quantum yields determined by the optical dilution method. d Absolute fluorescence quantum yield determined with an integrating sphere. | ||||
| 3 | 531 | 570 | 1300 | 0.42d |
| 4 | 463 | 520 | 2400 | 0.01d |
| 5 | 505 | 555 | 1800 | 0.63c |
| 7a | 610 | 637 | 700 | 0.68c |
| 7b | 604 | 632 | 700 | 0.71c |
| 7c | 608 | 638 | 800 | 0.71d |
| 8 | 527 | 534 | 200 | 0.9920 |
| 9 | 576 | 606 | 900 | 0.9620 |
The fluorescence properties of PTEs 4 and 5, PBIs 3 and 7a–c, as well as reference compounds are summarized in Table 1. The normalized absorption and fluorescence spectra of 5, 7a, 8, and 9 are shown in Fig. 2b (for other derivatives see also Fig. S3–S6, ESI†). The emission spectra of the tetraesters are devoid of vibronic structure. The fluorescence peak maximum of 5 is observed at 555 nm, which corresponds to the large Stokes shift of 1800 cm−1. Likewise, the emission spectra of 7a−c are rather structureless, in striking contrast to the emission spectrum of reference 8 characterized by well-resolved vibronic peaks. The emission maxima of 7a–c are bathochromically shifted (632–638 nm) compared to PBIs 8 and 9. The corresponding Stokes shifts (700–800 cm−1) are much lower than for PTE 5, which can be associated with the more rigid molecular framework of PBIs, thereby with less pronounced geometry relaxation upon photoexcitation. On the other hand, the Stokes shift values are much higher when compared to the parent PBI 8 demonstrating still significant excited state relaxations.
Fluorescence quantum yields of tetraesters 4 and 5 as well as PBIs 3, and 7a–c have been determined in DCM (Table 1). Tetramethoxy-PTE 5 is fluorescent with Φfl > 0.60. Remarkably, all the investigated tetramethoxy-PBIs, despite their significant bathochromic shift, still display high fluorescence quantum yields (Table 1), which indicates the minor impact of non-radiative processes. For comparison, 1,7-dipyrrolidino-PBI bearing the same imide substituents exhibits only a very low Φfl of 0.19 in DCM.20 The dramatic decrease in the fluorescence quantum yield of dipyrrolidino-PBI vs. unsubstituted PBI 8 was attributed to the deactivation pathway via charge transfer between the electron-rich substituents and the PBI core playing as an electron acceptor. Obviously, this effect is not yet as prominent for the fourfold methoxylated PBIs. For a better understanding and to evaluate the scope of tetramethoxy-PBIs for fluorescence-related applications we investigated nevertheless also the absorption and emission properties of PBI 7a in a set of solvents of diversified polarity: tetrahydrofuran (THF), acetonitrile (MeCN), and toluene. The data collected in Table 2 reveal a rather negligible solvent effect. The variations in the positions, intensities and shapes of the absorption spectra, as well as emission maxima are dependent marginally on the solvent polarity (Fig. S7, ESI†). Likewise, the fluorescence quantum yields are rather weakly influenced by the solvent polarity. In MeCN Φfl slightly decreases when compared to DCM, which may imply a charge transfer character.
Electrochemistry
The electronic consequences of the introduction of four methoxy groups into bay positions of PBIs were also investigated by cyclic voltammetry (CV), square wave voltammetry (SWV) and differential pulse voltammetry (DPV). Half-wave redox potentials obtained from CV measurements for target PBIs 7a and 7b and reference molecules 8 and 9 are summarized in Table 3. Cyclic voltammograms of 7a, 8, and 9 are shown in Fig. 3 and S8 (ESI†).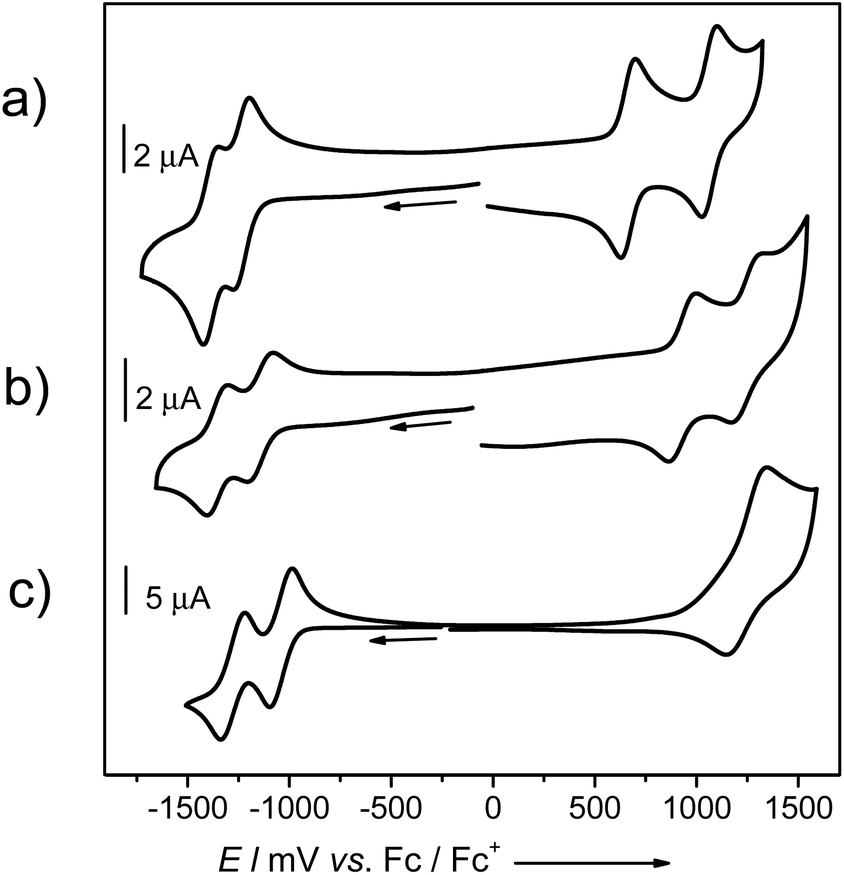 | ||
| Fig. 3 Cyclic voltammograms of (a) 7a (2.3 × 10−4 M), (b) 9 (2.2 × 10−4 M), and (c) 8 (1.10 × 10−3 M) in CH2Cl2 solution of Bu4NPF6 (0.1 M) at a scan rate of 100 mV s−1. The measurements were calibrated with an internal standard (ferrocene/ferrocenium). | ||
| Cpd | E 1/2 (Red2) [V] | E 1/2 (Red1) [V] | E 1/2 (Ox1) [V] | E 1/2 (Ox2) [V] |
|---|---|---|---|---|
| a Redox potentials vs. ferrocene/ferrocenium (Fc/Fc+) in CH2Cl2 solutions, using Bu4NPF6 (0.1 M) as a supporting electrolyte at a scan rate of 100 mV s−1. | ||||
| 7a | –1.39 | –1.24 | +0.66 | +1.06 |
| 7b | –1.43 | –1.30 | +0.62 | +1.08 |
| 9 | –1.35 | –1.14 | +0.93 | +1.25 |
| 8 | –1.26 | –1.02 | +1.27 | — |
All the investigated compounds exhibited two reversible reduction waves that can be correlated with the formation of radical anions and dianions. The relative electron-donating ability of a particular substituent is reflected in the values of both the oxidation and reduction potentials of PBIs. The tendency towards reduction decreases in the following order H > OPh > OMe. A voltammogram of non-substituted 8 displays two reduction waves at E1/2 of −1.02 and −1.26 V. Upon introduction of four phenoxy groups the first and the second reductions are shifted to lower potentials by ca. 0.1 V and occur at −1.14 and −1.35 V, respectively. A further shift of around 0.1–0.15 V was determined for PBIs bearing methoxy substituents to give first reduction potentials of −1.24 and −1.30 V for 7a and 7b, respectively. The corresponding E1/2 (Red2) waves were measured at −1.39 and −1.43 V. As expected, the substituent effect is more pronounced for the oxidation process of the series of PBIs leading to higher variations in E1/2 (Ox). For the parent PBI 8 only one reversible oxidation wave could be observed within the scanning range of the employed electrolyte (tetrabutyl ammonium hexafluorophosphate in dichloromethane) at a half-wave potential of +1.27 V. Upon introduction of the electron-donating substituents the oxidation processes became more feasible. Thus, voltammograms of all methoxy-substituted PBIs 7a and 7b as well as tetraphenoxy-PBI 9 displayed two well-defined reversible oxidation waves shifted to lower potentials vs. E1/2 of unsubstituted PBI. These processes correspond to the stepwise formation of radical cations and dications of perylene bisimides. As expected, due to the strong electron-releasing character of the methoxy group, PBIs 7a and 7b are quite easily oxidized at such low potentials as +0.66 and +0.62 V which are already in the range of common p-type seminconductors. The HOMO energy levels of PBIs 7a and 7b, calculated from CV measurements, with the energy level of Fc/Fc+ set to −4.8 eV vs. vacuum,21 are accordingly rather high for these PBIs: −5.46 and −5.42 eV. The second oxidations occur at +1.06 and +1.08 V, respectively. Similar to the trend observed for the reduction order of PBIs 7, 8, and 9, the first E1/2 (Ox1) of PBI 9 (+0.93 V) lies somewhere in the middle between the potentials of 7 and 8. For comparison, the oxidation of 1,7-dipyrrolidino-PBI1a is observed at a considerably lower potential of +0.03 V, which corresponds to the HOMO level of −4.83 eV.
It is noteworthy that four methoxy substituents do not lift the HOMO level as substantially as the two pyrrolidino groups attached to 1 and 7 bay carbon atoms. Thus, we could expect a higher (photo-)chemical stability of PBIs 7a–c. To shed light on the photostability of these dyes under ambient conditions we exposed the solutions of 7a and 7b in DCM to sunlight and measured the absorption spectra at time intervals of 2–3 h over the total time of 10 h. In contrast to pyrrolidino-substituted PBIs, neither shapes, positions, nor the intensity of absorption bands underwent any noticeable change, which demonstrates that tetramethoxy-PBIs are in the ‘safe’ range of potentials of robust and air-stable materials.
Conclusions
In this article we have shown the first example of copper-mediated cross-coupling for a 1,6,7,12-tetrabromo-perylene that gives rise to tetramethoxy-substituted perylene tetracarboxylic acid ester in a satisfactory yield. The latter serves as a key intermediate in the synthesis of the corresponding PBIs. These new members of the PBI family display interesting photophysical properties, such as bathochromically shifted absorption and emission (beyond 600 nm), and most notably high fluorescence quantum yields of around 70% in DCM. In particular, upon the introduction of the four OMe donating groups, a significant modulation of the electronic band gap was achieved, while still keeping Φfl at a decent level. These features, along with high stability under ambient conditions, make tetramethoxy-PBIs attractive building blocks for further functionalization that could give access to a large variety of PBIs for utilization in supramolecular photosystems and organic materials.Experimental
General
All reagents were purchased from commercial sources and used as received without further purification, unless otherwise stated. Reagent grade solvents were distilled prior to use. Dry methanol was prepared by treatment with magnesium turnings activated by iodine and fractional distillation. Ethyl acetate was kept over K2CO3 and fractionally distilled from P2O5. Column chromatography was performed on silica (silica gel, 230–400 mesh). 1H and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer and calibrated to the residual solvent signals. J values are given in Hz. The following abbreviations were used to designate multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet, q = quartet, sep = septet. High resolution mass spectra (ESI) were obtained by electrospray ionization (ESI) and were recorded on an ESI micrOTOF Focus spectrometer from Bruker Daltonics. Melting points were determined on an optical microscope and are uncorrected. Compounds 2 and 3 were prepared according to the literature procedures.22Steady-state UV/Vis absorption and fluorescence spectroscopy
All spectroscopy measurements were conducted with spectroscopic grade solvents (Uvasol®) from Merck (Hohenbrunn, Germany) by using conventional quartz cells (light path 1 cm). UV/Vis spectra were recorded on Perkin-Elmer UV/Vis spectrometers Lambda 35 or Lambda 950. Steady-state fluorescence emission spectra were recorded with a PTI QM-4/2003 spectrometer. Polarizers in the specified set-up applying magic angle conditions (54.7°) were used. Relative fluorescence quantum yields were determined for 5, 7a (in all the solvents used) and 7b by the optical dilution method (ODmax < 0.05) and were determined as the average value for three different excitation wavelengths using N,N′-bis-(2,6-di-isopropylphenyl)-1,6,7,12-tetraphenoxy-perylene-3,4:9,10-tetracarboxylic acid bisimide (Φfl (CHCl3) = 0.96)23 and N,N′-di-n-octyl-2-chloro-6-n-octylamino-1,4,5,8-naphthalenetetracarboxylic acid diimide (Φfl (CH2Cl2) = 0.58)24 as reference compounds. For compounds 3, 4, and 7c absolute fluorescence quantum yields were recorded on the Hamamatsu instrument equipped with an integrating sphere.Cyclic voltammetry (CV)
The CV measurements were performed on a standard, commercial electrochemical analyzer (EC epsilon; BAS Instruments, UK) in a three electrode single-compartment cell under an argon atmosphere. Dichloromethane (HPLC grade) was dried over calcium hydride under an argon atmosphere, distilled, and degassed prior to use. The supporting electrolyte NBu4PF6 was synthesized according to the literature,25 recrystallized from ethanol/water, and dried in a high vacuum. The measurements were carried out under the exclusion of air and moisture at a concentration of approximately 2.5 × 10−4 M with ferrocene as an internal standard for the calibration of the potential. Working electrode: glassy carbon or Pt disc; reference electrode: Ag/AgCl; auxiliary electrode: Pt wire.Synthetic procedures and product characterization
100), 401 nm (12300). 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 4H), 4.36 (m, 8H), 1.81 (m, 8H), 1.51 (m, 8H), 1.01 (t, J = 7.4 Hz, 12H). 13C NMR (101 MHz, CDCl3) δ 167.0, 135.1, 134.0, 130.6, 130.4, 123.8, 122.7, 60.2, 30.7, 19.4, 13.9. MS HR (ESI) [M + Na]+m/z calcd for C40H40Br4NaO8: 986.9349, found 986.9362. Fluorescence (CH2Cl2): λmax = 520 nm (λex = 450 nm); Φfl = 0.01.
:3) to give 5 as orange crystals (103 mg, 33%) and 5′ (77 mg, 26%). Compound 5: mp 271–272 °C. UV/Vis λmax (CH2Cl2, c = 2.0 × 10−5 M)/nm 505 (ε/dm3 mol−1 cm−1 25700), 388 nm (7100). 1H NMR (400 MHz, CDCl3) δ 7.80 (s, 4H), 4.01 (s, 12H), 3.93 (s, 12H). 13C NMR (101 MHz, CDCl3) δ 169.3, 155.0, 134.8, 128.9, 118.0, 116.6, 115.5, 56.5, 52.3. MS HR (ESI) m/z calcd for C32H28NaO12 [M + Na]+ 627.1473, found 627.1466. Fluorescence (CH2Cl2): λmax = 555 nm (λex = 475 nm); Φfl = 0.63 ± 0.02. Compound 5′: 1H NMR (400 MHz, CDCl3) δ 8.83 (d, J = 8.1 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.91 (s, 1H), 7.84 (s, 1H), 7.80 (s, 1H), 4.10 (s, 3H), 4.09–4.04 (m, 6H), 3.99–3.87 (m, 12H). 13C NMR (101 MHz, CDCl3) δ 169.5, 169.1, 169.0, 155.6, 155.2, 154.4, 134.1, 132.9, 131.2, 130.2, 129.2, 129.1, 128.1, 127.7, 126.3, 122.7, 118.9, 118.3, 117.7, 117.3, 117.0, 116.0, 115.8, 56.8, 56.5, 56.5, 52.4, 52.4, 52.3, 52.2. MS HR (ESI) m/z calcd for C31H26O11 M+ 574.1475, found 574.1470.
500), 248 (47500), 266 (34100), 283 (33500), 566 (32400), 442 (11400). 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 4H), 7.51 (t, J = 7.8 Hz, 2H), 7.36 (d, J = 7.8 Hz, 4H), 4.27 (s, 12H), 2.77 (sep, J = 6.8 Hz, 4H), 1.19 (d, J = 6.7 Hz, 24H). 13C NMR (101 MHz, CDCl3) δ 164.1, 156.9, 145.9, 132.7, 131.1, 129.7, 124.2, 122.3, 119.4, 119.0, 115.1, 57.0, 29.3, 24.2. MS HR (ESI) m/z calcd for C52H51N2O8 [M + H]+ 623.2393, found 623.2388. Fluorescence (CH2Cl2): λmax = 637 nm (λex = 540 nm); Φfl = 0.68 ± 0.05.
700), 246 (39300), 284 (33500), 265 (29400), 562 (28300), 441 (10300). 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 4H), 4.25 and 4.21 (overlapping t, J = 7.5 Hz, 4H and s, 12H), 1.82–1.68 (m, 4H), 1.53–1.40 (m, 4H), 1.00 (t, J = 7.4 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 164.1, 156.9, 132.3, 122.2, 119.2, 118.3, 114.7, 56.8, 40.5, 30.4, 20.5, 14.0. MS HR (ESI) m/z calcd for C36H34N2O8 [M + H]+ 623.2393, found 623.2388. Fluorescence (CH2Cl2): λmax = 632 nm (λex = 540 nm); Φfl = 0.71 ± 0.03.
000), 283 (32200), 266 (31400), 565 (29500), 441 (10200). 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 4H), 7.50 (m, 4H), 7.33 (m, 4H), 7.24 (m, 2H), 6.59 (q, J = 7.1 Hz, 2H), 4.17 (s, 12H), 2.02 (d, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 164.1, 156.8, 141.0, 132.2, 128.3, 127.07, 127.05, 122.4, 119.2, 118.3, 114.8, 56.8, 50.3, 16.4. MS HR (ESI) m/z calcd for C44H34N2O10 M+ 718.2310, found 718.2315. Fluorescence (CH2Cl2): λmax = 638 nm (λex = 560 nm); Φfl = 0.71.
Acknowledgements
We are grateful to the Alexander-von-Humboldt Foundation for the postdoctoral fellowships for P. L. and A. N.-K.Notes and references
- Selected reviews: (a) F. Würthner, Chem. Commun., 2004, 1564 RSC; (b) C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386 CrossRef CAS PubMed; (c) F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962 CrossRef PubMed.
- (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed; (b) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS PubMed.
- (a) L. Beverina and P. Salice, Eur. J. Org. Chem., 2010, 1207 CrossRef CAS; (b) U. Mayerhöffer, M. Gsänger, M. Stolte, B. Fimmel and F. Würthner, Chem. – Eur. J., 2013, 19, 218 CrossRef PubMed.
- Y. Zhao and M. R. Wasielewski, Tetrahedron Lett., 1999, 40, 7047 CrossRef CAS.
- (a) S. Nakazono, Y. Imazaki, H. Yoo, J. Yang, T. Sasamori, N. Tokitoh, T. Cédric, H. Kageyama, D. Kim, H. Shinokubo and A. Osuka, Chem. – Eur. J., 2009, 15, 7530 CrossRef CAS PubMed; (b) S. Nakazono, S. Easwaramoorthi, D. Kim, H. Shinokubo and A. Osuka, Org. Lett., 2009, 11, 5426 CrossRef CAS PubMed; (c) G. Battagliarin, C. Li, V. Enkelmann and K. Müllen, Org. Lett., 2011, 13, 3012 CrossRef CAS PubMed.
- Y. Avlasevich, C. Li and K. Müllen, J. Mater. Chem., 2010, 20, 3814 RSC.
- F. Würthner, Pure Appl. Chem., 2006, 78, 2341 CrossRef.
- (a) M.-J. Lin, B. Fimmel, K. Radacki and F. Würthner, Angew. Chem., Int. Ed., 2011, 50, 10847 CrossRef CAS PubMed; (b) M.-J. Lin, M. Schulze, K. Radacki and F. Würthner, Chem. Commun., 2013, 49, 9107 RSC.
- (a) L. Perrin and P. Hudhomme, Eur. J. Org. Chem., 2011, 5427 CrossRef CAS; (b) A. Fin, I. Petkova, D. A. Doval, N. Sakai, E. Vauthey and S. Matile, Org. Biomol. Chem., 2011, 9, 8246 RSC.
- W. Qiu, S. Chen, X. Sun, Y. Liu and D. Zhu, Org. Lett., 2006, 8, 867 CrossRef CAS PubMed.
- (a) H. Aalten, G. van Koten, D. M. Grove, T. Kuilman, O. G. Piekstra, L. A. Hulshof and R. A. Sheldon, Tetrahedron, 1989, 45, 5565 CrossRef CAS; (b) B. S. Ghanem, M. Hashem, K. D. Harris, K. J. Msayib, M. Xu, P. M. Budd, N. Chaukura, D. Book, S. Tedds, A. Waltone and N. B. McKeown, Macromolecules, 2010, 43, 5287 CrossRef CAS.
- W. Zhang, X. Zhou, Z. Xie, B. Yang, L. Liu and Y. Ma, Chem. Commun., 2013, 49, 11560 RSC.
- Y. Li, C. Wang, C. Li, S. Di Motta, F. Negri and Z. Wang, Org. Lett., 2012, 14, 5278 CrossRef CAS PubMed.
- S. Sengupta, R. K. Dubey, R. W. M. Hoek, S. P. P. van Eeden, D. D. Gunbaş, F. C. Grozema, E. J. R. Sudhölter and W. F. Jager, J. Org. Chem., 2014, 79, 6655 CrossRef CAS PubMed.
- A. J. Jiménez, M.-J. Lin, C. Burschka, J. Becker, V. Settels, B. Engels and F. Würthner, Chem. Sci., 2014, 5, 608 RSC.
- (a) P. Osswald and F. Würthner, J. Am. Chem. Soc., 2007, 129, 14319 CrossRef CAS PubMed; (b) M. A. Iron, R. Cohen and B. Rybtchinski, J. Phys. Chem. A, 2011, 115, 2047 CrossRef CAS PubMed.
- Y. Zagranyarski, L. Chen, D. Jänsch, T. Gessner, C. Li and K. Müllen, Org. Lett., 2014, 16, 2814 CrossRef CAS PubMed.
- Z. Chen, M. G. Debije, T. Debaerdemaeker, P. Osswald and F. Würthner, ChemPhysChem, 2004, 5, 137 CrossRef CAS PubMed.
- F. Würthner, A. Sautter and C. Thalacker, Angew. Chem., Int. Ed., 2000, 39, 1243 CrossRef.
- C. Hippius, I. H. M. van Stokkum, E. Zangrando, R. M. Williams, M. Wykes, D. Beljonne and F. Würthner, J. Phys. Chem. C, 2008, 112, 14626 CrossRef CAS.
- F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109 RSC.
- M. Queste, C. Cadiou, B. Pagoaga, L. Giraudet and N. Hoffmann, New J. Chem., 2010, 34, 2537 RSC.
- G. Seybold and G. Wagenblast, Dyes Pigm., 1989, 11, 303 CrossRef.
- C. Thalacker, C. Röger and F. Würthner, J. Org. Chem., 2006, 71, 8098 CrossRef CAS PubMed.
- A. J. Fry, in Laboratory Techniques in Electroanalytical Chemistry, ed. P. T. Kissinger and W. R. Heineman, Marcel Dekker Ltd, New York, 2nd edn, 1996, p. 481 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1438126. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00047a |
| This journal is © the Partner Organisations 2016 |