
Modulation of band gap and p- versus n-semiconductor character of ADA dyes by core and acceptor group variation†
Agnieszka
Nowak-Król
a,
Reinhard
Wagener
a,
Felix
Kraus
a,
Amaresh
Mishra
b,
Peter
Bäuerle
b and
Frank
Würthner
*a
aUniversität Würzburg, Institut für Organische Chemie & Center for Nanosystems Chemistry, Am Hubland, 97074 Würzburg, Germany. E-mail: wuerthner@chemie.uni-wuerzburg.de
bUniversität Ulm, Institute of Organic Chemistry II and Advanced Materials, Albert-Einstein-Allee 11, 89081 Ulm, Germany
First published on 19th February 2016
Abstract
A series of ADA dyes was synthesized by Knoevenagel condensation reactions between dialdehydes of the electron-rich dithienopyrrole (DTP), cyclopentadithiophene (CPDT), carbazole (CAR) and fluorene (FL) moieties and the electron-poor CH-acidic indandione (IND) or 1,1-dicyanomethylene-3-indanone (DCIND) units, respectively. The UV/vis absorption spectra of the dyes cover a wide range of the visible spectrum from 450 to almost 700 nm in solution. Likewise, owing to differently balanced donor and acceptor strengths they may be applicable either as p- or n-type organic semiconductor materials in transistor and solar cell devices. As a proof-of-concept for dye vacuum-deposited organic thin film transistors (OTFTs) with either hole or electron mobilities of ∼0.1 cm2 V−1 s−1 depending on the HOMO/LUMO levels could be demonstrated.
Introduction
Acceptor–Donor–Acceptor (ADA) dyes were probably the functional dyes that provided the largest impact to the field of small molecule based organic photovoltaics (OPV) materials during the last decade.1 According to this concept an electron-rich conjugated oligomer is functionalized at its ends with two acceptor units among which dicyanovinyl groups are the most widely utilized.1,2 Stronger heterocyclic acceptors were originally utilized less frequently despite their beneficial influence on the absorption properties, i.e. reducing the band gap and increasing the tinctorial strength. The reason is attributed to the fact that, if not balanced by a stronger electron donating subunit in the center, these strong acceptors would displace the frontier orbitals to such an extent that these dyes would not be compatible anymore with the commonly utilized fullerene acceptors in organic solar cells (OSCs). In particular, their LUMO level would be decreased too significantly as for the injection of electrons from photoexcited molecules into the fullerene LUMO. Nevertheless, most recently Chen and co-workers impressively demonstrated how stronger heterocyclic acceptors may be utilized in combination with extended oligothiophene chains such as twofold with 2-(1,1-dicyanomethylene)rhodanine-substituted heptathiophenes, giving power conversion efficiency (PCE) of 9.3% in combination with fullerene derivative PC61BM.3 It is noteworthy, however, that such large oligomeric compounds can only be processed from solution and not by vacuum sublimation anymore.Most recent exciting developments have opened, on the other hand, the door for the replacement of fullerenes by n-type semiconductors other than those based on fullerenes.4 For instance, the groups of Rajaram, Wang and Nuckolls succeeded in the development of perylene bisimide dimers which meanwhile in combination with p-type semiconducting polymers deliver OSCs with >6% PCE.5 Even more interestingly, reports by the groups of McCulloch, Park, Zhan and Yan introduced synthetically more easily accessible electron-poor ADA dyes which in combination with p-type semiconducting polymers also succeeded with PCEs of 4.1, 5.4, 6.8, and 7.3% respectively.6 This work motivated us to elucidate the fundamental optical and redox properties for a couple of ADA dyes with heterocyclic acceptor units, which we had formerly utililzed successfully in DA merocyanine dyes that afforded PCEs up to 6.2% in combination with C60 fullerene.7 As we will show in this article, these acceptors, now utilized in duplicate around an electron rich-central π-scaffold indeed shift the frontier orbitals to significantly lower levels compared to the formerly utilized DA dyes. Thus, whilst the DA dyes had ideal HOMO and LUMO levels for being utilized as donor component in combination with fullerene acceptors, the newly synthesized ADA dyes are most interesting candidates to replace the latter.
Results and discussion
Design and synthesis
In this study we focused our attention on ADA systems bearing indandione (IND) or indandione derivative 1,1-dicyanomethylene-3-indanone (DCIND)7,8 as acceptor moieties. We were particularly interested how a systematic variation of acceptor and donor components would influence photophysical and electrochemical properties, as well as secondary features being of high importance for the device preparation and operation. To this end, we designed two series of compounds defined by an acceptor moiety (see Chart 1). For the donor components, we selected four types of electron-rich aromatic systems consisting of a central five-membered ring fused to two flanking six- or five-membered aromatic rings. Two of them comprise bithienyl unit (compounds 1 and 2), while the other two biphenyl moiety (compounds 3 and 4). Another way to classify these molecules is by the type of a central ring. From this perspective, we can distinguish two cores containing heterocyclic rings with nitrogen as a heteroatom (compounds 1 and 3) and two systems with carbocyclic central rings (compounds 2 and 4). Both groups are equipped with solubilizing groups. The choice of the alkyl chains is crucial. On one hand, they should provide reasonable solubility to facilitate the synthesis, work-up and purification. On the other hand, their length should be balanced since an excessive increase in the length of alkyl substituents may have detrimental effect on the self-assembly propensity of the dyes and in turn – on the operation of devices.9 For our studies racemic ethylhexyl chains were selected. The proposed synthetic route was based on Knoevenagel condensation between electron-rich dialdehydes and electron-poor CH-acids in the key step (Scheme 1). For active methylene compounds indandione IND and DCIND were chosen. Both compounds feature rigid framework and additional benzene rings that should further shift absorption towards lower energies.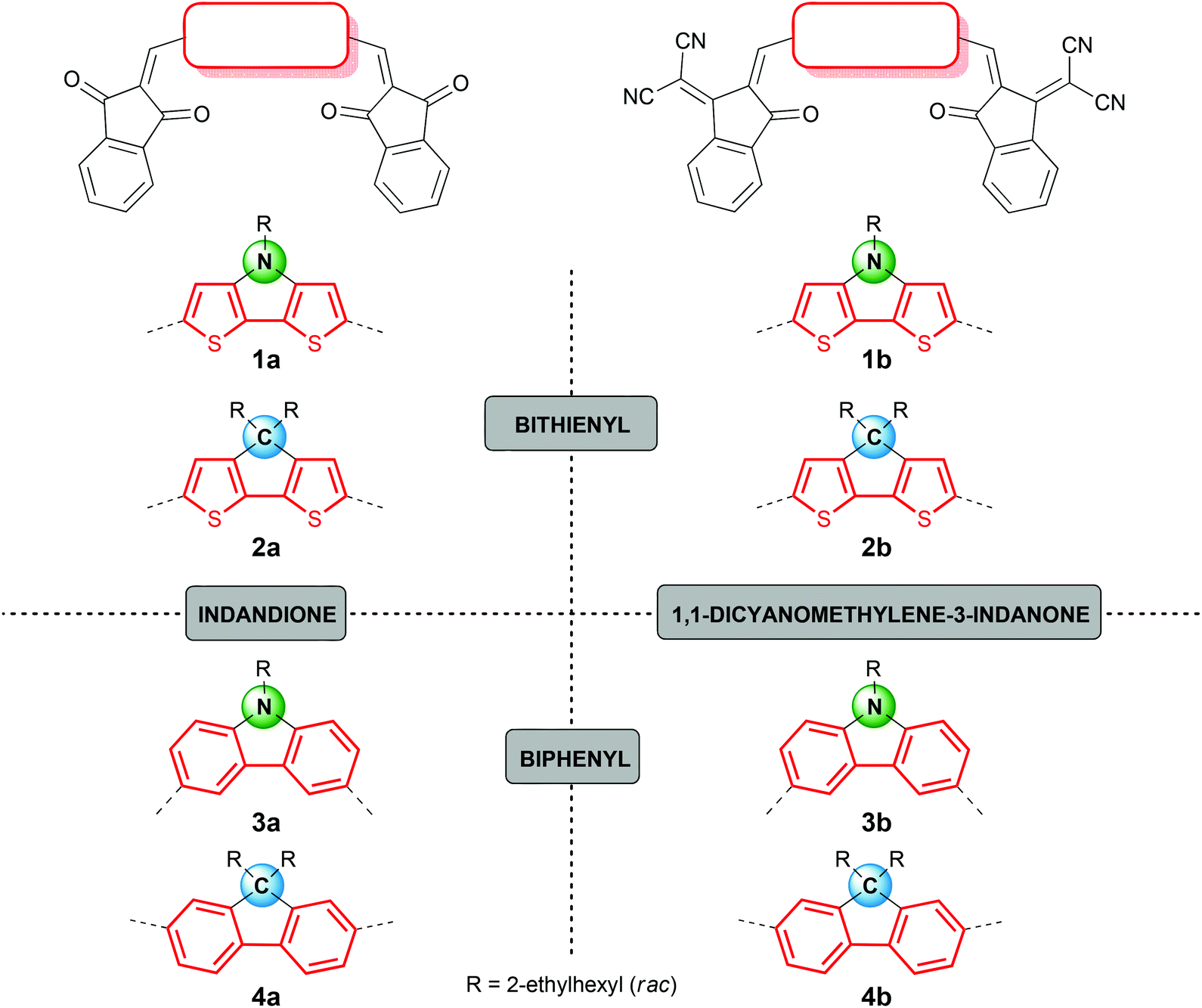 | ||
| Chart 1 Structures of target ADA molecules 1–4. | ||
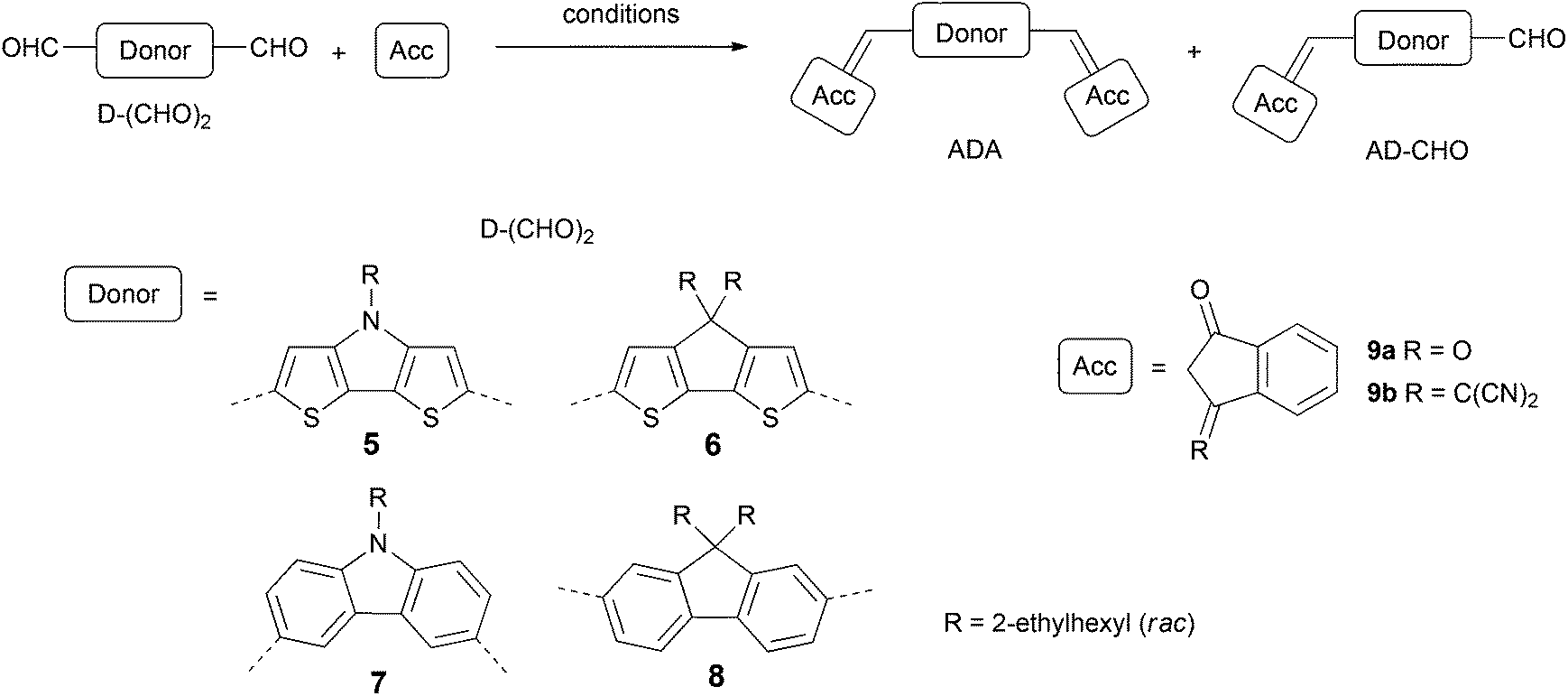 | ||
| Scheme 1 Synthesis of ADA and AD-CHO molecules. | ||
Dialdehyde starting materials as well as a DCIND acceptor were synthesized according to the literature-known procedures and are described in ESI.† Having in hand all these building blocks, we focused on Knoevenagel condensation between donor components and active methylene compounds. Conditions of selected experiments and yields of main products are gathered in Table 1. First, we reacted CH-acids 9a and 9b with DTP 5 using pyridine as a base. Both reactions were carried out at RT overnight producing corresponding dyes 1a and 1b in satisfactory yields (52 and 50%, respectively). By anology to the preparation of 1a and 1b, the condensation between structurally similar donor 6 and acceptor 9a was initially carried out under basic conditions. In the first hour the reaction was unsatisfactorily sluggish, even though an elevated temperature was applied. In addition, we observed the formation of unidentified side- and/or by-products in significant amounts. After 50 min, compound 2a was isolated in only 14% yield, together with a small amount of mono-product 2am (11%). In the next attempt we applied NaOAc and Ac2O affording 2a in 67% yield, along with product 2am in 32%. Not only was the latter reaction higher-yielding but it also proceeded cleaner, which simplified isolation of the target molecule. In striking contrast to the synthesis of 2a, the condensation between 6 and 9b with pyridine as a base proceeded smoothly. Within the first five minutes, 2b was formed as a major product, accompanied by a substantial amount of mono-product 2bm. It is important to note that only 1 h is absolutely sufficient for the full conversion of the starting material and to obtain dye 2b in a quantitative yield.
| Entry | D-(CHO)2 | Acc | ADA | Y (%) | AD-CHOb | Y (%) | Conditionsc |
|---|---|---|---|---|---|---|---|
| a Isolated yield. b m denotes a product of mono-condensation AD-CHO. c Conditions of selected reactions. d Product decomposed. | |||||||
| 1 | 5 | 9a | 1a | 52 | — | — | Pyridine, DCE, RT, 18 h |
| 2 | 5 | 9b | 1b | 50 | — | — | Pyridine, DCE, RT, 20 h |
| 3 | 6 | 9a | 2a | 67 | 2am | 32 | Ac2O, NaOAc, 60 °C, 2 h |
| 4 | 6 | 9a | 2a | 11 | 2am | 14 | Pyridine, DCE, 60 °C, 50 min |
| 5 | 6 | 9b | 2b | 99 | — | — | Pyridine, DCE, 60 °C, 1 h |
| 6 | 7 | 9a | 3a | 52 | 3am | 17 | Ac2O, NaOAc, 80 °C, 7 h |
| 7 | 7 | 9b | 3b | 38 | 3bm | 40 | Pyridine, DCE, 60 °C, 75 min |
| 8 | 7 | 9b | 3b | 20 | 3bm | 56 | Pyridine, DCE, 60 °C, 30 min |
| 9 | 8 | 9a | 4a | 34 | 4am | 8 | Piperidine, EtOH, 60 °C, 1.5 h |
| 10 | 8 | 9a | 4a | 6 | 4am | 11 | Ac2O, NaOAc, 80 °C, 8 h |
| 11 | 8 | 9b | 4b | —d | 4bm | —d | Pyridine, DCE, 60 °C, 50 min |
More intriguing were the preparations of the dyes consisting of a biphenyl unit in donor scaffolds (3 and 4). While a condensation between 7 and indandione (9a) in Ac2O was straightforward giving rise to a stable bis-product 3a in 52% yield, clear differences were observed for the synthesis of 3b. In the course of our studies on this reaction we established that molecule 3b was formed in all the following conditions employing a base: dichloromethane (DCM)/pyridine, dichloroethane (DCE)/pyridine or ethanol (EtOH)/piperidine, at RT as well as at elevated temperatures. Surprisingly, we encountered difficulties with the isolation of the target molecule. In all the cases when a product had an extensive contact with a stationary phase: silica gel, alumina or Bio-Beads and/or chlorinated solvents were employed, a decomposition occurred. This process was suppressed when a crude reaction mixture was purified by flash chromatography using a mixture of ethyl acetate (EtOAc) and pentane, followed by recrystallization. In this case we were able to isolate pure 3b in 20% and 3bm in 56% yields when the condensation was conducted for 0.5 h. Prolongation of the reaction up to 75 min allowed to increase the yield of 3b almost twofold. Further, we conjectured that a plausible reason for a decomposition was a retro-Knoevenagel reaction. It is worth noting that the information on this reaction is scarce in the literature. An interesting report was published by Lambert and co-workers10 who investigated a reversible aminolysis of electron-poor fulvene derivatives to cyclopentadienes and imines. To verify our hypothesis attributing a decomposition of a target molecule to a reverse reaction we took a sample of 3b, whose purity was confirmed by elemental analysis and NMR spectra, and submitted it to a column chromatography on silica gel in methylene chloride (DCM). Within a short time we observed a gradual conversion of a purple band corresponding to 3b into a red band of 3bm, which in turn partially transformed into a yellow one. All fractions were collected and the identities of particular compounds were ultimately confirmed by 1H NMR. This experiment brought a concrete evidence of our assumption that 3b undergoes a stepwise retro-Knoevenagel reaction when exposed to silica. On the other hand, compound 3b is stable in the solid state even on heating to high temperatures (Fig. S5, ESI†), and represents a decent stability in solution.
Even more demanding was the syntheses of fluorene derivatives. To prepare 4a, dialdehyde 8 was reacted with an excess of CH-acid 9a in Ac2O in the presence of sodium acetate at 80 °C. After 4 h, due to a low conversion of the starting material, another portion of indandione was added and the stirring was continued overnight. However, a prolonged heating resulted only in a decomposition of bis-product 4a. Therefore, in the next attempt the reaction was quenched after 8 h to give 4a in 6% and 4am in 11%. Low reaction yields prompted us to test basic conditions. Dry ethanol in combination with dry piperidine used as a base had a beneficial effect on the reaction outcome, increasing the yield of the desired product to 34% after only 1.5 h of heating at 60 °C. The analogous reaction as well as reactions employing pyridine as a base were executed for a DCIND acceptor. As expected, compounds 4b and 4bm featured lower stability than more robust indandione counterparts, similarly to carbazole derivatives. In the course of the reaction we observed the formation of an orange-colored dye, accompanied by a less polar yellow compound and an unreacted starting material. The first substance was identified by high resolution mass spectrometry (Fig. S29, ESI†) as bis-compound 4b ([M + K]+ 837.3565). In the electrospray ionization (ESI) spectrum of the yellow compound an ion at 623.36 m/z was found corresponding to the [M + H]+ ion of mono-product 4bm (Fig. S30, ESI†). Dye 4b, however, upon column chromatography on silica gel decomposed giving rise to 4bm and other compounds. Product 4bm could not be isolated either. All the attempts to purify this molecule for analyses resulted in a further decomposition, showing that retro-Knoevenagel reaction in this case is much more pronounced than for the carbazole derivative 3b. At this point we can draw two conclusions. Firstly, DTP and CPDT dyes show higher stability than CAR and FL counterparts. Secondly, DCIND derivatives are more prone to decomposition that IND systems. We attribute the latter observation to the stronger electron-withdrawing nature of DCIND substituents. High polarization of the exocyclic double bond, enabled by the stabilization of the negative charge by dicyanomethylidene and carbonyl groups, favors nucleophilic attack on the vinyl carbon atom. Therefore, the cleavage could occur even under mild conditions provided by an adsorbent. The superior stability of bithienyl containing molecules to CAR and FL derivatives can be explained by the lower sterical demands (enabling a fully planar π-scaffold)7b and a lower resonance energy of thiophene comparing to benzene. For these reasons, conjugation and charge delocalization are less pronounced in the latter systems and as a consequence the positive charge is more localized on the vinyl carbon atom and the molecules are more susceptible to nucleophilic attack.11 Nevertheless, all the molecules display high stability in the solid state and in solution at RT, as well as upon heating up to ca. 250–300 °C (Fig. S5, ESI†).
All the synthesized dyes were characterized by means of 1H, 13C NMR, 2D NMR, and high resolution MS. Importantly, compounds containing a central heterocyclic ring with one 2-ethylhexyl chain were obtained as racemic mixtures, whereas those equipped with two 2-ethylhexyl chains as mixtures of diastereomers. 1H and 13C NMR spectra of DCIND derivatives 1b, 3b, and 3bm as well as IND counterparts 1a, 3a, and 3am revealed only single sets of signals. The complexity of the NMR spectra increased upon introductions of two alkyl chains. This was reflected in the spectra of all dyes with the carbocyclic central rings, i.e.2a, 2am, 2b, and 4a showing more than one set of signals in both aliphatic and aromatic regions. Noteworthy, analogous complexity was observed in the spectra of the precursors, i.e. aldehydes 6 and 8. Hence, we can attribute this feature to the presence of diastereomers. Moreover, all the DCIND dyes were isolated as single geometrical isomers as proved by the combined NMR techniques. Owing to the lower steric congestion around the thiophene ring and the additional stabilization imparted by sulfur-oxygen interactions, the formation of the Z,Z-isomers is favored.7b,8,12 Likewise, due to the lower steric hindrance for Z,Z-isomer than E,E-isomer, we can expect the preference towards the first stereoisomer, which is consistent with the structure of CAR-DCIND DA system published elsewhere.13 In terms of solubility, the carbon-bridged molecules comprising CPDT and fluorene equipped with two alkyl chains surpass CAR and DTP molecules. The latter feature a relatively poor solubility with compound 1b showing the lowest solubility within the whole series.
Absorption and redox properties
The optical properties of ADA molecules 1–4 were investigated in solution by steady-state absorption and fluorescence spectroscopy. Fig. 1a presents absorption spectra of target molecules in DCM. The optical properties are summarized in Table 2. Compounds 1a and 2a show a comparable absorption behavior. Solutions of these dyes exhibits purple color in DCM, which corresponds to the absorption maxima at 582 nm and 584 nm, respectively, characterized by high molar absorption coefficients (εmax above 1.2 × 105 M−1 cm−1). As other compounds with DTP or CPDT core moiety,2a they show additional vibronic shoulders at shorter wavelengths of 542 and 544 nm, respectively. When the DTP and CPDT donors are combined with DCIND units, redshifts in absorption of about 80 nm (∼2100 cm−1) are observed, while keeping similar lineshapes of the spectra. Compounds 2a and 2b are blue in DCM solution. The large bathochromic shifts reflect more pronounced electronic interactions between electron-rich cores of DTP and DTCP and strong electron-withdrawing DCIND substituents. The absorption maxima of 3a and 4a are located at 478 nm and 466 nm, respectively, showing blue-shifted absorption comparing to 1a and 2a, which confirms the fact that CAR and FL are weaker donors than DTP and CPDT. Moreover, they are weaker absorbers than the latter dyes, which is expressed in lower εmax values (70 × 103 – 76 × 103 M−1 cm−1), as well as in lower square transition dipole moments μeg2 of 75 and 102 D2 for 3a, and 4avs. 147 and 138 D2 for 1a and 2a, respectively. The lowest-energy absorption band of DCIND derivative 3b is bathochromically shifted by 64 nm (∼2500 cm−1) compared to IND counterpart. Dyes 4a, 3a, and 3b exhibit yellow, orange, and red colors in solution, respectively. In principle, all dyes bearing DCIND moieties are stronger absorbers than the corresponding IND derivatives based on both μeg2 and εmax. The only exception in this set of compounds is 3b that displays much lower εmax than its IND counterpart 3a due to a broadened absorption. Conversely, square transition dipole moment for 3b is higher than for 3a, similarly to other dyes possessing the same donor core but different acceptors. Furthermore, the spectra of all ADA systems studied here display vibronic progressions at shorter wavelengths.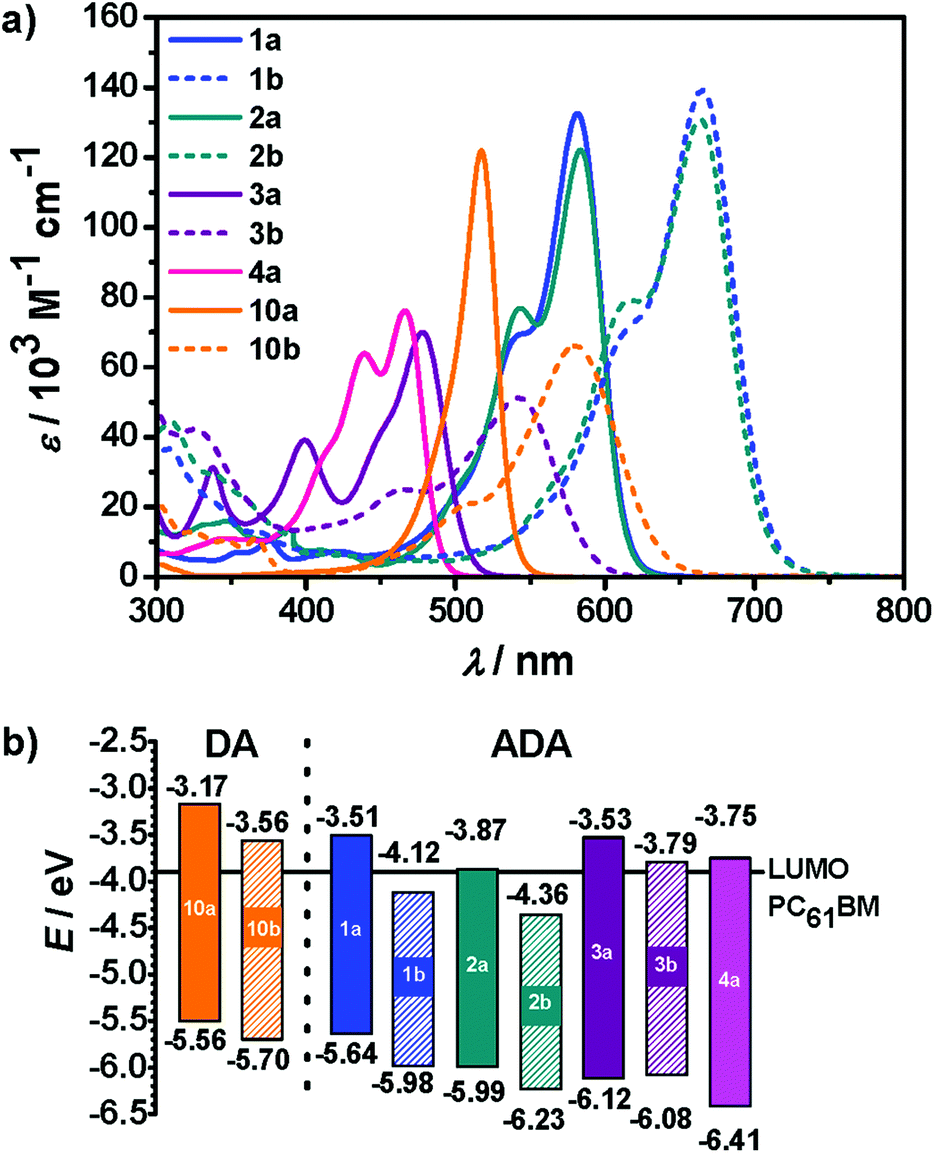 | ||
| Fig. 1 (a) Absorption spectra of ADA dyes 1a, 1b, 2a, 2b, 3a, 3b, 4a, and DA dyes 10a, and 10b in CH2Cl2 (c ∼10−5 M). (b) HOMO and LUMO levels defining the band gap (solid area) of the dyes. The solid black line represents the LUMO energy level of PC61BM (−3.9 eV). | ||
| Dye |
λ
abs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a [nm ] a [nm ] |
ε
maxb [103 M−1 cm−1] |
μ
eg
2c [D2] |
λ
emd [nm] |
Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) e [cm−1] e [cm−1] |
E
red1/2f [V] |
E
ox1/2f [V] |
|---|---|---|---|---|---|---|---|
| a Absorption maximum in CH2Cl2. b Molar absorption coefficient in CH2Cl2. c Square transition dipole moments calculated from the measured data for the lowest-energy absorption band. d Emission maximum in CH2Cl2. e Stokes shift. f Redox potentials. Measurements were carried out in dry CH2Cl2 (c ∼10−4 – 10−5 M); scan rate 100 mV s−1; supporting electrolyte Bu4NPF6 (c = 0.1 M); calibrated vs. ferrocene/ferrocenium (Fc/Fc+) as an internal standard. g Peak potential. | |||||||
| 1a | 582 | 133 | 147 | 611 | 800 | −1.52g | +0.49 |
| 1b | 665 | 139 | 175 | 697 | 700 | −0.96g | +0.83g |
| 2a | 584 | 122 | 138 | 610 | 700 | −1.30 | +0.84 |
| 2b | 664 | 131 | 167 | 693 | 600 | −0.84g | +1.08 |
| 3a | 478 | 70 | 75 | 518 | 1600 | −1.76g | +0.97g |
| 3b | 542 | 51 | 89 | 582 | 1300 | −1.25g | +0.93 |
| 4a | 466 | 76 | 102 | 497 | 1300 | −1.51g | +1.26g |
| 10a 7b | 517 | 144 | 82 | — | — | −1.86g | +0.41g |
| 10b 7b | 577 | 78 | 97 | — | — | −1.53 | +0.55 |
Accordingly, the spectral features of these ADA dyes show similarities with DA dyes. For the latter it was demonstrated that tinctorial strength (∼μeg2) and εmax values are maximized at the so-called ‘cyanine limit’ where the π-system is most delocalized with equal contributions of the two resonance structures depicted in Scheme 2. Likewise, a similar ‘cyanine limit’ appears to exist for ADA dyes leading to sharp and intense absorption bands. Based on the shape of the absorption bands given in Fig. 1, this limit is, however, not reached yet for our ADA dyes, albeit the bithienyl derivatives are much closer to it than the biphenyl counterparts.
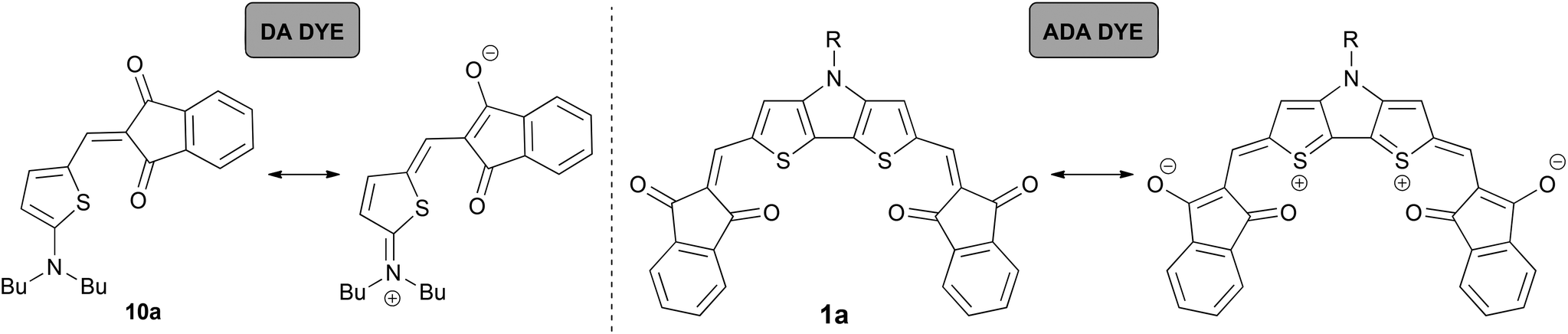 | ||
| Scheme 2 Valence structure description for DA dye 10a and ADA dye 1a. | ||
For comparison, we chose two merocyanine dyes 10a and 10b studied previously in our group.7b,14 Notably, 10b, which was introduced as EL86, is among the best performing DA dyes in bulk heterojunction solar cells.7b Both DA compounds consist of 2-aminothiophene as a donor, and IND or DCIND as an acceptor unit (Chart 2). The spectrum of 10a reveals a sharp transition, lacking a vibronic progression. In contrast, absorption of 10b in terms of lineshapes resembles closely the spectrum of compound 3b. The main absorption bands positioned at 517 and 577 nm for 10a and 10b, respectively, lie in between the absorption bands of the DTP/CPDT and CAR/FL ADA compounds bearing the same acceptor units. The two merocyanine dyes display square transition dipole moments of 82 and 97 D2, which are comparable to the those of 3a, 3b and 4a. Nevertheless, the ADA dyes equipped with weaker donor cores (3a, 3b, 4a) show inferior absorption properties to the DA dyes (10a, 10b), which consist of a strong donor. Thus, the latter compounds feature a comparable absorption strength but smaller band gaps, while possessing only one acceptor moiety. In contrast to the ADA molecules 3a, 3b, and 4a, the ADA molecules with stronger donor moieties (1a, 1b, 2a, 2b) display a significant increase in absorption strength and smaller optical band gaps, making these dyes most promising for application in organic solar cells. This results clearly show that to extend the effective π-conjugation the choice of the right donor and acceptor combination with regards to the donor/acceptor strength is vital.
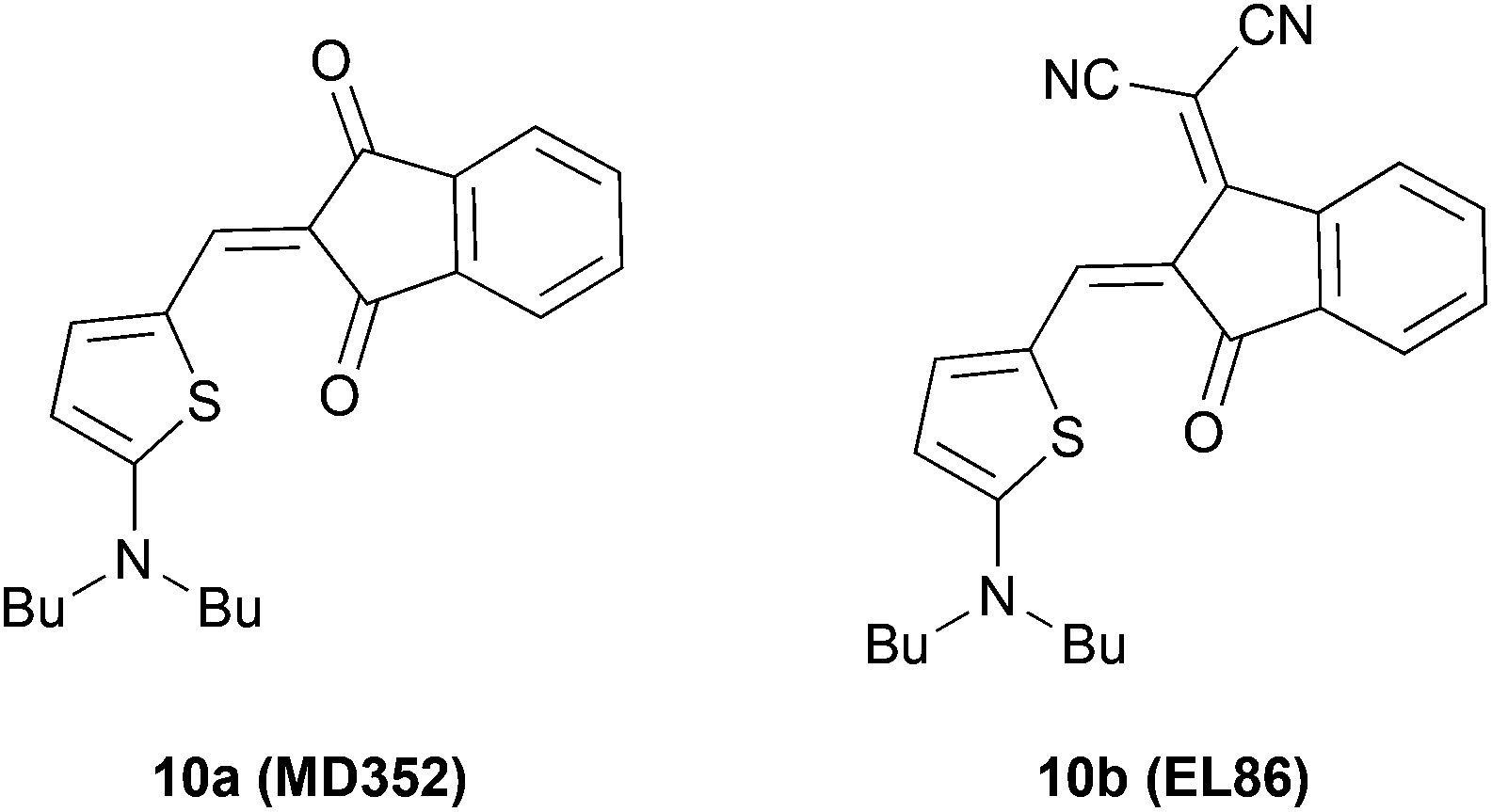 | ||
| Chart 2 Structures of the reference DA systems originally introduced7b as MD352 and EL86 bearing IND and DCIND acceptors. | ||
As expected, the investigated molecules are weakly emissive with Φfl typically below 5%. The emission maxima range between 497 nm for 4a and 697 nm for 1b and, similarly to absorption maxima, cover a wide spectral range. The fluorescence spectra of all the ADA molecules are devoid of vibronic structures. The normalized absorption and emission spectra are depicted in Fig. S1 (ESI†). The Stokes shifts of ADA chromophores are moderate, which indicates a relatively small reorganization in the excited state as expected for dyes with a cyanine-type electronic structure. Replacement of IND groups with DCIND moieties leads to a decrease in Stokes shifts by ca. 100–300 cm−1. Furthermore, a clear difference can be observed between molecules containing bithienyl and biphenyl units. For DTP or CPDT derivatives Stokes shifts are significantly lower (ca. 600–800 cm−1) than for the molecules with CAR or FL cores (ca. 1300–1600 cm−1) in accordance with a better delocalization and a more cyanine-type character of the former molecules.
Further, to obtain information on the valence and conduction band edges that are related to the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) cyclic voltammetry was performed in DCM for all ADA dyes. The measured redox potentials are included in Table 2 and the HOMO/LUMO levels are illustrated in Fig. 1b. The calculated electrochemical and optical band gaps, as well as the calculated HOMO and LUMO levels are given in Table S2 (ESI†). Dyes 1a, 2a, 2b, and 3b show reversible behavior on oxidation. On the other hand, most of the dyes, except 2a, exhibit irreversible or quasi-reversible reduction waves. In the latter case a well-resolved reduction wave at −1.30 V is observed, corresponding to the formation of a rather stable radical anion. The voltammograms of 3a and 4a show the second reduction processes associated with the formation of dianions. The reduction potentials of all indandione derivatives are negatively shifted by ca. 0.5 V vs. their DCIND counterparts (see Table 2), which is consistent with the stronger electron-withdrawing nature of the dicyanomethylidene substituents. For the same reason substitution with the DCIND groups entails a positive shift of the oxidation potentials by around 0.3 V for 1b and 2b. The HOMO energy levels were estimated from the oxidation potentials, assuming an energy level of Fc/Fc+ at 5.15 eV below vacuum.15
Fig. 1b illustrates the band gaps and the positions of the frontier molecular orbitals (FMOs) of ADA compounds with regard to the LUMO level of PC61BM. Within the presented set of compounds the HOMO energies vary by 0.85 eV, while the LUMO levels by 0.77 eV. The optical band gaps of the dyes are, with the exception of 3a and 4a, quite low (1.86 eV to 2.29 eV). Therefore, they are capable of absorbing light in the most relevant photon-rich range of solar radiation. All these dyes exhibit low-lying HOMO/LUMO energies, which implies high electron affinity. When we compare dyes 1a and 3a, both containing a nitrogen atom in the 5-membered ring of donor cores, with their carbocyclic counterparts 2a and 4a, a decrease by about 0.3 eV in HOMO/LUMO levels can be observed for the latter dyes. From this observation we can conclude that the nitrogen acts as an electron donor like in pyrrole, making the central unit more electron-rich. For this reason the replacement of the bridging methylene unit by nitrogen is quite useful if p-type semiconductors and donor components in solar cells are desired.2a,16
Additionally, the exchange of indandione for DCIND affects mainly LUMO levels which are decreased by up to 0.6 eV. This entails the decrease in the band gaps by around 0.3 eV for DCIND derivatives. Most interestingly, LUMO levels of compounds 1b (−4.12 eV) and 2b (−4.36 eV) lie even below the LUMO level of PC61BM fullerene commonly used as an acceptor in BHJ solar cells.17 Fullerenes show, however, some deficiencies, such as limited tunability of the LUMO level and low absorption in the visible region.18 Hence, intense absorption together with tunable FMO levels make some ADA dyes studied here (e.g.1b and 2b) most promising candidates to replace PC61BM. Despite the fact that DA dyes 10a and 10b show relatively low band gaps (2.39 eV and 2.14 eV), comparable to that of 1a, their HOMO and LUMO levels are still high and do not meet criteria for efficient acceptor materials in solar cells. Conversely, they were successfully utilized as donors in combination with PC61BM.7b Noteworthy, positions of the FMO levels of 1a basically overlap with those of 10b. Therefore, this molecule is likewise rather a candidate for a donor than an acceptor. Similar behavior can be also expected from 3a.
Thus, by varying a central donor unit and the strength of flanking acceptors, we were able to tune the levels of FMOs as well as the optical properties of ADA systems, and hence optical band gaps, over the entire visible spectrum. More importantly, the combination of only moderately electron-releasing donor cores with very strong acceptors in ADA systems provided low-band-gap materials whose LUMO levels fall below the LUMO level of PC61BMs.
Quantum chemical calculations
To get insight into the electronic structures and molecular energy levels of all synthesized ADA systems, we performed quantum chemical calculations for the gas phase by using density functional theory (DFT) at the B3LYP/def2-SVP level for simplified model compounds with branched alkyl chains replaced by methyl groups. The optimized molecular geometries, as well as orbital contour plots of HOMO and LUMO orbitals are depicted in Fig. 2 for compounds 2a and 2b, and in Fig. S4† for the other dyes. The molecules feature good planarity and high symmetry beneficial for π–π interactions in bulk materials. The HOMO electron densities are mainly localized around the central donor components including the conjugated carbonyl and dicyanomethylidene groups. In the LUMOs π-electrons are redistributed towards acceptor moieties. The distribution of HOMO and LUMO of 1a,b closely resembles that of their counterparts 2a,b containing a carbocyclic central ring. The calculated MO energy levels are listed in Table S3 (ESI†). The calculated HOMO levels correlate well with the HOMO levels obtained from CV and UV/vis, whereas calculated LUMO energies are higher, which entails the difference in energy gaps of 0.34–0.65 eV. Both experimental and theoretical data indicate that the synthesized molecules are suitable for application in photovoltaics/organic electronics.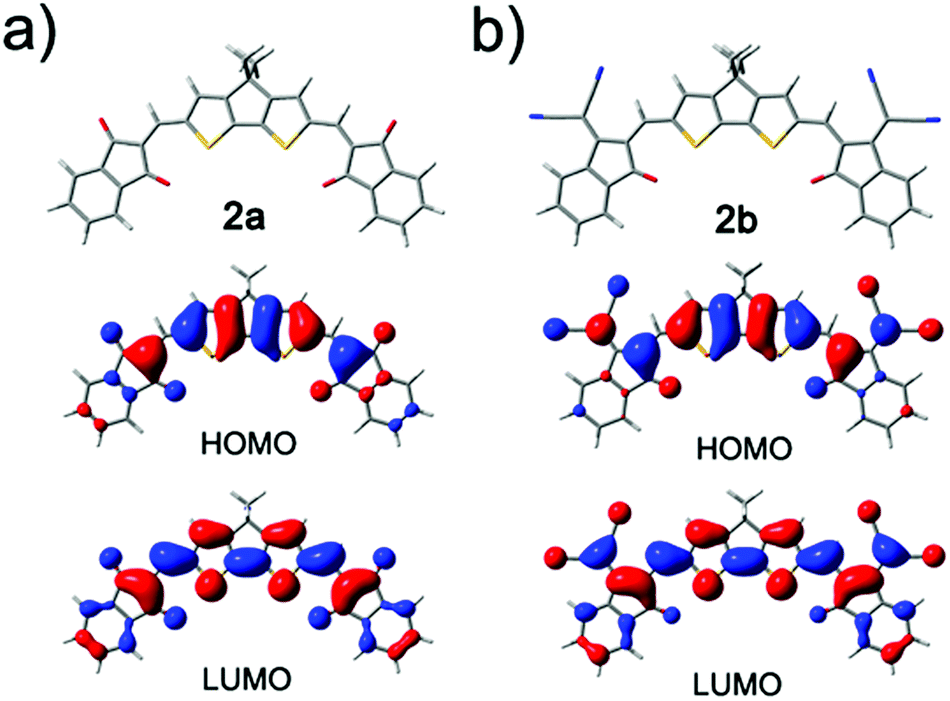 | ||
| Fig. 2 Geometry-optimized structures (with B3LYP/def2-SVP) as well as orbital contour plots of HOMO and LUMO orbitals for model compounds of 2a and 2b (ethylhexyl substituents are replaced with methyl groups). | ||
Organic transistor devices
To provide a further support for the transition from p- to n-type semiconductors within the given series of ADA dyes we prepared transistor devices for dyes 2a and 2b. Organic thin film transistors (OTFTs) with bottom-gate, top-contact configuration (channel width W = 200 μm; channel length L = 100 μm) were fabricated via vacuum deposition (30 nm) on n-tetradecylphosphonic acid (TPA) modified Si/SiO2 substrates (100 nm) and characterized under ambient conditions. Transfer curves of dyes 2a and 2b were measured under ambient conditions (Fig. 3a and b) and the mean values of five devices with each of these dyes were calculated. 2a showed an average hole mobility of 0.12 cm2 V−1 s−1 [μmax = 0.126 cm2 V−1 s−1], a current on/off ratio of 4 × 104 and a threshold voltage of about −1 V. At a gate voltage of about −10 V, the device showed a tilt in the |−IDS|0.5–UGS curve, which is accompanied by a decrease in the mobility (0.002 cm2 V−1 s−1), according to the equation |−IDS|0.5 = (μCiW/(2L) UGS − UT), where μ is the charge carrier mobility, Ci the capacitance of the gate dielectric, W the width, L the length of the transistor channel and UT the threshold voltage. When 2b was used as an active material, electron mobilities as high as 0.073 cm2 V−1 s−1 [μmax = 0.102 cm2 V−1 s−1], a current on/off ratio of 3 × 104 and a threshold voltage of 4 V were measured. The atomic force microscopy (AFM) images of 2a and 2b thin films (Fig. 3c and d) show large crystallites with a rod-like and a terrace-like morphology, respectively. These results demonstrate that by alternating the strength of the acceptor substituents, we were able to switch the π-scaffold from p-type semiconductor to n-type semiconductor in full compliance with our expectations based on redox potentials and calculated HOMO and LUMO levels (Fig. 1b). Thus, compound 2a (as well as 1a and 3a) constitutes a potential donor dye for conventional BHJ solar cells with fullerenes as acceptors, whilst compound 2b (as well as 1b) shows potential as a novel n-type fullerene replacement component for BHJ solar cells. Several of these dyes might function as p- or n-type components depending on the choice of the second material in BHJ.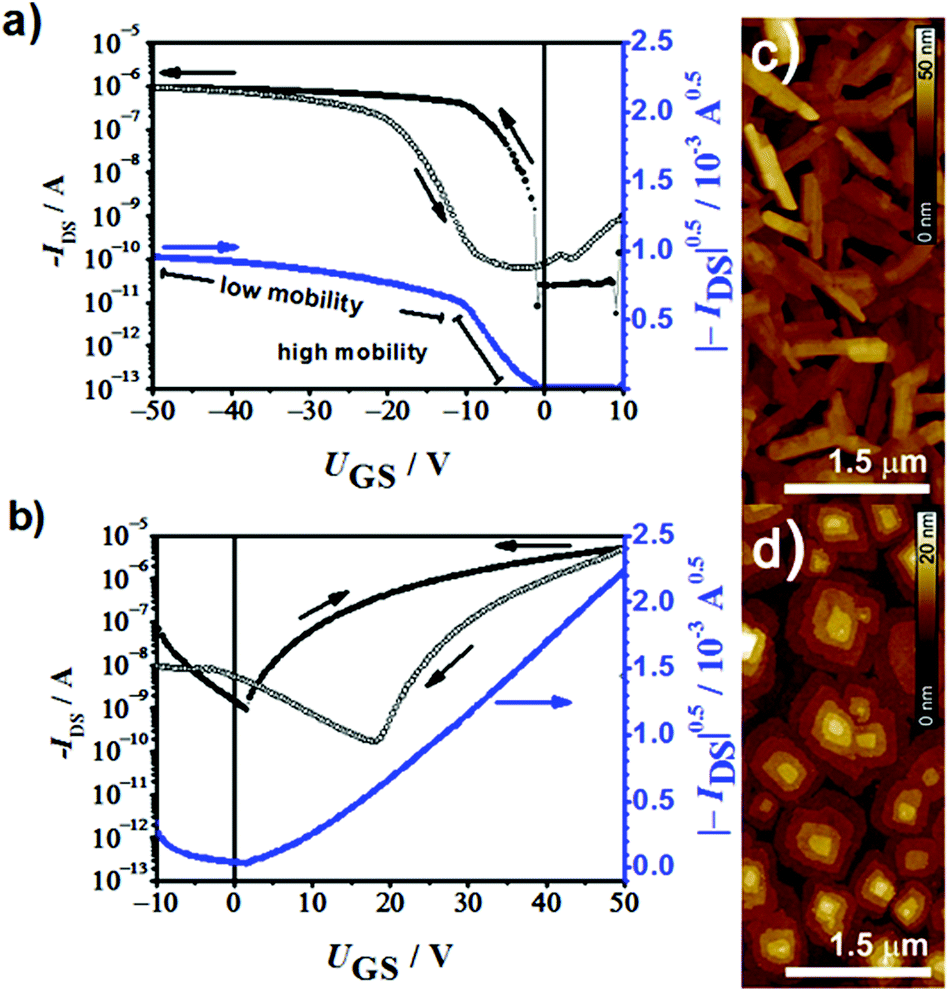 | ||
| Fig. 3 OTFT transfer curves of (a) 2a and (b) 2b on TPA-modified substrates in air. Corresponding AFM images of (c) 2a and (d) 2b thin films. | ||
Conclusions
In this article we have described a series of ADA dyes being a combination of four donor cores and two acceptors. By varying the electron-releasing character of the central unit and the strength of flanking acceptor components, we were able to modulate systematically the positions of the frontier molecular orbitals as well as optical band gaps, covering a broad range of the visible spectrum. More interestingly, the combination of bithienyl cores with DCIND in ADA systems converted the molecules into low-band-gap materials (i.e.1b, 2b) whose LUMO levels fall below the LUMO level of PC61BMs. This aspect was elucidated in detail for dyes 1b and 2b where alteration of the acceptor unit switched the function of these compounds from p-semiconductor 2a with hole mobility of 0.12 cm2 V−1 s−1 to n-semiconductor 2b with electron mobility as high as 0.073 cm2 V−1 s−1.Experimental
General
All reagents were purchased from commercial sources and used as received without further purification, unless otherwise stated. Reagent grade solvents were distilled prior to use. Column chromatography was performed on silica (silica gel, 230–400 mesh). 1H and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer at room temperature, unless otherwise noted, and calibrated to the residual solvent signals or TMS. J values are given in Hz. The following abbreviations were used to designate multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet. High resolution mass spectra were obtained by electrospray ionization (ESI) and were recorded on an ESI micrOTOF Focus spectrometer from Bruker Daltonics. Melting points of ADA systems were determined on an optical microscope and are uncorrected. The syntheses of building blocks are described in ESI.†Synthetic procedures and product characterization
:CH2Cl2, 1:4, then CH2Cl2 to CH2Cl2 + 5% MeOH) and recrystallized (CH2Cl2/hexane) to give 1a (180 mg, 52%) as green crystals. M.p. 278–280 °C (CH2Cl2/hexane). UV/vis (CH2Cl2, c ∼ 10−5 M): λmax/nm 582 (ε/M−1 cm−1 132600), 544 (69500), 422 (7400), 376 (10400), 354 (7000). 1H NMR (400 MHz, CDCl2CDCl2, 125 °C) δ 8.43 (s, 2H), 8.11–7.98 (m, 4H), 7.96 (s, 2H), 7.89–7.77 (m, 4H), 4.29 (d, J = 7.1 Hz, 2H), 2.26–2.06 (m, 1H), 1.61–1.34 (m, 8H), 1.08 (t, J = 7.4 Hz, 3H), 0.98 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl2CDCl2, 125 °C) δ 189.3, 188.5, 150.1, 142.4, 140.5, 140.1, 136.0, 134.6, 134.4, 126.1, 125.5, 122.7, 121.5, 51.9, 40.5, 31.1, 28.7, 24.5, 22.6, 13.4, 10.5. MS HR (ESI) m/z calcd for C36H30NO4S2 [M + H]+ 604.1616, found 604.1611. Anal. calcd for C36H29NO4S2: C, 71.62; H, 4.84; N, 2.32; S, 10.62. Found: C, 71.51; H, 4.72; N, 2.48; S, 10.25.
:CH2Cl2, 1:4, then CH2Cl2 to CH2Cl2 + 10% MeOH) and recrystallized (CH2Cl2/hexane) to give 1b (200 mg, 50%) as green-golden crystals. M.p. 307–310 °C (CH2Cl2/hexane). UV/vis (CH2Cl2, c ∼ 10−5 M): λmax /nm 665 (ε/M−1 cm−1 139300) with a shoulder at 367 (12900), 308 (36700). 1H NMR (400 MHz, CDCl2CDCl2, 125 °C) δ 8.80 (s, 2H), 8.65 (d, J = 7.6 Hz, 2H), 7.96–7.94 (m, 4H), 7.81–7.68 (m, 4H), 4.15 (d, J = 7.0 Hz, 2H), 2.08–1.97 (m, 1H), 1.51–1.23 (m, 8H), 0.95 (t, J = 7.3 Hz, 3H), 0.91–0.82 (m, 3H). 13C NMR (101 MHz, CDCl2CDCl2, 125 °C) δ 187.4, 160.4, 150.7, 141.1, 140.0, 137.8, 137.1, 135.3, 134.7, 128.9, 125.2, 124.8, 124.1, 123.7, 114.5, 70.2, 52.1, 40.4, 31.0, 28.7, 24.5, 22.9, 13.8, 10.7. MS HR (ESI) m/z calcd for C42H29N5O2S2 M+ 699.1763, found 699.1757.
:1) to obtaind pure 2a and a mixed fraction. Consecutive chromatography (silica, pentane/EtOAc 19:1) afforded additional portion of 2a and mono-product 2am. Both products were recrystallized (CH2Cl2/pentane) to afford 2a (105 mg, 67%) as green crystals and 2am (41 mg, 32%) as orange-red crystals. Compounds 2a and 2am were obtained as mixtures of stereoisomers. Compound 2a: m.p. 270–272 °C (CH2Cl2/pentane). UV/vis (CH2Cl2, c ∼ 10−5 M): λmax/nm 584 (ε/M−1 cm−1 122200), 544 (76900), 411 (7700), 387 (13500), 346 (16000), 333 (15100). 1H NMR (400 MHz, CD2Cl2) δ 8.06–7.89 (m, 8H), 7.85–7.74 (m, 4H), 2.12–1.97 (m, 4H), 1.08–0.84 (m, 16H), 0.77–0.55 (m, 14H). 13C NMR (101 MHz, CD2Cl2) δ 190.48, 190.47, 190.46, 190.01, 189.96, 189.92, 163.0, 162.9, 162.8, 150.83, 150.77, 150.72, 142.73, 142.70, 142.66, 141.0, 136.5, 136.4, 136.3, 135.9, 135.6, 135.4, 124.94, 124.89, 124.83, 123.3, 43.6, 35.9, 34.8, 28.97, 28.96, 27.9, 23.3, 14.3, 10.9, signal of one carbon atom is overlapping with the solvent signal. MS HR (ESI) m/z calcd for C45H47O4S2 [M + H]+ 715.2916, found 715.2910. Anal. calcd for C45H46O4S2: C, 75.60; H, 6.49; S, 8.97. Found: C, 75.23; H, 6.58; S, 8.91. Compound 2am: m.p. 136–139 °C (CH2Cl2/pentane). UV/vis (CH2Cl2, c ∼ 10−5 M): λmax/nm 507 (ε/M−1 cm−1 70100) with a shoulder, 324 (9700), 312 (11300). 1H NMR (400 MHz, CD2Cl2) δ 9.91 (s, 1H), 8.07–7.87 (m, 4H), 7.86–7.75 (m, 2H), 7.68–7.67 (m, 1H), 2.17–1.88 (m, 4H), 1.16–0.82 (m, 16H), 0.81–0.66 (m, 6H), 0.66–0.53 (m, 8H). 13C NMR (101 MHz, CD2Cl2) δ 190.48, 190.47, 190.46, 190.04, 190.00, 189.95, 183.53, 183.46, 183.39, 162.27, 162.22, 162.18, 150.38, 150.32, 150.26, 147.4, 146.05, 146.03, 146.00, 142.7, 141.89, 141.88, 141.85, 141.0, 136.7, 136.6, 136.5, 136.03, 136.00, 135.6, 135.4, 131.1, 131.03, 130.97, 124.81, 124.76, 124.72, 123.32, 123.28, 54.78, 43.5, 35.90, 35.87, 34.8, 34.7, 29.03, 29.02, 28.94, 28.92, 27.94, 27.86, 23.25, 23.24, 14.4, 14.3, 10.93, 10.89, 10.88. MS HR (ESI) m/z calcd for C36H43O3S2 [M + H]+ 587.2654, found 587.2648.
900), 617 (79200), 337 (30400), 310 (44400). 1H NMR (400 MHz, CD2Cl2) δ 8.92 (s, 2H), 8.74–8.67 (m, 2H), 8.01–7.94 (m, 2H), 7.86–7.73 (m, 6H), 2.13–1.96 (m, 4H), 1.11–0.85 (m, 16H), 0.81–0.67 (m, 8H), 0.67–0.58 (m, 6H). 13C NMR (101 MHz, CD2Cl2) δ 188.77, 188.75, 188.72, 163.4, 163.34, 163.28, 160.65, 160.62, 160.60, 153.62, 153.60, 153.57, 143.3, 140.6, 138.2, 138.1, 138.0, 137.90, 137.85, 137.80, 137.5, 135.9, 135.2, 125.9, 124.4, 124.2, 115.2, 115.09, 115.06, 115.04, 70.68, 70.66, 70.64, 43.6, 36.1, 34.6, 28.91, 28.90, 27.91, 27.89, 23.3, 14.4, 10.88, 10.86, signal of one carbon atom is overlapping with the solvent signal. MS HR (ESI) m/z calcd for C51H47N4O2S2 [M + H]+ 811.3140, found 811.3135. Anal. calcd for C51H46N4O2S2: C, 75.52; H, 5.72; N, 6.91; S, 7.91. Found: C, 75.77; H, 5.73; N, 7.06; S, 7.60.
:4) provided mono-substituted 3am and bis-substituted 3a that were recrystallized (CH2Cl2/pentane) to give 3am (35 mg, 17%) as yellow crystals and 3a (137 mg, 52%) as orange crystals. Compound 3a: m.p. 168–170 °C (CH2Cl2/pentane). UV/vis (CH2Cl2, c ∼ 10−5 M): λmax/nm 478 (ε/M−1 cm−1 70100) with a shoulder, 399 (39100), 338 (31300). 1H NMR (400 MHz, CD2Cl2) δ 9.35 (d, J = 1.6 Hz, 2H), 8.67 (dd, J = 8.8, 1.6 Hz, 2H), 8.04–7.98 (m, 2H), 7.96 (s, 2H), 7.95–7.90 (m, 2H), 7.84–7.74 (m, 4H), 7.45 (d, J = 8.7 Hz, 2H), 4.16 (d, J = 7.3 Hz, 2H), 2.08–1.99 (m, 1H), 1.50–1.17 (m, 8H), 0.92 (t, J = 7.4 Hz, 3H), 0.85 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CD2Cl2) δ 190.9, 189.9, 147.9, 145.1, 143.0, 140.5, 135.5, 135.4, 134.1, 128.9, 127.1, 126.7, 124.1, 123.5, 123.3, 110.5, 48.5, 40.1, 31.5, 29.3, 24.9, 23.6, 14.3, 11.2. MS HR (ESI) m/z calcd for C40H33NNaO4 [M + Na]+ 614.2307, found 614.2302. Compound 3am: m.p. 99–102 °C (CH2Cl2/pentane). UV/vis (CH2Cl2, c ∼ 10−5 M): λmax/nm 442 (ε/M−1 cm−1 37900) with a shoulder, 332 (16000). 1H NMR (400 MHz, CD2Cl2) δ 10.07 (s, 1H), 9.48 (d, J = 1.6 Hz, 1H), 8.65 (d, J = 1.1 Hz, 1H), 8.61 (dd, J = 8.8, 1.6 Hz, 1H), 8.03–7.97 (m, 2H), 7.97–7.87 (m, 2H), 7.79–7.71 (m, 2H), 7.50 (d, J = 8.7 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 4.19 (d, J = 7.4 Hz, 2H), 2.10–1.94 (m, 1H), 1.43–1.09 (m, 8H), 0.86 (t, J = 7.4 Hz, 3H), 0.77 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 191.9, 191.1, 190.1, 148.0, 145.8, 145.3, 143.0, 140.6, 135.7, 135.5, 134.3, 130.4, 128.7, 128.0, 127.3, 126.5, 124.7, 124.0, 123.8, 123.5, 123.4, 110.8, 110.6, 48.6, 40.0, 31.5, 29.3, 24.9, 23.5, 14.3, 11.2. MS HR (ESI) m/z calcd for C31H30NO3 [M + H]+ 464.2226, found 464.2220.
:4 to 3:7) to obtain pure fraction of 3b and a mixed fraction. Consecutive chromatography (silica, EtOAc/pentane 1:3) afforded pure products, which were recrystallized (CH2Cl2/pentane) to give 3b (78 mg, 38%) and 3bm (61 mg, 40%) as dark red and maroon crystals, respectively. Compound 3b: m.p. 210–211 °C (CH2Cl2/pentane). UV/vis (CH2Cl2, c ∼ 10−5 M): λmax/nm 542 (ε/M−1 cm−1 51200), 466 (25000), 327 (42300). 1H NMR (400 MHz, CD2Cl2) δ 9.28 (d, J = 1.6 Hz, 2H), 8.79 (s, 2H), 8.70 (dd, J = 7.0, 0.9 Hz, 2H), 8.47 (dd, J = 8.8, 1.6 Hz, 2H), 8.02–7.93 (m, 2H), 7.88–7.75 (m, 4H), 7.56 (d, J = 8.8 Hz, 2H), 4.26 (d, J = 7.2 Hz, 2H), 2.16–2.01 (m, 1H), 1.51–1.21 (m, 8H), 0.95 (t, J = 7.4 Hz, 3H), 0.87 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 187.3, 163.1, 148.9, 145.5, 140.3, 138.0, 135.9, 135.3, 134.7, 129.2, 127.9, 126.3, 125.6, 124.6, 124.0, 115.2, 115.0, 110.6, 71.5, 48.7, 40.1, 31.5, 29.3, 24.9, 23.5, 14.3, 11.2. MS HR (ESI) m/z calcd for C46H33N5NaO2 [M + Na]+ 710.2532, found 710.2527. Anal. calcd for C46H33N5O2: C, 80.33; H, 4.84; N, 10.18. Found: C, 79.98; H, 4.79; N, 10.06. Compound 3bm: m.p. 188–191 °C (CH2Cl2/pentane). UV/vis (CH2Cl2, c ∼ 10−5 M): λmax/nm 502 (ε/M−1 cm−1 27100), 320 (28300). 1H NMR (400 MHz, CD2Cl2) δ 10.11 (s, 1H), 9.28 (d, J = 1.8 Hz, 1H), 8.75 (s, 1H), 8.68–8.63 (m, 2H), 8.42 (dd, J = 8.8, 1.8 Hz, 1H), 8.05 (dd, J = 8.5, 1.6 Hz, 1H), 7.97–7.90 (m, 1H), 7.84–7.73 (m, 2H), 7.54 (t, J = 8.9 Hz, 2H), 4.24 (d, J = 7.5 Hz, 2H), 2.17–1.98 (m, 1H), 1.51–1.17 (m, 8H), 0.93 (t, J = 7.4 Hz, 3H), 0.85 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 191.8, 187.3, 163.0, 148.9, 145.8, 145.6, 140.2, 137.9, 135.8, 135.3, 134.6, 130.6, 129.2, 128.2, 127.6, 125.9, 125.6, 124.6, 124.5, 123.9, 123.8, 115.2, 115.0, 110.9, 110.5, 71.3, 48.6, 40.0, 31.4, 29.3, 24.9, 23.5, 14.3, 11.2. MS HR (ESI) m/z calcd for C34H29N3NaO2 [M + Na]+ 534.2157, found 534.2152. Anal. calcd for C34H29N3O2: C, 79.82; H, 5.71; N, 8.21. Found: C, 79.70; H, 5.70; N, 8.21.
:1, then CH2Cl2). Consecutive chromatography (silica, pentane/CH2Cl2 4:1) afforded mono-product (10 mg, 8%) as a light-yellow solid and bis-product (53 mg, 34%) as a yellow solid. Compounds 4a and 4am were obtained as mixtures of stereoisomers. Compound 4a: m.p. 169–171 °C. UV/vis (CH2Cl2, c ∼ 10−5 M): λmax/nm 466 (ε/M−1 cm−1 76100), 439 (63900) with a shoulder, 346 (11000). 1H NMR (400 MHz, CD2Cl2) δ 8.94–8.77 (m, 2H), 8.57–8.44 (m, 2H), 8.08–7.99 (m, 4H), 7.99–7.94 (m, 4H), 7.89–7.81 (m, 4H), 2.29–2.19 (m, 4H), 0.99–0.66 (m, 16H), 0.61–0.46 (m, 14H). 13C NMR (101 MHz, CD2Cl2) δ 190.77, 190.75, 190.73, 189.55, 189.52, 189.49, 153.24, 153.18, 153.12, 147.13, 147.11, 145.73, 145.72, 145.71, 143.2, 140.6, 135.9, 135.7, 134.9, 134.8, 134.6, 133.65, 133.63, 133.60, 130.62, 130.57, 130.52, 129.46, 129.44, 129.42, 123.7, 123.6, 121.5, 56.0, 44.8, 35.4, 34.2, 28.47, 28.46, 27.7, 23.2, 14.3, 10.63, 10.62. MS HR (ESI) m/z calcd for C49H50NaO4 [M + Na]+ 725.3607, found 725.3601. Anal. calcd for C49H50O4: C, 83.73; H, 7.17. Found: C, 83.83; H, 7.25. Compound 4am: m.p. 91–95 °C. UV/vis (CH2Cl2, c ∼ 10−5 M): λmax/nm 403 (ε/M−1 cm−1 45400) with a shoulder. 1H NMR (400 MHz, CD2Cl2) δ 10.08 (s, 1H), 8.97–8.72 (m, 1H), 8.50 (m, 1H), 8.07–7.89 (m, 7H), 7.89–7.80 (m, 2H), 2.31–2.07 (m, 4H), 0.98–0.66 (m, 16H), 0.66–0.60 (m, 3H), 0.60–0.53 (m, 3H), 0.53–0.38 (m, 8H). 13C NMR (101 MHz, CD2Cl2) δ 192.49, 192.42, 192.36, 190.71, 190.69, 190.68, 189.53, 189.50, 189.47, 153.49, 153.45, 153.40, 153.02, 152.95, 152.87, 147.05, 147.04, 147.03, 146.64, 146.63, 145.34, 145.33, 145.31, 143.2, 140.6, 136.5, 136.0, 135.8, 134.8, 134.7, 134.5, 133.64, 133.62, 133.60, 130.56, 130.51, 130.46, 130.25, 130.22, 130.20, 129.58, 129.55, 129.53, 125.5, 123.7, 123.6, 121.74, 121.72, 121.6, 55.9, 44.79, 44.75, 35.39, 35.36, 34.2, 34.13, 34.11, 28.54, 28.53, 28.43, 28.41, 27.6, 27.53, 27.50, 23.2, 14.3, 14.2, 10.63 (2 carbons), 10.55, 10.53. MS HR (ESI) m/z calcd for C40H47O3 [M + H]+ 575.3525, found 575.3520.
Acknowledgements
We are grateful to the Alexander-von-Humboldt Foundation for a postdoctoral fellowship for A. N.-K. and to the Bavarian State Ministry for of Science, Research, and the Arts for financial support within the Collaborative Research Network “Solar Technologies go Hybrid”.Notes and references
- Selected reviews: (a) A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020 CrossRef CAS PubMed; (b) Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470 RSC; (c) W. Ni, X. Wan, M. Li, Y. Wang and Y. Chen, Chem. Commun., 2015, 51, 4936 RSC.
- (a) A. Yassin, T. Rousseau, P. Leriche, A. Cravino and J. Roncali, Sol. Energy Mater. Sol. Cells, 2011, 95, 462 CrossRef CAS; (b) R. Fitzner, E. Mena-Osteritz, A. Mishra, G. Schulz, E. Reinold, M. Weil, C. Körner, H. Ziehlke, C. Elschner, K. Leo, M. Riede, M. Pfeiffer, C. Uhrich and P. Bäuerle, J. Am. Chem. Soc., 2012, 134, 11064 CrossRef CAS PubMed; (c) A. Mishra, D. Popovic, A. Vogt, H. Kast, T. Leitner, K. Walzer, M. Pfeiffer, E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2014, 26, 7217 CrossRef CAS PubMed; (d) H. Kast, A. Mishra, G. L. Schulz, M. Urdanpilleta, E. Mena-Osteritz and P. Bäuerle, Adv. Funct. Mater., 2015, 25, 3414 CrossRef CAS.
- Q. Zhang, B. Kan, F. Liu, G. Long, X. Wan, X. Chen, Y. Zuo, W. Ni, H. Zhang, M. Li, Z. Hu, F. Huang, Y. Cao, Z. Liang, M. Zhang, T. P. Russell and Y. Chen, Nat. Photonics, 2015, 9, 35 CrossRef CAS.
- (a) J. E. Anthony, Chem. Mater., 2011, 23, 583 CrossRef CAS; (b) A. F. Eftaiha, J. P. Sun, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 1201 RSC.
- (a) S. Rajaram, R. Shivanna, S. K. Kandappa and K. S. Narayan, J. Phys. Chem. Lett., 2012, 3, 2405 CrossRef CAS PubMed; (b) Y. Zang, C.-Z. Li, C.-C. Chueh, S. T. Williams, W. Jiang, Z.-H. Wang, J.-S. Yu and A. K.-Y. Jen, Adv. Mater., 2014, 26, 5708 CrossRef CAS PubMed; (c) Y. Zhong, M. T. Trinh, R. Chen, W. Wang, P. P. Khlyabich, B. Kumar, Q. Xu, C.-Y. Nam, M. Y. Sfeir, C. Black, M. L. Steigerwald, Y.-L. Loo, S. Xiao, F. Ng, X.-Y. Zhu and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 15215 CrossRef CAS PubMed; (d) J. Zhao, Y. Li, H. Lin, Y. Liu, K. Jiang, C. Mu, T. Ma, J. Y. Lin Lai, H. Hu, D. Yu and H. Yan, Energy Environ. Sci., 2015, 8, 520 RSC; (e) D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156 CrossRef CAS PubMed.
- (a) S. Holliday, R. S. Ashraf, C. B. Nielsen, M. Kirkus, J. A. Röhr, C.-H. Tan, E. Collado-Fregoso, A.-C. Knall, J. R. Durrant, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 898 CrossRef CAS PubMed; (b) O. K. Kwon, J.-H. Park, D. W. Kim, S. K. Park and S. Y. Park, Adv. Mater., 2015, 27, 1951 CrossRef CAS PubMed; (c) Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170 CrossRef CAS PubMed; (d) H. Lin, S. Chen, Z. Li, J. Y. L. Lai, G. Yang, T. McAfee, K. Jiang, Y. Li, Y. Liu, H. Hu, J. Zhao, W. Ma, H. Ade and H. Yan, Adv. Mater., 2015, 27, 7299 CrossRef CAS PubMed.
- (a) N. M. Kronenberg, M. Deppisch, F. Würthner, H. W. A. Lademann, K. Deing and K. Meerholz, Chem. Commun., 2008, 6489 RSC; (b) H. Bürckstümmer, E. V. Tulyakova, M. Deppisch, M. R. Lenze, N. M. Kronenberg, M. Gsänger, M. Stolte, K. Meerholz and F. Würthner, Angew. Chem., Int. Ed., 2011, 50, 11628 CrossRef PubMed; (c) A. Arjona-Esteban, J. Krumrain, A. Liess, M. Stolte, L. Huang, D. Schmidt, V. Stepanenko, M. Gsänger, D. Hertel, K. Meerholz and F. Würthner, J. Am. Chem. Soc., 2015, 137, 13524 CrossRef CAS PubMed.
- (a) H. Bai, Y. Wang, P. Cheng, J. Wang, Y. Wu, J. Hou and X. Zhan, J. Mater. Chem. A, 2015, 3, 1910 RSC; (b) Y. Lin, Z.-G. Zhang, H. Bai, J. Wang, Y. Yao, Y. Li, D. Zhu and X. Zhan, Energy Environ. Sci., 2015, 8, 610 RSC; (c) Y. Lin and X. Zhan, Acc. Chem. Res., 2016, 49, 175 CrossRef CAS PubMed.
- X. Guo, S. Wang, V. Enkelmann, M. Baumgarten and K. Müllen, Org. Lett., 2011, 13, 6062 CrossRef CAS PubMed.
- J. S. Bandar, R. W. Coscia and T. H. Lambert, Tetrahedron, 2011, 67, 4364 CrossRef CAS.
- (a) N. Kleinhenz, L. Yang, H. Zhou, S. C. Price and W. You, Macromolecules, 2011, 44, 872 CrossRef CAS; (b) P. Coppo and M. L. Turner, J. Mater. Chem., 2005, 15, 1123 RSC.
- F. Würthner, S. Yao, J. Schilling, R. Wortmann, M. Redi-Abshiro, E. Mecher, F. Gallego-Gomez and K. Meerholz, J. Am. Chem. Soc., 2001, 123, 2810 CrossRef.
- A. Singh, C.-K. Lim, Y.-D. Lee, J.-h. Maeng, S. Lee, J. Koh and S. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 8881 CAS.
- A. Zitzler-Kunkel, M. R. Lenze, T. Schnier, K. Meerholz and F. Würthner, Adv. Funct. Mater., 2014, 24, 4645 CrossRef CAS.
- (a) N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed; (b) W. N. Hansen and G. J. Hansen, Phys. Rev. A, 1987, 36, 1396 CrossRef CAS PubMed; (c) C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS PubMed.
- C. Wetzel, A. Mishra, E. Mena-Osteritz, A. Liess, M. Stolte, F. Würthner and P. Bäuerle, Org. Lett., 2014, 16, 362 CrossRef CAS PubMed.
- C.-Z. Li, H.-L. Yip and A. K.-Y. Jen, J. Mater. Chem., 2012, 22, 4161 RSC.
- (a) S. M. McAfee, J. M. Topple, A.-J. Payne, J.-P. Sun, I. G. Hill and G. C. Welch, ChemPhysChem, 2015, 16, 1190 CrossRef CAS PubMed; (b) Y. Kim, C. E. Song, E.-J. Ko, D. Kim, S.-J. Moon and E. Lim, RSC Adv., 2015, 5, 4811 RSC; (c) H. Bai, Y. Wu, Y. Wang, Y. Wu, R. Li, P. Cheng, M. Zhang, J. Wang, W. Ma and X. Zhan, J. Mater. Chem. A, 2015, 3, 20758 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
- A. J. Fry, in Laboratory Techniques in Electroanalytical Chemistry, ed. P. T. Kissinger and W. R. Heineman, Marcel Dekker Ltd, New York, 2nd edn, 1996, p. 481 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00046k |
| This journal is © the Partner Organisations 2016 |