
Tartrate-derived iminophosphorane catalyzed asymmetric hydroxymethylation of 3-substituted oxindoles with paraformaldehyde†
Xing
Gao
ab,
Jianwei
Han
*b and
Limin
Wang
*a
aKey Laboratory of Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, 130 Mei Long Road, Shanghai 200237, P. R. China. E-mail: wanglimin@ecust.edu.cn
bShanghai-Hong Kong Joint Laboratory in Chemical Synthesis, Shanghai Institute of Organic Chemistry, The Chinese Academy of Sciences, 345 Ling Ling Road, Shanghai, 200032, P. R. China. E-mail: jianweihan@sioc.ac.cn
First published on 10th March 2016
Abstract
The enantioselective synthesis of 3-hydroxymethyl-2-oxindoles was achieved through organocatalysis by a tartrate-derived chiral iminophosphorane in 81–98% yields and up to 94% ee under mild conditions. Of note is that readily available and easily usable paraformaldehyde was employed as a hydroxymethylation C1 unit in the reaction.
Oxindole, a well-known scaffold, has been found in a number of medicinally important compounds.1 Because of the structural diversity and biological activities of oxindole based small molecules, such as NSC635473, YK-4-279, donaxaridine, convolutamydine A, dioxibrassinin, SU11248 (Sutent) etc. (Fig. 1), they have continued to attract the attention of chemists and pharmacologists.2 Therefore, inspired by the pharmaceutical importance and the synthetic value of oxindole derivatives, intense efforts have been made toward the construction of enantiomerically enriched chiral 3,3-disubstituted oxindoles in recent years.3
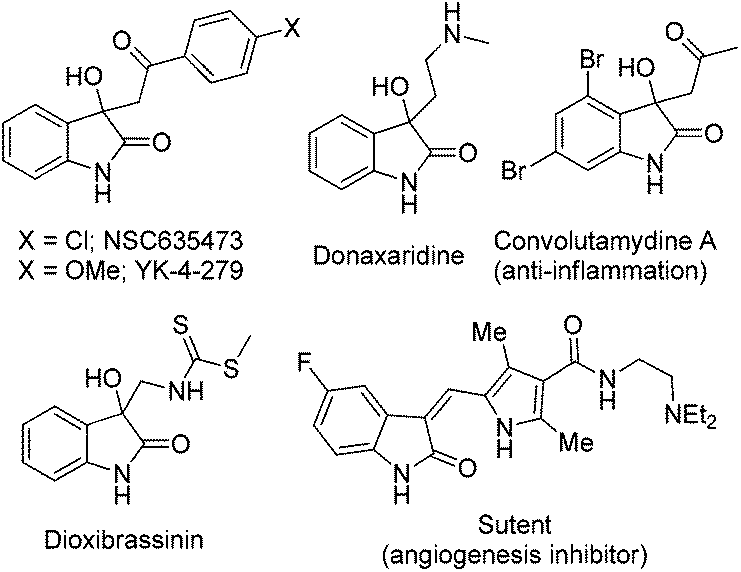 | ||
| Fig. 1 Examples of biologically active oxindole-based molecules. | ||
On the other hand, the hydroxymethylation reaction, one of the most important reactions in organic synthesis due to its rich chemistry of functional group conversion with the hydroxyl group, is a powerful and atom-economical one-carbon extension method.4 In this regard, catalytic asymmetric hydroxymethylations of silyl enolates,5 α-cyanopropionates,6 β-keto esters7 and ketones8 have been intensively investigated in the last decade.9 Particularly, in 2010, Kobayashi et al. tried one example of enantioselective hydroxymethylation of N-Boc-3-methyl-2-oxindole with formalin (37% formaldehyde in aqueous solution) using a chiral bipyridine–ScIII complex, which gave the desired product in 59% yield with 82% ee (Scheme 1).8c Subsequently, Yuan and co-workers reported a bifunctional thiourea-catalyzed asymmetric hydroxymethylation of N-Boc-oxindoles using paraformaldehyde, and the adducts were afforded in 80–99% yields and 5–91% ee.8d Since then, the Feng group described an enantio-selective synthesis of 1,3-bis(hydroxymethyl)-2-oxindoles with formalin, in up to 93% yield and 99% ee by using a recyclable NdIII–N,N′-dioxide complex.8e Interestingly, Bisai and co-workers employed this strategy involving a thiourea catalyzed enantioselective hydroxymethylation reaction for the total synthesis of both spiro(pyrrolidinyl-oxindole) and hexahydropyrrolo[2,3-b]indole alkaloids.8f However, there are still some limitations in terms of the substrate scope and enantioselectivity in the asymmetric hydroxymethylation of 3-disubstituted-2-oxindoles using these catalytic systems. Recently, chiral Brønsted base catalysis has achieved great advances in the growing branch of asymmetric organocatalysis.10,11 As such, given the abundant availability and ready diversification of tartaric acid and its derivatives thereof, we have developed a novel family of chiral iminophosphoranes featuring a tartrate-derived skeleton (Fig. 2); these iminophosphoranes showed remarkable catalytic activity toward the asymmetric chlorination of 3-substituted oxindoles.11b To further expand the utility of this library of iminophosphoranes, herein we describe the asymmetric hydroxymethylation of 3-substituted oxindoles with paraformaldehyde as a useful C1 unit in high yields (81–98%) and excellent stereocontrol (up to 94% ee).
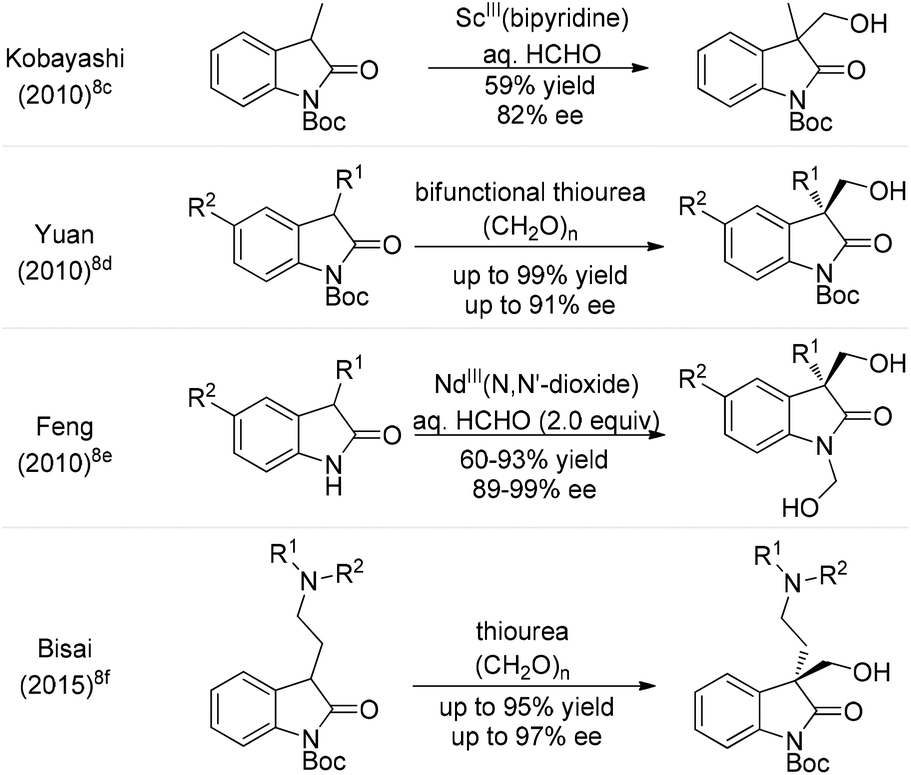 | ||
| Scheme 1 Established asymmetric hydroxymethylation of 3-substituted oxindoles. | ||
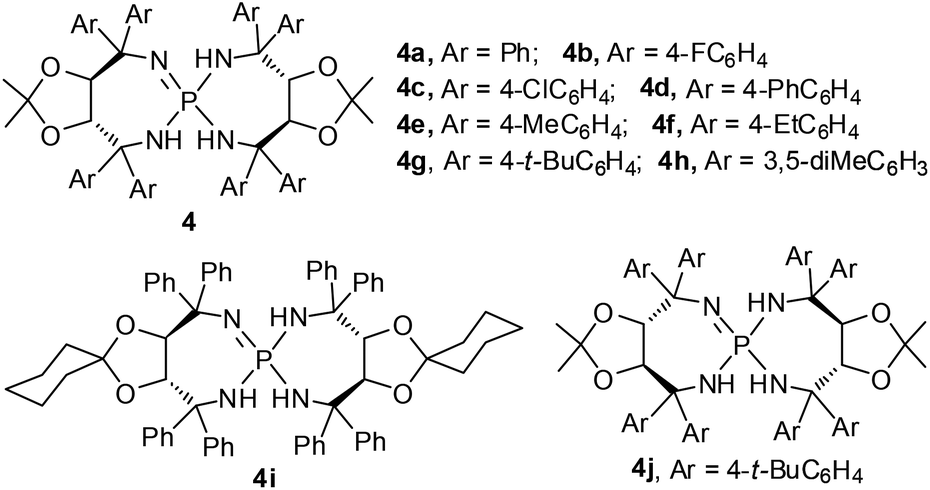 | ||
| Fig. 2 The iminophosphorane catalysts used in this work. | ||
Initially, we studied the hydroxymethylation of N-Boc-3-benzyloxindole 1a with paraformaldehyde 2 to optimize the reaction conditions and catalysts at room temperature (Table 1). Firstly, given the easy availability of iminophosphorane 4a (Fig. 2), we used 4a as the catalyst (10 mol%) to screen various solvents. In chlorinated solvents, dichloromethane (DCM) or 1,2-dichloroethane (DCE), the reaction afforded excellent yields and moderate ee values in 15 hours (Table 1, entries 1 and 2, 97% yield and 46% ee, 98% yield and 43% ee, respectively). In polar solvents, although high yields were obtained, the enantiomeric excess was quite low. For example, when the reaction was conducted in acetone (Table 1, entry 3), methanol (MeOH) (Table 1, entry 4) or acetonitrile (MeCN) (Table 1, entry 5), it could afford 85–98% yields but only 19% ee, 12% ee, or −5% ee, respectively. Next, screening of non-polar solvents revealed that not only excellent yields could be obtained, but also the enantiomeric excess was significantly improved (Table 1, entries 6–9, 91–95% yields, 73–88% ee). Mesitylene was demonstrated to be superior to the other solvents; it could provide the desired product 3a in 95% yield and 88% ee (Table 1, entry 9). Regarding the formaldehyde reactant, we also tried formalin as an aldehyde source instead of (CH2O)n in mesitylene solution, even though the yield was still excellent, 96%, the enantiomeric excess was obviously decreased to 67% (Table 1, entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Cat. 4 | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.1 mmol, 1.0 equiv.) and catalyst 4 (10 mol%) were dissolved in solvent (2 mL), then 2 (3 equiv.) was added into the stirring solution at 25 °C. After 15 h, the solution mixture was purified directly by silica gel column chromatography to afford product 3a. b Isolated yield. c Enantiopurity of the products was determined by HPLC analysis using a chiral column with hexane/isopropanol as the solvent. d Formalin (25 μL, 3 equiv.) was used instead of (CH2O)n. e A pre-made HCl salt of 4a was used. f 5 mol% of 4g was used. g 1 mol% of 4g was used and the reaction time was 30 h. | ||||
| 1 | 4a | DCM | 97 | 46 |
| 2 | 4a | DCE | 98 | 43 |
| 3 | 4a | Acetone | 85 | 19 |
| 4 | 4a | MeOH | 89 | 12 |
| 5 | 4a | MeCN | 98 | −5 |
| 6 | 4a | n-Hexane | 91 | 73 |
| 7 | 4a | Cyclohexane | 92 | 83 |
| 8 | 4a | Toluene | 93 | 76 |
| 9 | 4a | Mesitylene | 95 | 88 |
| 10d | 4a | Mesitylene | 96 | 67 |
| 11 | 4b | Mesitylene | 88 | 73 |
| 12 | 4c | Mesitylene | 89 | 72 |
| 13 | 4d | Mesitylene | 85 | 90 |
| 14 | 4e | Mesitylene | 98 | 89 |
| 15 | 4f | Mesitylene | 97 | 92 |
| 16 | 4g | Mesitylene | 98 | 94 |
| 17 | 4h | Mesitylene | 97 | 38 |
| 18 | 4i | Mesitylene | 96 | 87 |
| 19 | 4j | Mesitylene | 97 | −95 |
| 20e | 4a·HCl | Mesitylene | Trace | N.D. |
| 21f | 4g | Mesitylene | 90 | 94 |
| 22g | 4g | Mesitylene | 91 | 95 |
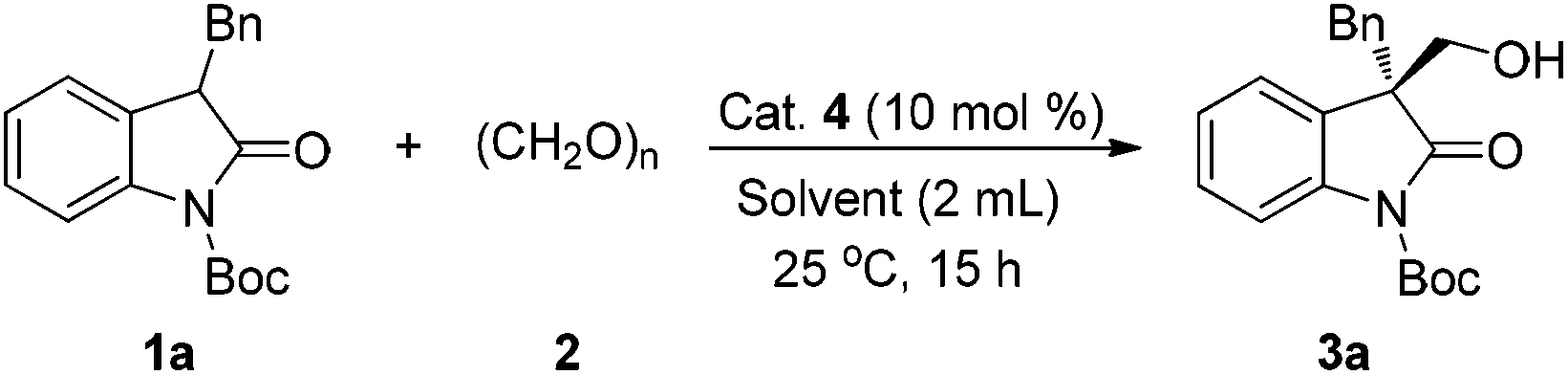
With mesitylene as the optimal solvent, we were pleased to discover that all iminophosphoranes 4 (Fig. 2) bearing diverse aryl groups could catalyze the direct hydroxymethylation of 3-benzyl substituted oxindole 1a with paraformaldehyde, providing 3a in good yields with a quaternary stereocenter as shown in Table 1, entries 11–17. When 4b (Ar = 4-FC6H4) or 4c (Ar = 4-ClC6H4) was used as the catalyst, comparable results were obtained (Table 1, entries 11 and 12, 88% yield and 73% ee, 89% yield and 72% ee, respectively). However, when 4d (Ar = 4-PhC6H4) was applied in the reaction, both the yield and enantiomeric excess were slightly increased (Table 1, entry 13, 85% yield, 90% ee) compared to the result of 4a (Table 1, entry 9). Furthermore, the catalytic performance of the alkyl-substituted Ar group of iminophosphoranes was exploited in the model reaction, and the desired product 3a was achieved in excellent yields, and the enantiomeric excess was increased along with the bulk of alkyl chains substituted at the C4 position of the Ar group (Table 1, entries 9, 14–16, 95–98% yield, 88–94% ee). In particular, catalyst 4g was superior to the other catalysts 4a–f in terms of enantioselectivity (Table 1, entries 16 vs. 9, 11–15). In sharp contrast, catalyst 4h, bearing 3,5-dimethylphenyl groups, provided excellent yield but low ee (Table 1, entry 17, 97% yield, 38% ee). The ketal moiety of the iminophosphorane catalyst 4i with a cyclohexanone unit afforded 96% yield of 3a together with 87% ee (Table 1, entry 18). Additionally, as the enantiomer of catalyst 4g, catalyst 4j was employed to this reaction and it afforded the product with 97% yield and −95% ee (Table 1, entry 19 was compared with entry 16). Notably, when the HCl salt of 4a was employed as the catalyst, only trace 3a was observed by thin-layer chromatography (Table 1, entry 20). The enantioselectivity did not decrease at all with the decline of catalytic loading, and we could obtain 95% ee even in the case of 1 mol% 4g as the catalyst but the reaction rate slowed down (Table 1, entries 21 and 22).
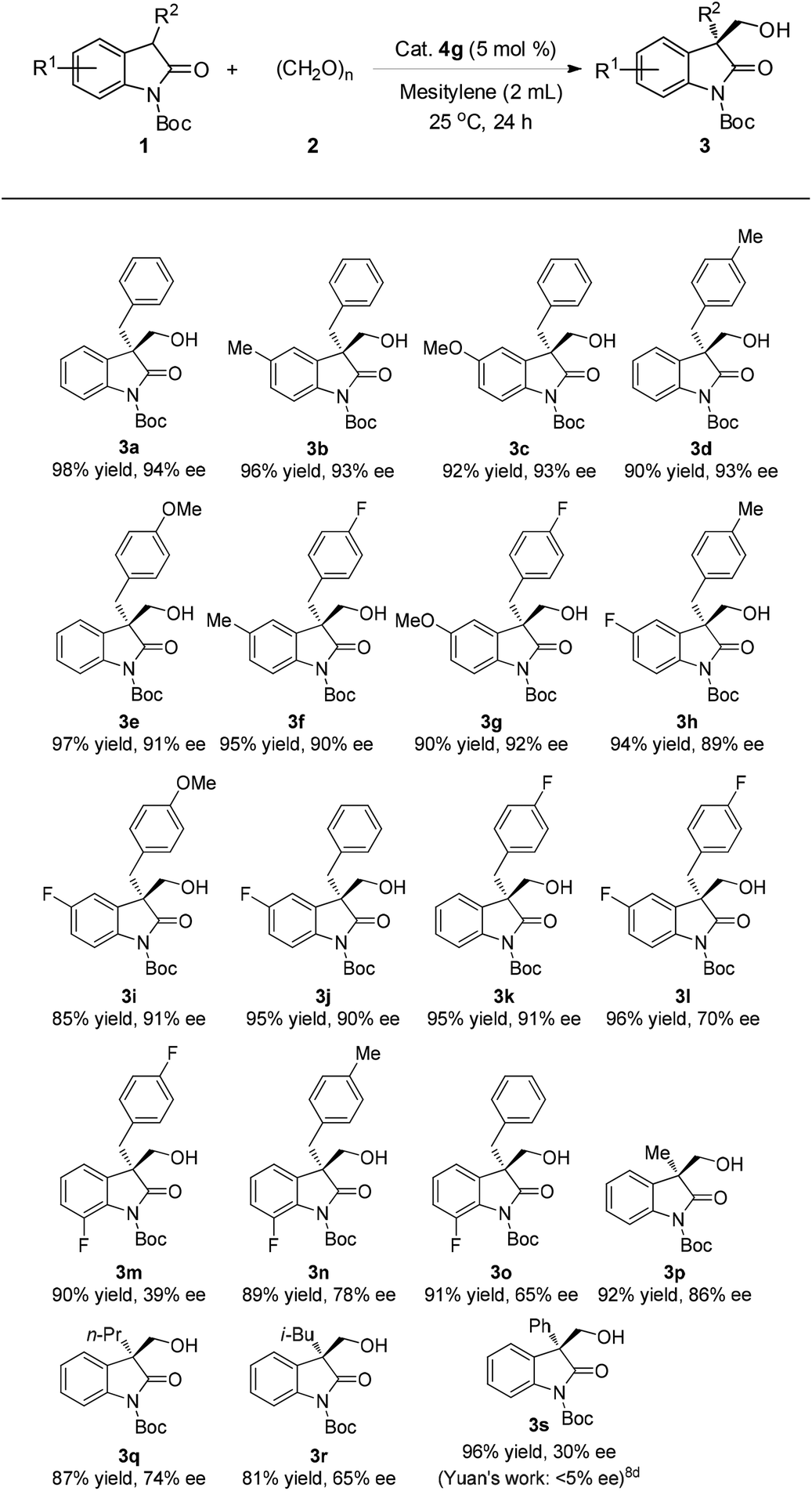
Having established the optimal reaction conditions, we then explored the generality of the substrate scope by varying the structure of N-Boc-protected oxindoles 1 as summarized in Table 2. A wide array of oxindoles were proved to be suitable substrates. For example, an electron-rich (3b, 96% yield, 93% ee, and 3c, 92% yield, 93% ee) or electron-poor (3j, 95% yield, 90% ee) group substituted at the C5 position or 4-methyl (3d, 90% yield, 93% ee), 4-methoxyl (3e, 97%, 91% ee), 4-flouro (3k, 95%, 91% ee) substituted on the 3-benzyl group were accommodated in this reaction. In addition, oxindoles bearing both electron-rich and electron-poor groups on the aryl rings respectively were also well tolerated (3f–i, 85–95% yields, 89–92% ee). However, the difluoro-substituted or 7-fluoro-substituted oxindoles such as 3m, 3n and 3o were not well compatible with this method (Table 2). C3-aliphatic substituents led to moderate to good enantioselectivity despite the good yields (3p–r, 81–92% yield, 65–86% ee). It is noteworthy that, in the case of 3-phenyl oxindole 1s as the substrate, the corresponding adduct 3s was obtained in 30% ee which however has been the best result so far in comparison with a previous report (95% yield, <5% ee with a bifunctional thiourea–tertiary amine as the catalyst).8d
| a Conditions: 1 (0.1 mmol, 1.0 equiv.),and cat. 4g (0.005 mmol, 5 mol%) were added to a tube with mesitylene (2 mL) and a magnetic stirrer at 25 °C, then 2 (0.3 mmol, 3.0 equiv.) was added to the stirring solution. After stirring for 24 h at 25 °C, the reaction mixture was purified directly by silica gel column chromatography to yield product 3. The ee of product 3 was determined by chiral HPLC analysis. |
|---|
|
The stereochemical rationale for our hypothesized catalytic hydroxymethylation process in the presence of iminophosphorane is shown in Fig. 3. Oxindole 1 would be initially deprotonated by chiral iminophosphorane 4 which is known as a Brønsted base, the resulting chiral phosphonium and enolate of 1 could form a structured ion pair (I) and activate the nucleophile 1, allowing the highly stereoselective addition to formaldehyde (electrophile) and giving the enantioenriched adducts 3.
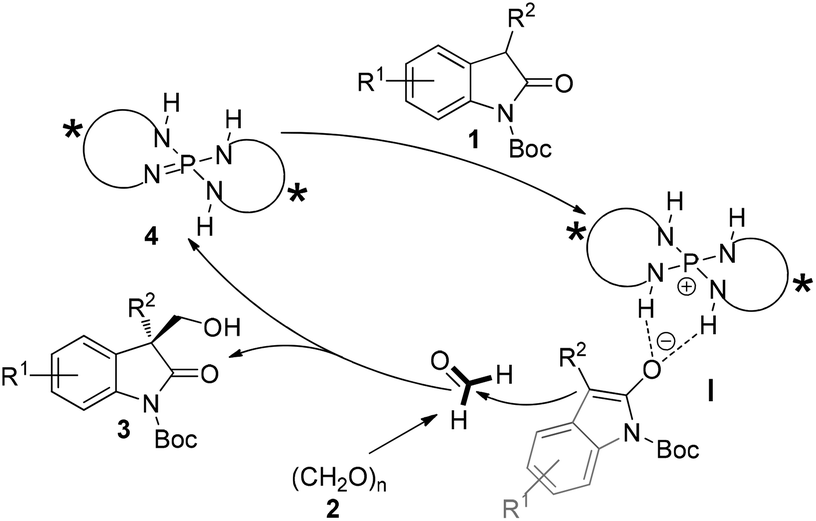 | ||
| Fig. 3 Mechanism hypothesis for the asymmetric hydroxymethylation. | ||
In conclusion, we have developed a new catalytic asymmetric approach for the hydroxymethylation of 3-substituted oxindoles. With the suitable choice of a chiral tartrate-derived iminophosphorane catalyst, a range of various substituted oxindoles bearing quaternary stereocenters were synthesized with high efficiency (81–98% yield) and good to excellent enantioselectivity (up to 94% ee). Further investigations on this catalytic system by using iminophosphoranes to produce important compounds are currently underway in our laboratory.
Acknowledgements
This work was supported by grants from the National Nature Science Foundation of China (NSFC no. 21472213, 21202186), as well as by Croucher Foundation (Hong Kong) in the form of a CAS-Croucher Foundation Joint Laboratory Grant. This research program was performed under the auspices of Professor Henry N. C. Wong, whom we thank for helpful discussions and generous support.References
- (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed; (b) C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2209 CrossRef CAS; (c) S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 CrossRef CAS.
- (a) T. Ueda, M. Inada, I. Okamoto, N. Morita and O. Tamura, Org. Lett., 2008, 10, 2043 CrossRef CAS PubMed; (b) T. Itoh, H. Ishikawa and Y. Hayashi, Org. Lett., 2009, 11, 3854 CrossRef CAS PubMed; (c) S. Lin, Z. Yang, B. H. B. Kwok, M. Koldobskiy and C. M. Crews, J. Am. Chem. Soc., 2004, 126, 6347 CrossRef CAS PubMed; (d) H. B. Rasmussen and J. K. Macleod, J. Nat. Prod., 1997, 60, 1152 CrossRef CAS; (e) H. V. Erkizan, Y. Kong, M. Merchant, S. Schlottmann, J. S. Barber-Rotenberg, L. Yuan, O. D. Abaan, T. Chou, S. Dakshanamurthy, M. L. Brown, A. Üren and J. A. Toretsky, Nat. Med., 2009, 15, 750 CrossRef CAS PubMed; (f) V. U. Khuzhaev, I. Zhalolov, K. K. Turgunov, B. Tashkhodzhaev, M. G. Levkovich, S. F. Arpova and A. S. Shashkov, Chem. Nat. Compd., 2004, 40, 269 CrossRef CAS; (g) J. I. Jimenez, U. Huber, R. E. Moore and G. M. L. Patterson, J. Nat. Prod., 1999, 62, 569 CrossRef CAS PubMed; (h) J. S. Carle and C. Christophersen, J. Org. Chem., 1981, 46, 3440 CrossRef CAS; (i) H. Prenen, J. Cools, N. Mentens, C. Folens, R. Sciot, P. Schoffski, A. Van Oosterom, P. Marynen and M. Debiec-Rychter, Clin. Cancer Res., 2006, 8, 2622 CrossRef PubMed; (j) R. J. Motzer, M. D. Michaelson, B. G. Redman, G. R. Hudes, G. Wilding, R. A. Figlin, M. S. Ginsberg, S. T. Kim, C. M. Baum, S. E. DePrimo, J. Z. Li, C. L. Bello, C. P. Theuer, D. J. George and B. I. Rini, J. Clin. Oncol., 2006, 24, 16 CrossRef CAS PubMed; (k) R. J. Roskoski, Biochem. Biophys. Res. Commun., 2007, 356, 323 CrossRef CAS PubMed; (l) P. Sharma, K. R. Senwar, M. K. Jeengar, T. S. Reddy, V. G. M. Naidu, A. Kamal and N. Shankaraiah, Eur. J. Med. Chem., 2015, 104, 11 CrossRef CAS PubMed.
- For selected examples, see: (a) G. Luppi, P. G. Cozzi, M. Monari, B. Kaptein, Q. B. Broxterman and C. Tomasini, J. Org. Chem., 2005, 70, 7418 CrossRef CAS PubMed; (b) T. Ishimaru, N. Shibata, J. Nagai, S. Nakamura, T. Toru and S. Kanemasa, J. Am. Chem. Soc., 2006, 128, 16488 CrossRef CAS PubMed; (c) Y. Kato, M. Furutachi, Z. Chen, H. Mitsunuma, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 9168 CrossRef CAS PubMed; (d) T. Bui, N. R. Candeias and C. F. Barbas III, J. Am. Chem. Soc., 2010, 132, 5574 CrossRef CAS PubMed; (e) T. Zhang, L. Cheng, S. Hameed, L. Liu, D. Wang and Y. J. Chen, Chem. Commun., 2011, 47, 6644 RSC; (f) C. Curti, G. Rassu, V. Zambrano, L. Pinna, G. Pelosi, A. Sartori, L. Battistini, F. Zanardi and G. Casiraghi, Angew. Chem., Int. Ed., 2012, 51, 6200 CrossRef CAS PubMed; (g) L. Li, W. Chen, W. Yang, Y. Pan, H. Liu, C. H. Tan and Z. Jiang, Chem. Commun., 2012, 48, 5124 RSC; (h) B. Zhu, W. Zhang, R. Lee, Z. Han, W. Yang, D. Tan, K. W. Huang and Z. Jiang, Angew. Chem., Int. Ed., 2013, 52, 6666 CrossRef CAS PubMed; (i) B. D. Cui, W. Y. Han, Z. J. Wu, X. M. Zhang and W. C. Yuan, J. Org. Chem., 2013, 78, 8833 CrossRef CAS PubMed; (j) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed; (k) F. Zhou, Y. L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS; (l) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef CAS PubMed; (m) X. Z. Bao, B. M. Wang, L. C. Cui, G. D. Zhu, Y. L. He, J. P. Qu and Y. M. Song, Org. Lett., 2015, 17, 5168 CrossRef CAS PubMed.
- For reviews, see: (a) J. L. G. Fierro, Catal. Lett., 1993, 22, 67 CrossRef CAS; (b) J. S. Lee, J. C. Kim and Y. G. Kim, Appl. Catal., 1990, 57, 1 CrossRef CAS.
- (a) N. Ozawa, M. Wadamoto, K. Ishihara and H. Yamamoto, Synlett, 2003, 2219 Search PubMed; (b) K. Manabe, S. Ishikawa, T. Hamada and S. Kobayashi, Tetrahedron, 2003, 59, 10439 CrossRef CAS; (c) S. Ishikawa, T. Hamada, K. Manabe and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 12236 CrossRef CAS PubMed; (d) S. Kobayashi, T. Ogino, H. Shimizu, S. Ishikawa, T. Hamada and K. Manabe, Org. Lett., 2005, 7, 4729 CrossRef CAS PubMed; (e) M. Kokubo, C. Ogawa and S. Kobayashi, Angew. Chem., Int. Ed., 2008, 47, 6909 CrossRef CAS PubMed; (f) C. Mukherjee, T. Kitanosono and S. Kobayashi, Chem. – Asian J., 2011, 6, 2308 CrossRef CAS PubMed.
- (a) R. Kuwano, H. Miyazaki and Y. Ito, Chem. Commun., 1998, 71 RSC; (b) C. B. Ji, Y. L. Liu, Z. Y. Cao, Y. Y. Zhang and J. Zhou, Tetrahedron Lett., 2011, 52, 6118 CrossRef CAS; (c) S. Shirakawa, K. Ota, S. J. Terao and K. Maruoka, Org. Biomol. Chem., 2012, 10, 5753 RSC.
- (a) I. Fukuchi, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2007, 349, 509 CrossRef CAS; (b) S. Mouri, Z. H. Chen, S. Matsunaga and M. Shibasaki, Chem. Commun., 2009, 5138 RSC.
- (a) H. Torii, M. Nakadai, K. Ishihara, S. Saito and H. Yamamoto, Angew. Chem., Int. Ed., 2004, 43, 1983 CrossRef CAS PubMed; (b) J. Casas, H. Sunden and A. Cordova, Tetrahedron Lett., 2004, 45, 6117 CrossRef CAS; (c) S. Kobayashi, M. Kokubo, K. Kawasumi and T. Nagano, Chem. – Asian J., 2010, 5, 490 CrossRef CAS PubMed; (d) X. L. Liu, Y. H. Liao, Z. J. Wu, L. F. Cun, X. M. Zhang and W. C. Yuan, J. Org. Chem., 2010, 75, 4872 CrossRef CAS PubMed; (e) K. Shen, X. H. Liu, W. T. Wang, G. Wang, W. D. Cao, W. Li, X. L. Hu, L. L. Lin and X. M. Feng, Chem. Sci., 2010, 1, 590 RSC; (f) S. De, M. K. Das, S. Bhunia and A. Bisai, Org. Lett., 2015, 17, 5922 CrossRef CAS PubMed; (g) A. Q. Chen, J. Xu, W. Chiang and C. L. L. Chai, Tetrahedron, 2010, 66, 1489 CrossRef CAS; (h) M. Pasternak, J. Paradowska, M. Rogozinska and J. Mlynarski, Tetrahedron Lett., 2010, 51, 4088 CrossRef CAS; (i) C. Gioia, A. Ricci, L. Bernardi, K. Bourahla, N. Tanchoux, M. Robitzer and F. Quignard, Eur. J. Org. Chem., 2013, 588 CrossRef CAS; (j) N. Mase, A. Inoue, M. Nishio and K. Takabe, Bioorg. Med. Chem. Lett., 2009, 19, 3955 CrossRef CAS PubMed.
- For direct aldol reactions using formaldehyde as a C1 unit, see: (a) T. Smejkal, H. Han, B. Breit and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 10366 CrossRef CAS PubMed; (b) R. K. Boeckman Jr. and J. R. Miller, Org. Lett., 2009, 11, 4544 CrossRef PubMed; (c) M. Priede, M. Kazak, T. Kalnins, K. Shubin and E. Suna, J. Org. Chem., 2014, 79, 3715 CrossRef CAS PubMed; (d) R. K. Boeckman Jr., K. F. Biegasiewicz, D. J. Tusch and J. R. Miller, J. Org. Chem., 2015, 80, 4030 CrossRef PubMed.
- (a) T. Ishikawa, Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts, Wiley, New York, 2009 Search PubMed. For reviews on catalysis by chiral organobases, see: (b) T. Ishikawa and T. Kumamoto, Synthesis, 2006, 737 CrossRef CAS; (c) D. Leow and C. H. Tan, Chem. – Asian J., 2009, 4, 488 CrossRef CAS PubMed; (d) D. Leow and C. H. Tan, Synlett, 2010, 1589 CAS; (e) T. Ishikawa, Chem. Pharm. Bull., 2010, 58, 1555 CrossRef CAS PubMed; (f) X. Fu and C. H. Tan, Chem. Commun., 2011, 47, 8210 RSC; (g) P. Selig, Synthesis, 2013, 703 CrossRef CAS; (h) H. Krawczyk, M. Dzięgielewski, D. Deredas, A. Albrecht and Ł. Albrecht, Chem. – Eur. J., 2015, 21, 10268 CrossRef CAS PubMed. For selected examples on catalysis by chiral organobases, see: (i) E. J. Corey and M. J. Grogan, Org. Lett., 1999, 1, 157 CrossRef CAS PubMed; (j) T. Ishikawa, Y. Araki, T. Kumamoto, H. Seki, K. Fukuda and T. Isobe, Chem. Commun., 2001, 245 RSC; (k) B. M. Nugent, R. A. Yoder and J. N. Johnston, J. Am. Chem. Soc., 2004, 126, 3418 CrossRef CAS PubMed; (l) M. Terada, H. Ube and Y. Yaguchi, J. Am. Chem. Soc., 2006, 128, 1454 CrossRef CAS PubMed; (m) D. Uraguchi, S. Sakaki and T. Ooi, J. Am. Chem. Soc., 2007, 129, 12392 CrossRef CAS PubMed; (n) T. A. Davis, J. C. Wilt and J. N. Johnston, J. Am. Chem. Soc., 2010, 132, 2880 CrossRef CAS PubMed; (o) Y. Sohtome, B. Shin, N. Horitsugi, R. Takagi, K. Noguchi and K. Nagasawa, Angew. Chem., Int. Ed., 2010, 49, 7299 CrossRef CAS PubMed; (p) T. A. Davis and J. N. Johnston, Chem. Sci., 2011, 2, 1076 RSC; (q) D. Uraguchi, K. Yoshioka, Y. Ueki and T. Ooi, J. Am. Chem. Soc., 2012, 134, 19370 CrossRef CAS PubMed; (r) M. T. Corbett, D. Uraguchi, T. Ooi and J. S. Johnson, Angew. Chem., Int. Ed., 2012, 51, 4685 CrossRef CAS PubMed; (s) J. S. Bandar and T. H. Lambert, J. Am. Chem. Soc., 2012, 134, 5552 CrossRef CAS PubMed; (t) T. Misaki, N. Jin, K. Kawano and T. Sugimura, Chem. Lett., 2012, 41, 1675 CrossRef CAS; (u) T. E. Shubina, M. Freund, S. Schenker, T. Clark and S. B. Tsogoeva, Beilstein J. Org. Chem., 2012, 8, 1485 CrossRef CAS PubMed; (v) T. Takeda and M. Terada, J. Am. Chem. Soc., 2013, 135, 15306 CrossRef CAS PubMed; (w) J. S. Bandar and T. H. Lambert, J. Am. Chem. Soc., 2013, 135, 11799 CrossRef CAS PubMed; (x) A. M. Goldys, M. G. Núñez and D. J. Dixon, Org. Lett., 2014, 16, 6294 CrossRef CAS PubMed; (y) J. S. Bandar, A. Barthelme, A. Y. Mazori and T. H. Lambert, Chem. Sci., 2015, 6, 1537 RSC; (z) D. Uraguchi, K. Yamada and T. Ooi, Angew. Chem., Int. Ed., 2015, 54, 9954 CrossRef CAS PubMed.
- (a) M. Işik, M. Y. Unver and C. Tanyeli, J. Org. Chem., 2015, 80, 828 CrossRef PubMed; (b) X. Gao, J. Han and L. Wang, Org. Lett., 2015, 17, 4596 CrossRef CAS PubMed; (c) A. J. M. Farley, C. Sandford and D. J. Dixon, J. Am. Chem. Soc., 2015, 137, 15992 CrossRef CAS PubMed; (d) G. P. Robertson, A. J. M. Farley and D. J. Dixon, Synlett, 2016, 21 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00074f |
| This journal is © the Partner Organisations 2016 |