
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conformations of large macrocycles and ring-in-ring complexes†
Jeremy K.
Klosterman‡
a,
Janis
Veliks
a,
Derik K.
Frantz
a,
Yoshizumi
Yasui
a,
Michael
Loepfe
a,
Eli
Zysman-Colman§
a,
Anthony
Linden
a and
Jay S.
Siegel
*ab
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland
bSchool of Pharmaceutical Science and Technology, Tianjin University, 92 Wejin Road, Nankai District Tianjin, 300072, P. R. China. E-mail: dean_spst@tju.edu.cn
First published on 23rd March 2016
Abstract
A kinetically directed, stepwise approach towards molecular Borromean links enabled the isolation and structural characterization of synthetic intermediates along the way. Here we report the synthesis and crystal structures of three flexible macrocyclic intermediates and a new ring-in-ring complex, anchored together through ruthenium(II) centers, which contains open terpyridine caps in the inner Ring II. Terpyridines circumvent the conformational cis/trans limitations of bipyridines and the new ring-in-ring complex forms tetrametallic complexes with Zn(II), Pt(II) and Ru(III) metal ions. Analysis of the four macrocyclic structures provides a good foundation for the conformational flexibility in these complexes and demonstrates the robust applicability of the terpyridine design elements towards the engineered synthesis of ring-in-ring topologies.
Introduction
Thanks to a variety of chemical template methodologies,1 ring-in-ring threaded molecules (catenanes) are now routine design elements and products of chemical synthesis;2 however, directed inclusion of one macrocycle within another to form non-intertwined ring-in-ring complexes remains an interesting and challenging molecular target. In one strategy for the synthesis of such complexes, a rigid open-host ring, such as a cyclodextrin,3 curcurbituril,4 calixarene,5 pyridinium6 or metal–organic7 macrocycle houses a guest macrocycle. In another strategy, metal–ligand complexes serve as anchoring points between the two macrocycles or curved ligands.8 Phenomenal examples of ring-in-ring elements within higher order structures include: the thermodynamic assembly of a Borromean link (B-link),9 in which three rings form a topological link without concatenation, Borromean networks,10 and the assembly of curved-aromatic carbon materials in onion-like ring and shell hierarchies.11Focusing on B-links as topologically special targets of molecular synthesis,12 a stabilized single ring-in-ring element could serve as a key intermediate in a general design strategy for a kinetic, stepwise synthesis of B-links (Scheme 1).13 One embodiment of this intermediate comprises an outer macrocycle with two inward pointing (endo) terpyridine (terpy) elements and an inner macrocycle with two outward (exo) pointing terpy elements, plus two bipyridine (bipy) elements (2-bipy) (Scheme 2). Failed attempts to extend this entity into a B-link motivate structural studies to better understand these intermediates. This work details such studies and reports the synthesis of 2-terpy with two open terpyridine moieties.
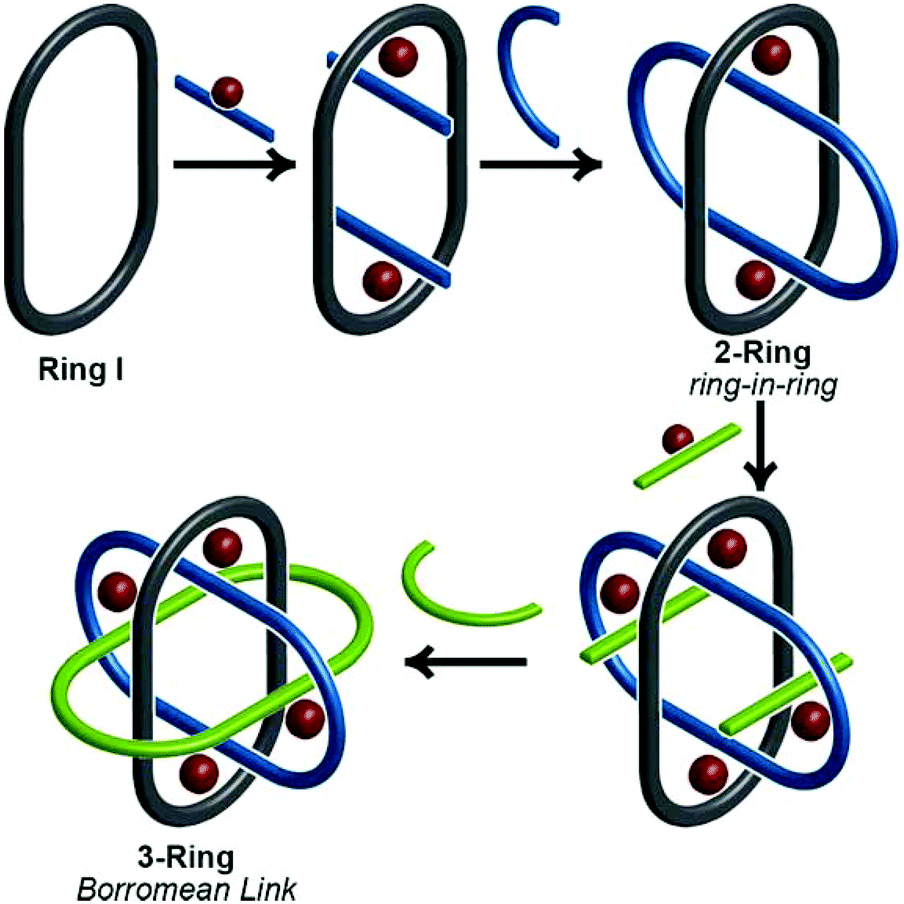 | ||
| Scheme 1 A conceptual stepwise kinetic strategy for an interlocked Borromean link that begins with a single macrocycle (Ring I) which templates formation of the second ring (Ring II) as the key ring-in-ring intermediate (2-Ring). | ||
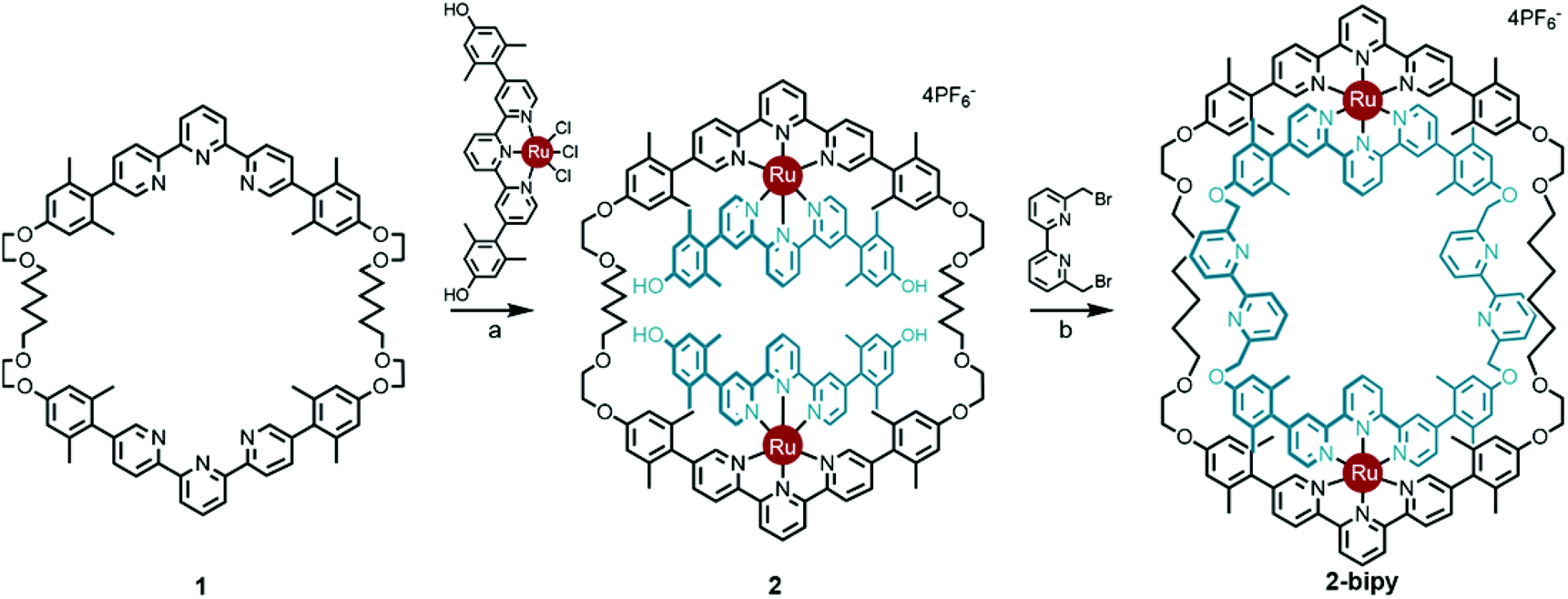 | ||
Scheme 2 Synthesis of ring-in-ring structure 2-bipyvia the doubly threaded Ring I intermediate 2. Reaction conditions: (a) 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 CH2Cl2:EtOH:ethylene glycol, reflux, 12 h, 65%; (b) Cs2CO3, acetonitrile, reflux, 72 h, 49%.13 :1:1 CH2Cl2:EtOH:ethylene glycol, reflux, 12 h, 65%; (b) Cs2CO3, acetonitrile, reflux, 72 h, 49%.13 | ||
Results
A thermodynamically controlled approach to ring-in-ring structures employs the spontaneous assembly of two macrocycles, stabilized by the strength of non-covalent bonding between them. A kinetically controlled approach13 to ring-in-ring structures differs by employing the stepwise construction of each ring and requires a virtually irreversible anchoring point (e.g. a [Ru(terpy)2]2+ complex) to hold the pieces together. One reduction to practice of this latter strategy starts by forming a macrocycle 1 with endo terpy segments (Ring I). Treatment of 1 with two equivalents of monoleptic [Ru(terpy)Cl3] reagent, generates threaded structure 2 with two heteroleptic terpy/terpy′Ru complexes in which the two terpy′ units are threaded within Ring I and lie proximal to each other (Scheme 2). In the final step, the two terpy′ units are connected to form an inner macrocycle (Ring II) and afford the representative ring-in-ring complex. Specifically, 2,2′-bipyridine units were used to link the terpy′ units and form Ring II, in the hope of iterating the process to form a B-link.13Macrocycle 1 was first presented as the key intermediate in a stepwise synthetic approach to a B-link topology. Although macrocycle 1 is a quite flexible and large ring – formally 66-membered – its immediate tetraalkyne precursor forms in high overall yield by Eglington macrocyclization of two halves (A) assisted by metal coordination.13 The assumption has been that the intermediate is a figure-eight shaped metal complex CuA2, which then springs open to release the metal after ring formation; subsequent hydrogenation yields 1. A kinetically inert cognate of the putative figure-eight intermediate (1a) forms during the reaction of 1 with one equivalent of Ru(II)Cl2(DMSO)4 (Scheme 3). Structural elucidation of 1a shows the expected overall structure and a linear crankshaft conformation of the hexamethylene unit, where the linear methylenediynemethylene unit would have been in the intermediate, thus providing little evidence of any specific strain that could be the “loaded spring”. Nonetheless, the formation of a preorganized complex that reduces the degrees of freedom and favours cyclization seems reasonable.
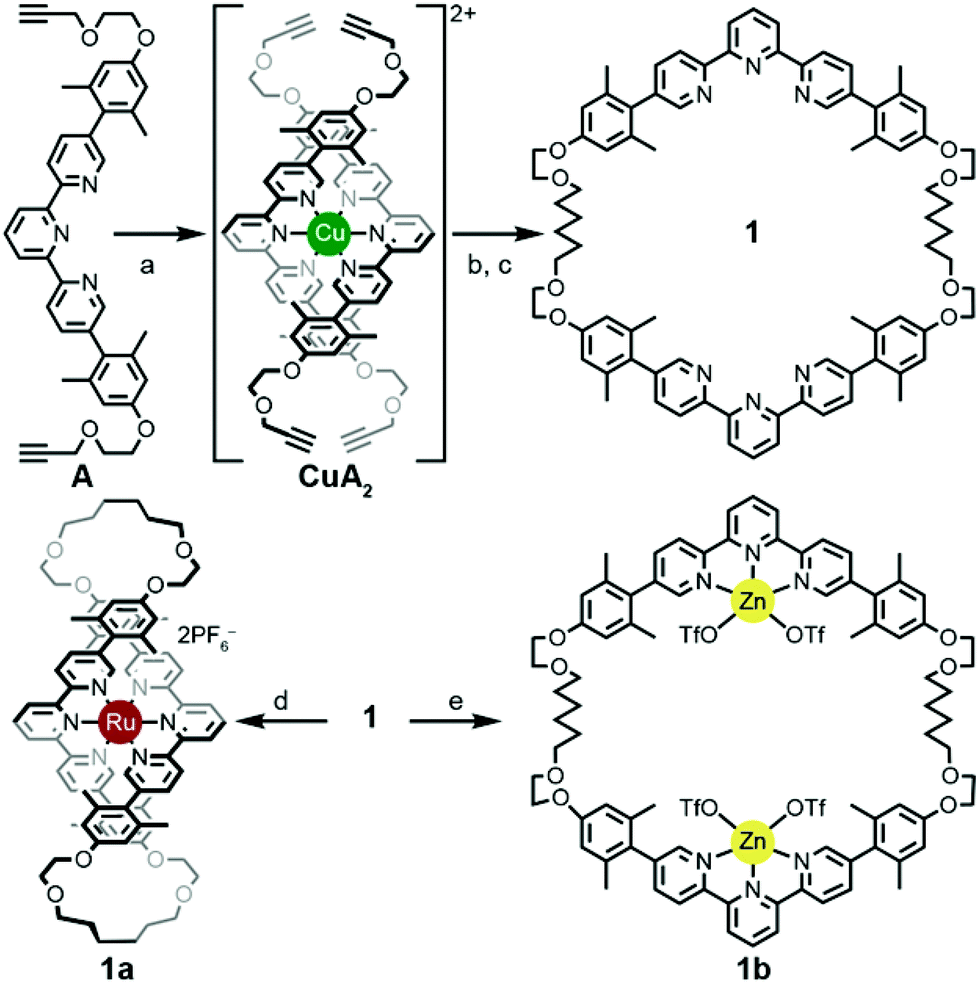 | ||
| Scheme 3 Synthesis of bis-terpyridine macrocycle 1 and metal complexes 1a and 1b. Reaction conditions: (a) Cu(OAc)2·H2O (0.5 equiv.), ethanol, RT, 1 h; (b) Cu(OAc)2·H2O (10 equiv.), ethanol, reflux, 72 h, 91%; (c) H2, 80 psi, Pd/C (10%), 1:1 ethanol:CH2Cl2, 6 h, 91%; (d) Ru(DMSO)4Cl2, 10:4:1 ethylene glycol:dichloroethane:ethanol, reflux, 16 h, 96%; (e) Zn(OTf)2, 1:1 ethanol:CH2Cl2, RT, 6 h, 97%. | ||
In contrast, treating macrocycle 1 with one equivalent of Zn(II)OTf2, a more kinetically-labile metal, gave a complex mix of spectroscopic signatures, while increasing the amount of metal to greater than a two-fold excess of the Zn(II)OTf2 reagent transformed 1 cleanly into 1b (Scheme 3). The ease of formation of complexes 1a/b and their crystal structure geometries support the notion of a conformationally flexible macrocycle 1.
The terpy elements in macrocycle 1 are 5,5′′-diaryl substituted and sometimes denoted as V-terpys in reference to their overall shape. An alternative form is the 4,4′′-diaryl substitution, which then becomes a W-terpy by analogy. The reaction of macrocycle 1 with two equivalents of an appropriate W-terpyRuCl3 complex gives the doubly threaded ring-in-ring precursor 2 (Scheme 2). The structure of 2 further exemplifies the conformational manifold accessible to the alkyl chains in these systems. From the crystal structure of 2, one also sees that the strategy of using V-shaped and W-shaped terpy derivatives to direct the ends of the segments makes ring-selective coupling more feasible. Combined with the insight from the structure 1b, one gets a feeling for the dynamic range of metrics suitable for a coupling bridge.
Previously, intermediate 2 was shown to react with 6,6′-bis-bromomethyl-2,2′-bipyridine to give the bipy ring-in-ring complex 2-bipy (Scheme 2).13 In the present context, one can see that although the metrics of the trans conformation of 6,6′-bis-bromomethyl-2,2′-bipyridine fit the structural model well, the cis-conformation is too short; therefore the idea of using the bipy bridge for further coordination complex formation was ill-fated, as was the use of this bridge in a strategic synthesis of the B-link.
The V-terpy bridged ring-in-ring structure 2-terpy removes the shape ambiguity associated with s-cis/s-trans bipy conformations. s-cis/s-trans conformations of the V-terpy leave the linking vectors essentially unchanged in their position and direction. The 5-to-5′′ distance in 5,5′′-dimethylterpy fits the structural model and was added as a new design element. The new ring-in-ring structure was thus prepared from precursor 2 and 5,5′′-bis(bromomethyl)-terpyridine under Williamson ether synthesis conditions in dimethylformamide (Scheme 4). Addition of aqueous potassium hexafluorophosphate resulted in a red precipitate, which was purified by column chromatography to provide pure 2-terpy in 56% yield. Characteristic benzylic signals corresponding to the terpyridine units appeared in the complex but highly symmetrical (D2h) 1H and 13C NMR spectra (Fig. S6 and S7, ESI†). The robust ring-in-ring complex was stable under ESI-MS conditions and the sequential loss of PF6− counter-ions was observed. Final structural confirmation was supplied by single crystal X-ray analysis using synchrotron radiation (Fig. 1).
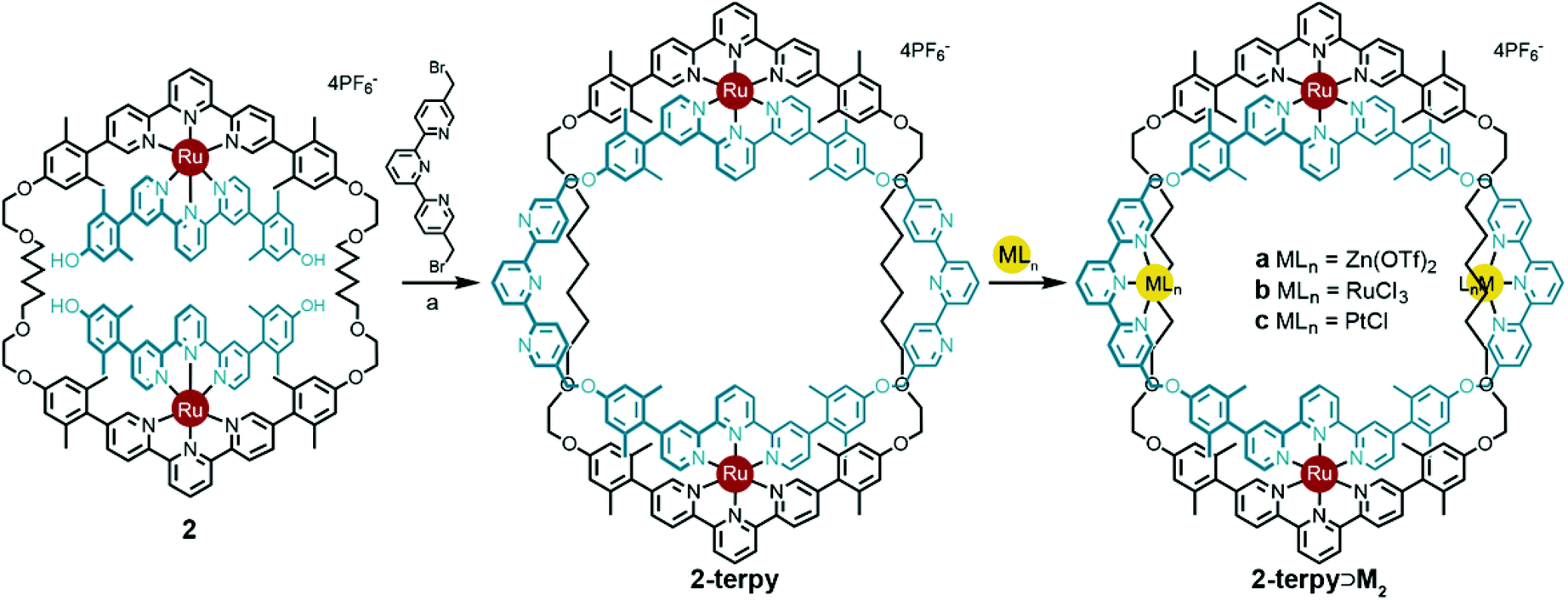 | ||
| Scheme 4 Synthesis of new ring-in-ring structure 2-terpy and the subsequent coordination of metal ions to give tetrametallic two-ring structures 2-terpy⊃Ma–c. Reaction conditions: (a) Cs2CO3, dimethylformamide, reflux, 4 h, 56%; (b) Zn(OTf)2, acetonitrile, RT, 3 h, 94%; (c) RuCl3·3H2O, 1:1 ethanol:1,2-dichloroethane, 80 °C, 16 h, 99%; (d) PtCl2(COD), AgBF4, acetonitrile, RT, 30 min, 70%. | ||
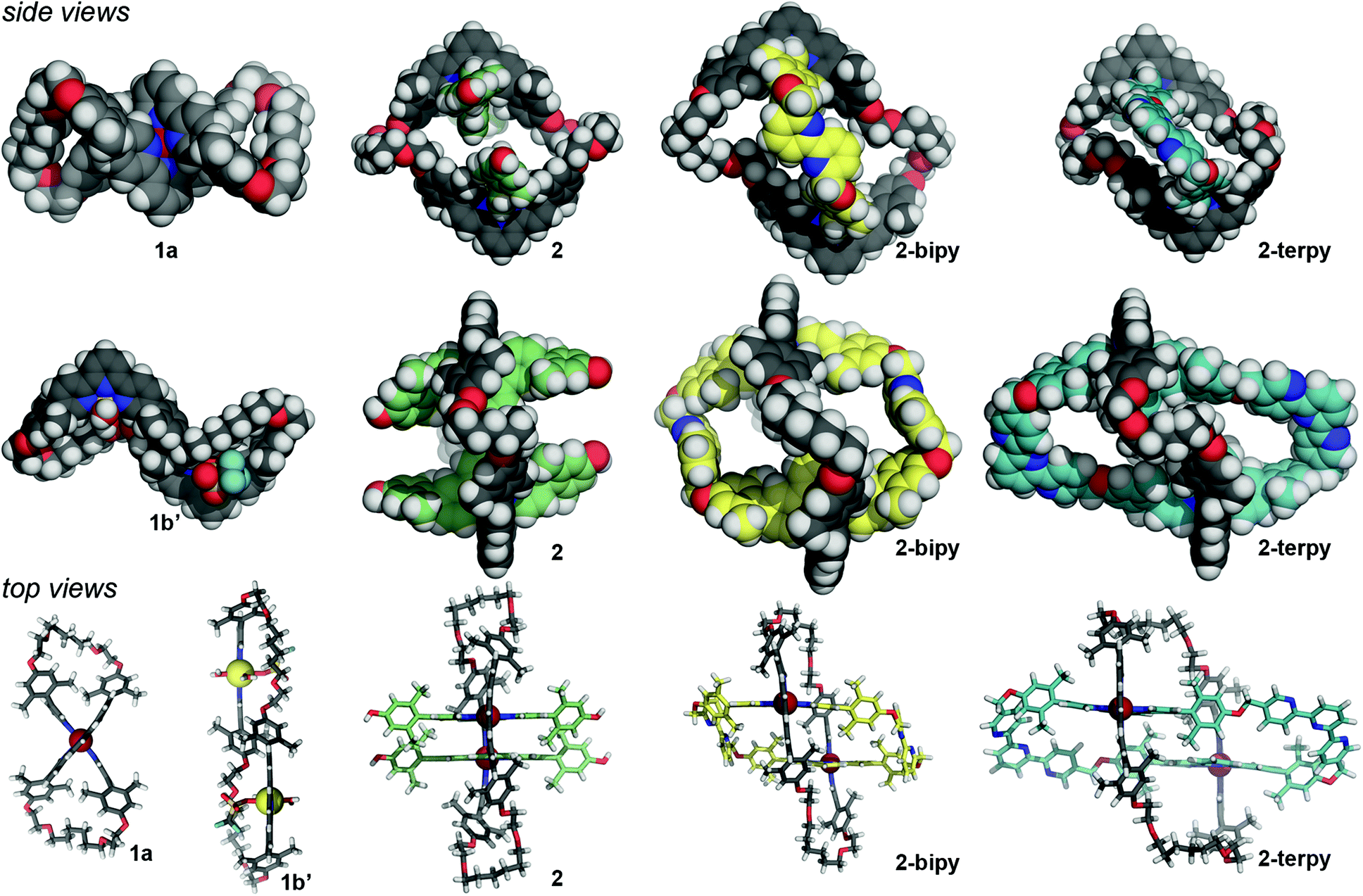 | ||
| Fig. 1 Side and top views of macrocyclic cation structures from crystal diffraction analyses emphasizing the conformational flexibility of macrocycle 1 (black) in complexes 1a Ru(II); 1b′ Zn(II); doubly threaded complex 2; and ring-in-ring structures 2-bipy and 2-terpy with Ring II highlighted in yellow and blue, respectively. Disordered atoms, solvent molecules, and anions removed for the sake of clarity. | ||
Circumventing the previous conformational issues arising from s-cis/s-trans isomerisation of 2,2′-bipyridine in 2-bipy, enabled ring-in-ring structure 2-terpy to coordinate additional metal ions. To demonstrate this, treatment of ring-in-ring complex 2-terpy with two equivalents of Zn(II), Ru(II), or Pt(II) gave high yields of the mixed tetra-metal complexes 2-terpy⊃Ma–c, respectively (Scheme 4).
Structural studies
X-ray diffraction quality crystals of 1a, 1b′, 2, and 2-terpy were grown using solvent diffusion techniques.¶14 The crystal structure of macrocyclic complex 1a confirmed the figure-eight helical structure wound upon a central Ru(II) ion (centrosymmetric space group P21/n) (Fig. 1). The kinetically inert character of the Ru(II) complex should impose configurational stability in the molecule. As such, it should be possible to resolve these complexes into the pure, time-averaged D2 symmetric enantiomers, analogous to Prelog's vesperines.15Macrocycle complex 1b crystallizes as 1b′ in space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with each macrocyclic cation resting across a crystallographic centre of inversion. Each terpyridine subunit binds a single Zn(II) cation with the remaining coordination sites occupied by one triflate anion and two water molecules that displaced the weakly bound, second triflate anion of 1b. The macrocycle adopts a collapsed conformation with the two Zn(II) cations offset; the Zn⋯Zn distance is ca. 13 Å (Fig. 1). This slipped structure is further characterized by intramolecular and anti-parallel offset aromatic–aromatic contacts between the manisyl groups of opposing V-terpy elements.16 Whereas the Ru(II) in 1a holds the opposing V-terpy planes of Ring I roughly orthogonal, the planes of the V-terpy units in 1b′ are parallel but offset (Fig. 1). Complex 1b′ is achiral by virtue of a centre of inversion in the crystal; in solution, the dynamic symmetry consistent with the NMR data is time-averaged D2h.
, with each macrocyclic cation resting across a crystallographic centre of inversion. Each terpyridine subunit binds a single Zn(II) cation with the remaining coordination sites occupied by one triflate anion and two water molecules that displaced the weakly bound, second triflate anion of 1b. The macrocycle adopts a collapsed conformation with the two Zn(II) cations offset; the Zn⋯Zn distance is ca. 13 Å (Fig. 1). This slipped structure is further characterized by intramolecular and anti-parallel offset aromatic–aromatic contacts between the manisyl groups of opposing V-terpy elements.16 Whereas the Ru(II) in 1a holds the opposing V-terpy planes of Ring I roughly orthogonal, the planes of the V-terpy units in 1b′ are parallel but offset (Fig. 1). Complex 1b′ is achiral by virtue of a centre of inversion in the crystal; in solution, the dynamic symmetry consistent with the NMR data is time-averaged D2h.
The crystal structure of threaded two-ring precursor 2 adopts space group P with the macrocycle again sitting across a crystallographic centre of inversion. The two W-terpyRu(II) elements clearly thread Ring I, forming a double pseudo-rotaxane (Fig. 1). Outer Ring I adopts an expanded conformation with ca. 25 Å between the apical 4′-terpyridine H's. The planes of the two terpyridine units in Ring I are parallel but slightly offset avoiding steric congestion of the two 4′-hydrogen atoms. As a result, the two Ru(II) centres are ca. 13 Å apart and, as expected, the phenol oxygen atoms of the threaded terpyridines are preorganized suggesting a favoured macrocyclization O⋯O distance of around 8 Å.
The crystal structure of the bipyridine-containing two-ring complex 2-bipy has been reported previously, but is discussed here for comparison (space group P).13 Macrocyclization with 6,6′-substituted 2,2′-bipyridines resulted in an expanded Ring I as the bridging bipyridines adopt the extended trans-conformation (Fig. 1). Rings I and II adopt a chair-like conformation where the planes defined by the two V-terpy (in Ring I) and two W-terpy (in Ring II) elements are parallel relative to each other, but offset roughly by 5 Å. As a result, the expanse of Ring I is on the order of 26 Å and the two Ru(II) centres are 16 Å apart.
The crystal structure of 2-terpy adopts space group Pnnn and confirmed the new ring-in-ring topology. The limited quality of the diffraction data precludes a detailed analysis of the geometry,17 but an overview of the cation topology, which is not unlike that of 2-bipy, is depicted in Fig. 1. The new tetra-terpyridine Ring II (blue) assumes a rhomboidal shape tilted relative to the z-axis of Ring I. The two coordinated exotopic terpyridine subunits are offset, but the octahedral geometries of the Ru(II) centres ensure a parallel orientation relative to the long z-axis of Ring I. Concomitantly, Ring I distorts into a pronounced S or Z-shape where coordinated V-terpy subunits are parallel but displaced. The newly installed V-terpy elements of Ring II span the distance between the W-terpy elements well, consistent with the structural design model.
Overall, the expanse of the new ring-in-ring structure 2-terpy along the arbitrary z-axis of Ring I is on the order of 25 Å, and is comparable to the two-ring structure 2-bipy. V-terpy-bridged Ring II in 2-terpy is a 64-atom macrocycle, 10 atoms larger than Ring II in 2-bipy (Fig. S1, ESI†), and although distorted, the semi-rigid structure of Ring II appears to generate two large pockets suggestive of binding sites for the coordination of subsequent metal ions; unlike in the bipy elements of 2-bipy the flanking pyridines of the V-terpy elements in 2-terpy can simply rotate into the requisite s-cis-binding conformation with minimal structural rearrangement (Scheme 4).
Discussion and conclusions
The general ring-in-ring strategy for a kinetically directed synthesis of the B-link is enhanced by the ability to isolate and structurally characterize synthetic intermediates along the way. The analysis of four macrocyclic structures has provided a good foundation for the dimensional scope in these complexes and the V-terpy/W-terpy design elements are now robust in terms of their chemical synthesis and their applicability as design elements toward an engineered synthesis of ring-in-ring topologies. Remarkably, even with the apparent space available within the open V-terpy loops of 2-terpy and the ability to bind additional simple ligated metals, additional bipy or terpy metal complexes have not been successful threading partners for advancing to the final phase of the B-link synthesis. New strategies are needed, among them building the loops with the threading elements already in place.18 This strategy may be facilitated by the design of newer laterally extended terpy analogues.19Overall, the future for directed synthesis of higher-order topological molecular structures is positive. Soon chemists will have the ability to design in local active sites in these topological molecules. The prospect of catalysis with biological activity and specificity in this class of supramolecules seems well within reach of our design capabilities.
Acknowledgements
This work was supported by the Swiss National Science Foundation. We thank Dr Meitian Wang of the Swiss Light Source (Paul Scherrer Institute) for access to time on a PX beamline.Notes and references
- (a) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260 CrossRef CAS PubMed; (b) Templated organic synthesis, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, New York, 2000 Search PubMed; (c) Modern supramolecular chemistry: strategies for macrocycle synthesis, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110 CrossRef PubMed; (b) C. Schouwey, J. J. Holstein, R. Scopelliti, K. O. Zhurov, K. O. Nagornov, Y. O. Tsybin, O. S. Smart, G. Bricogne and K. Severin, Angew. Chem., Int. Ed., 2014, 53, 11261 CrossRef CAS PubMed; (c) D. A. Leigh, R. G. Pritchard and A. J. Stephens, Nat. Chem., 2014, 6, 978 CrossRef CAS PubMed; (d) S. P. Black, A. R. Stefankiewicz, M. M. J. Smulders, D. Sattler, C. A. Schalley, J. R. Nitschke and J. K. M. Sanders, Angew. Chem., Int. Ed., 2013, 52, 5749 CrossRef CAS PubMed; (e) S. Freye, J. Hey, A. Torras-Galán, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191 CrossRef CAS PubMed; (f) Molecular catenanes, rotaxanes, and knots: a journey through the world of molecular topology, ed. J.-P. Sauvage and C. Dietrich-Buchecker, Wiley-VCH, Weinheim, New York, 1999 Search PubMed.
- (a) S.-Y. Kim, I.-S. Jung, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 2119 CrossRef CAS; (b) S. Kamitori, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1987, 109, 2409 CrossRef CAS; (c) S. Kamitori, K. Hirotsu and T. Higuchi, J. Chem. Soc., Chem. Commun., 1986, 690 RSC; (d) F. Vögtle and W. M. Müller, Angew. Chem., Int. Ed. Engl., 1979, 18, 623 CrossRef.
- (a) Y. Liu, Tetrahedron Lett., 2007, 48, 3871 CrossRef CAS; (b) T. Iwanaga, R. Nakamoto, M. Yasutake, H. Takemura, K. Sako and T. Shinmyozu, Angew. Chem., Int. Ed., 2006, 45, 3643 CrossRef CAS PubMed; (c) A. I. Day, R. J. Blanch, A. P. Arnold, S. Lorenzo, G. R. Lewis and I. Dance, Angew. Chem., Int. Ed., 2002, 41, 275 CrossRef CAS.
- S. J. Dalgarno, J. Fisher and C. L. Raston, Chem. – Eur. J., 2006, 12, 2772 CrossRef CAS PubMed.
- (a) R. S. Forgan, C. Wang, D. C. Friedman, J. M. Spruell, C. L. Stern, A. A. Sarjeant, D. Cao and J. F. Stoddart, Chem. – Eur. J., 2012, 18, 202 CrossRef CAS PubMed; (b) R. S. Forgan, J. M. Spruell, J.-C. Olsen, C. L. Stern and J. F. Stoddart, J. Mex. Chem. Soc., 2009, 53, 134 CAS; (c) S.-H. Chiu, A. R. Pease, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2002, 41, 270 CrossRef CAS.
- (a) V. Vajpayee, Y. H. Song, T. R. Cook, H. Kim, Y. Lee, P. J. Stang and K.-W. Chi, J. Am. Chem. Soc., 2011, 133, 19646 CrossRef CAS PubMed; (b) R. S. Forgan, D. C. Friedman, C. L. Stern, C. J. Bruns and J. F. Stoddart, Chem. Commun., 2010, 46, 5861 RSC.
- (a) J.-H. Fu, Y.-H. Lee, Y.-J. He and Y.-T. Chan, Angew. Chem., Int. Ed., 2015, 54, 6231 CrossRef CAS PubMed; (b) M. Schmittel, A. Ganz and D. Fenske, Org. Lett., 2002, 4, 2289 CrossRef CAS PubMed.
- (a) S.-L. Huang, Y.-J. Lin, Z.-H. Li and G.-X. Jin, Angew. Chem., Int. Ed., 2014, 53, 11218 CrossRef CAS PubMed; (b) S.-L. Huang, Y.-J. Lin, T. S. A. Hor and G.-X. Jin, J. Am. Chem. Soc., 2013, 135, 8125 CrossRef CAS PubMed; (c) K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308 CrossRef CAS PubMed.
- (a) F. L. Thorp-Greenwood, A. N. Kulak and M. J. Hardie, Nat. Chem., 2015, 7, 526 CrossRef CAS PubMed; (b) M. Pan and C.-Y. Su, CrystEngComm, 2014, 16, 7847 RSC; (c) L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2003, 5, 269 RSC.
- (a) T. Kawase, Y. Nishiyama, T. Nakamura, T. Ebi, K. Matsumoto, H. Kurata and M. Oda, Angew. Chem., Int. Ed., 2007, 46, 1086 CrossRef CAS PubMed; (b) T. Kawase, K. Tanaka, N. Shiono, Y. Seirai and M. Oda, Angew. Chem., Int. Ed., 2004, 43, 1722 CrossRef CAS PubMed.
- (a) R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434 CrossRef CAS PubMed; (b) S. J. Cantrill, K. S. Chichak, A. J. Peters and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 1 CrossRef CAS PubMed; (c) J. S. Siegel, Science, 2004, 304, 1256 CrossRef CAS PubMed.
- J. C. Loren, M. Yoshizawa, R. F. Haldimann, A. Linden and J. S. Siegel, Angew. Chem., Int. Ed., 2003, 42, 5702 CrossRef CAS PubMed.
- For a detailed analysis of crystal packing see: J. K. Klosterman, PhD Thesis, Universität Zürich, 2007.
- (a) G. Haas and V. Prelog, Helv. Chim. Acta, 1969, 52, 1202 CrossRef CAS; (b) G. Haas, P. B. Hulbert, W. Klyne, V. Prelog and G. Snatzke, Helv. Chim. Acta, 1971, 54, 491 CrossRef CAS; (c) O. S. Mills, et al. CrossRef CAS, unpublished results cited in: V. Prelog and D. Bedeković, Helv. Chim. Acta, 1979, 62, 2285 CrossRef CAS.
- J. K. Klosterman, A. Linden, D. K. Frantz and J. S. Siegel, Dalton Trans., 2010, 39, 1519 RSC.
- The crystals of ring-in-ring complex 2-terpy were very weakly diffracting. The data were recorded on a protein beamline at the Swiss Light Source where only a single phi scan was possible. The limited data quality and completeness led to a model that is far from the quality usually obtained for routine small-molecule crystal structure determinations; however, for 2-terpy at ca. 4 kD we believe these results provide a reliable overall impression of the morphology and topology of the cation. Full details are in the deposited CIF.
- J. Veliks, H. M. Seifert, D. K. Frantz, J. K. Klosterman, J.-C. Tseng, A. Linden and J. S. Siegel, Org. Chem. Front., 2016 10.1039/c6qo00025h.
- (a) J. Veliks, J.-C. Tseng, K. I. Arias, F. Weisshar, A. Linden and J. S. Siegel, Chem. Sci., 2014, 5, 4317 RSC; (b) J. Veliks, O. Blacque and J. S. Siegel, Inorg. Chem., 2014, 53, 12122 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details and characterization. CCDC 1437407–1437409 and 1432774. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00024j |
| ‡ Current address: Department of Chemistry and Center for Photochemical Sciences, Bowling Green State University, Bowling Green, OH 43403, USA. |
| § Current address: Organic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife, KY16 9ST, UK. |
| ¶ Crystal data for 1a: C82H90F12N6O8P2Ru·5CH3CN, Mr = 1883.87, red prism: 0.07 × 0.20 × 0.32 mm, monoclinic, P21/n, a = 20.3614(3), b = 15.5902(3), c = 30.5936(5) Å, β = 94.4504(9)°, V = 9682.3(3) Å3, Z = 4, ρ = 1.292 g cm−3, Mo Kα radiation, F(000) = 3920, μ = 0.276 mm−1, T = 160 K, 2θmax = 50°, 141740 measured reflections, 17083 unique reflections used, 9660 with Io > 2σ(Io), Rint = 0.120, 1455 parameters, 4603 restraints, GoF = 1.032, R(F) = 0.1001 [Io > 2σ(Io) reflections], wR(F2) = 0.3174 (all reflections), 2.05 > Δρ > −0.70 e Å−3. Crystal data for 1b′: C86H98F12N6O24S4Zn2·4CH3CN·2C4H10O, Mr = 2399.14, colourless plate: 0.13 × 0.30 × 0.30 mm, triclinic, P, a = 11.0301(2), b = 12.3939(4), c = 21.8715(7) Å, α = 76.878(1), β = 78.788(2), γ = 87.305(2)°, V = 2856.3(1) Å3, Z = 1, ρ = 1.395 g cm−3, Mo Kα radiation, F(000) = 1252, μ = 0.587 mm−1, T = 160 K, 2θmax = 50°, 43549 measured reflections, 10053 unique reflections used, 7931 with Io > 2σ(Io), Rint = 0.060, 783 parameters, 153 restraints, GoF = 1.035, R(F) = 0.0495 [Io > 2σ(Io) reflections], wR(F2) = 0.1256 (all reflections), 0.70 > Δρ >−0.59 e Å−3. Crystal data for 2: C144H144F24N12O12P4Ru2·11CH2Cl2, Mr = 3950.91, red plate: 0.05 × 0.32 × 0.35 mm, triclinic, P, a = 15.0040(5), b = 15.1525(4), c = 21.5611(7) Å, α = 98.402(2), β = 98.012(1), γ = 108.321(2)°, V = 4513.0(2) Å3, Z = 1, ρ = 1.454 g cm−3, Mo Kα radiation, F(000) = 2014, μ = 0.611 mm−1, T = 160 K, 2θmax = 50°, 77052 measured reflections, 15898 unique reflections used, 11033 with Io > 2σ(Io), Rint = 0.103, 930 parameters, 271 restraints, GoF = 1.033, R(F) = 0.0627 [Io > 2σ(Io) reflections], wR(F2) = 0.1674 (all reflections), 0.59 > Δρ > −0.45 e Å−3. Crystal data for 2-terpy: C178H166F24N18O12P4Ru2·12C6H6, Mr = 4468.59, red plate, orthorhombic, Pnnn, a = 22.824(1), 22.583(1), 47.813(1) Å, V = 24645(2) Å3, Z = 4, ρ = 1.204 g cm−3, λ = 0.8000 Å, F(000) = 9296, μ = 0.309 mm−1, T = 100 K, 2θmax = 55°, 93055 measured reflections, 13465 unique reflections used, 9226 with Io > 2σ(Io), Rint = 0.048, 1087 parameters, 852 restraints, GoF = 2.178, R(F) = 0.1536 [Io > 2σ(Io) reflections], wR(F2) = 0.4940 (all reflections), 1.19 > Δρ > −0.65 e Å−3. |
| This journal is © the Partner Organisations 2016 |