
Cp*Co(III)-catalyzed vinylic C–H bond activation under mild conditions: expedient pyrrole synthesis via (3 + 2) annulation of enamides and alkynes†
Dhanaji M.
Lade
ab and
Amit B.
Pawar
*a
aDivision of Natural Product Chemistry, CSIR-Indian Institute of Chemical Technology, Hyderabad 500007, India. E-mail: amitorgchem@gmail.com
bAcademy of Scientific and Innovative Research (AcSIR), New Delhi, India
First published on 11th May 2016
Abstract
Cobalt(III)-catalyzed (3 + 2) oxidative annulation of enamides and alkynes for the synthesis of pyrroles has been developed under exceedingly mild conditions. The reaction works well with various internal alkynes with broad scope and functional group tolerance. In most cases, the reaction proceeds at room temperature leading to N-acetyl pyrroles in excellent yields. Synthetically useful N-H pyrroles can also be obtained at elevated temperature.
Introduction
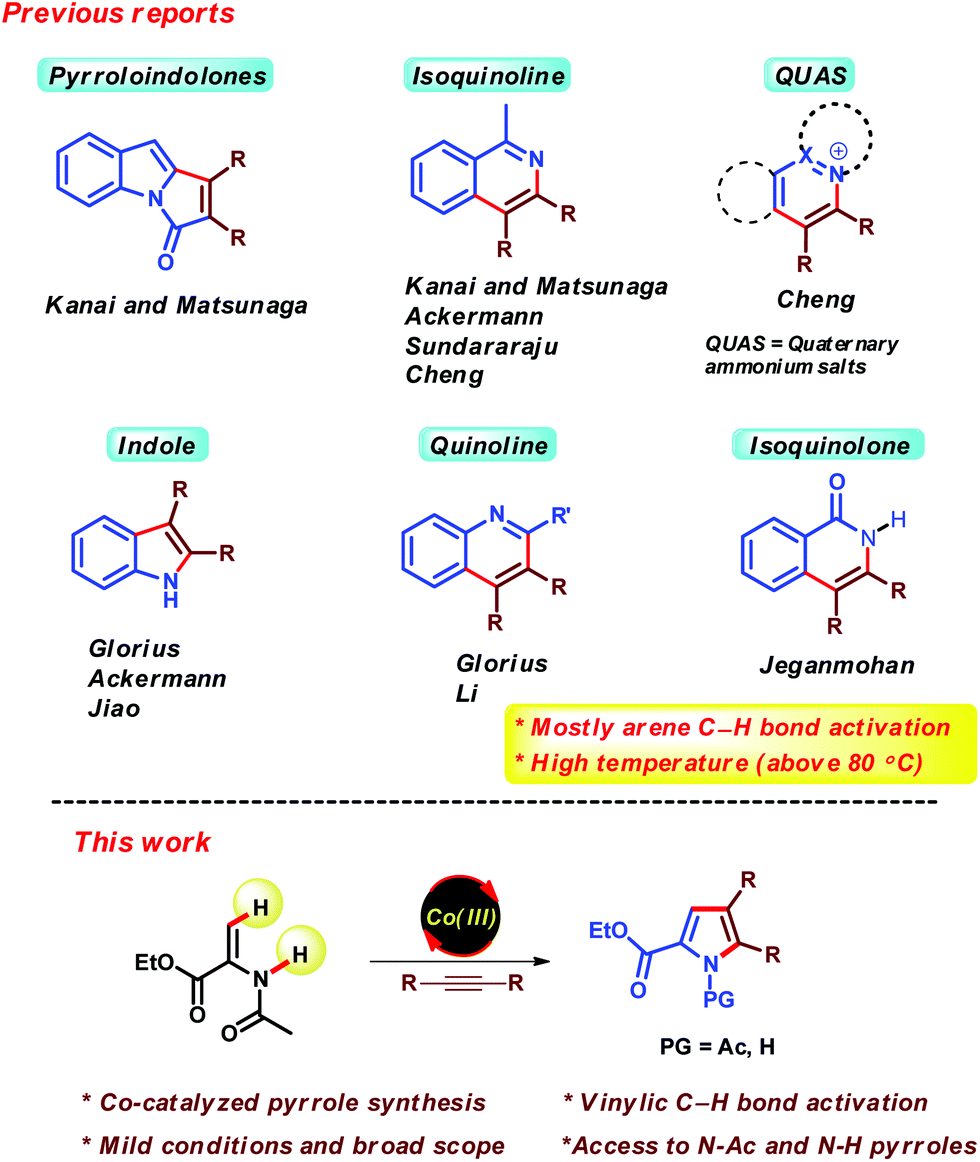
Recent years have witnessed tremendous developments in the area of Cp*Co(III) catalysis for C–H functionalization.1,2 After the seminal work of the Kanai/Matsunaga group,3 various other groups including those of Ackermann,4 Glorius,5 Chang,6 Ellman7 and others8 have also contributed to further advancing this field. The major advantages of Cp*Co(III) catalysis are (i) the use of earth abundant and less toxic first row transition metal,9 (ii) similar catalytic activity like precious transition metals such as Ru, Rh and Ir, and (iii) easily synthesized and air stable Co(III) complexes. Due to these advantages; Cp*Co(III) catalysis has became very popular within a short period of time.On the other hand, nitrogen containing heterocycles are privileged structures in organic chemistry.10 Most of them are found in wide range of natural products, pharmaceuticals and agrochemicals. Recently, Cp*Co(III) catalysis has been extensively applied in the preparation of various important heterocyclic compounds such as pyrroloindolones,3d isoquinolines,11 quaternary ammonium salts of heterocyclic nitrogen compounds,12 indoles,13 quinolines13b,14 and isoquinolones,15 which involves the annulation of alkynes with appropriate aromatic nitrogen compounds (Scheme 1). However, most of these reports are based on arene C–H bond functionalization and require higher temperatures for generation of the desired products (above 80 °C). Moreover, Cp*Co(III)-catalyzed pyrrole synthesis via the (3 + 2) annulation of enamides and alkynes via olefinic C–H bond functionalization has not been studied.16 There are several examples of Co(III)-catalyzed olefinic C–H bond functionalization. However, to the best of our knowledge, there is only one report of Cp*Co(III)-catalyzed vinylic C–H bond activation at room temperature.8b
 | ||
| Scheme 1 Cp*Co(III)-catalyzed synthesis of various nitrogen containing heterocycles. | ||
Pyrrole represents one of the most ubiquitous structural motifs in organic chemistry, which is often encountered in various bioactive natural products and pharmaceuticals.17 So far, transition metals such as Rh,18a–c Ru18d,e and Pd18f,g has been employed for the synthesis of pyrroles via the (3 + 2) annulation of enamides and alkynes. The major drawbacks of these methods are the high cost of the metal catalysts and in case of Ru and Pd, the reaction being carried out at higher temperatures. Therefore, it is highly desirable to develop a much milder protocol for these scaffolds using an inexpensive catalyst. In continuation of our interest in Cp*Co(III) catalysis,19 herein we report a Co(III)-catalyzed pyrrole synthesis to afford N-Ac and N-H pyrroles. The developed reaction conditions are extremely mild, furnishing a number of N-Ac pyrroles even at room temperature.
Results and discussion
We started our studies with enamide 1a as our starting substrate and diphenylacetylene 2a as a coupling partner (Table 1). When enamide 1a was treated with diphenylacetylene 2a in the presence of Cp*Co(CO)I2 (5 mol%) and Cu(OAc)2·H2O (50 mol%) in TFE at room temperature for 14 h, it did not furnish any product (Table 1, entry 1).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive | Oxidant | Solvent | Temp (°C) | Yieldb (%) | |
| 3aa | 4aa | |||||
| a Reaction conditions: 1a (0.10 mmol), 2a (1.2 equiv.), Cp*Co(CO)I2 (5 mol%), additive (10 mol%), oxidant (1.0 equiv.) in solvent (0.6 mL) for 14 h. b Yields are based 1H NMR analysis of the crude reaction mixture (internal standard: 1,1,2,2 tetrachloroethane). c Cu(OAc)2·H2O (50 mol%). n.d. = Not detected. TFE = 2,2,2-trifluoroethanol. | ||||||
| 1c | — | Cu(OAc)2·H2O | TFE | rt | n.d | n.d |
| 2c | AgSbF6 | Cu(OAc)2·H2O | TFE | rt | 67 | n.d. |
| 3 | AgSbF 6 | Cu(OAc) 2 ·H 2 O | TFE | rt | 95 | 4 |
| 4 | AgOAc | Cu(OAc)2·H2O | TFE | rt | 36 | n.d. |
| 5 | Ag2CO3 | Cu(OAc)2·H2O | TFE | rt | 38 | n.d. |
| 6 | AgSbF6 | AgOAc | TFE | rt | 8 | n.d. |
| 7 | AgSbF6 | Ag2CO3 | TFE | rt | 12 | n.d. |
| 8 | AgSbF6 | Cu(OAc)2·H2O | MeOH | rt | n.d. | n.d. |
| 9 | AgSbF6 | Cu(OAc)2·H2O | t AmOH | rt | 33 | n.d. |
| 10 | AgSbF6 | Cu(OAc)2·H2O | 1,2-DCE | rt | 80 | n.d. |
| 11 | AgSbF6 | Cu(OAc)2·H2O | TFE | 80 | 36 | 54 |
| 12 | AgSbF6 | Cu(OAc)2·H2O | TFE | 100 | 16 | 78 |
| 13 | AgSbF 6 | Cu(OAc) 2 ·H 2 O | TFE | 120 | n.d | 92 |
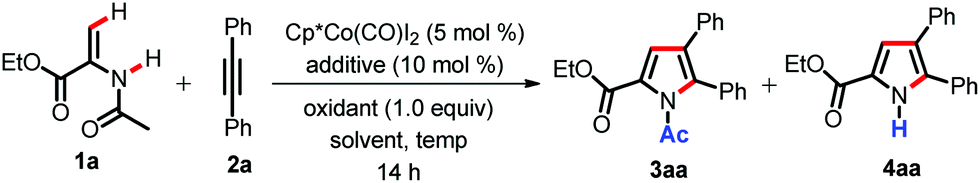
We were pleased to see that pyrrole 3aa formed in 67% yield after the introduction of 10 mol% AgSbF6 (entry 2). This indicates the role of cationic cobalt species in the reaction.3c When the amount of Cu(OAc)2·H2O was increased to 1.0 equiv., pyrrole 3aa was formed in almost quantitative yield along with traces of 4aa (entry 3). The replacement of AgSbF6 with other silver salts such as AgOAc and Ag2CO3 resulted in poor yields (entries 4 and 5). Instead of Cu(OAc)2·H2O, silver based oxidants were examined (AgOAc and Ag2CO3). However, they showed diminished yields of the product 3aa (entries 6 and 7). The use of other protic solvents such as MeOH or tAmOH gave poor yields of the pyrrole (entries 8 and 9). However, 1,2-DCE also furnished the pyrrole in good yield, but less compared to TFE (entry 10). Since we have observed the formation of a trace amount of 4aa at room temperature, we envisioned that an increase in reaction temperature might lead to an increase in the yield of N-H pyrrole 4aa. Indeed, we were excited to see that the amount of 4aa was gradually increased as the reaction temperature was increased (entries 11 and 12). We could observe the formation of 4aa in 92% yield at 120 °C (entry 13). Thus, by simple tuning of the reaction temperature we could be able to obtain both N-Ac and N-H pyrroles under identical catalytic conditions.
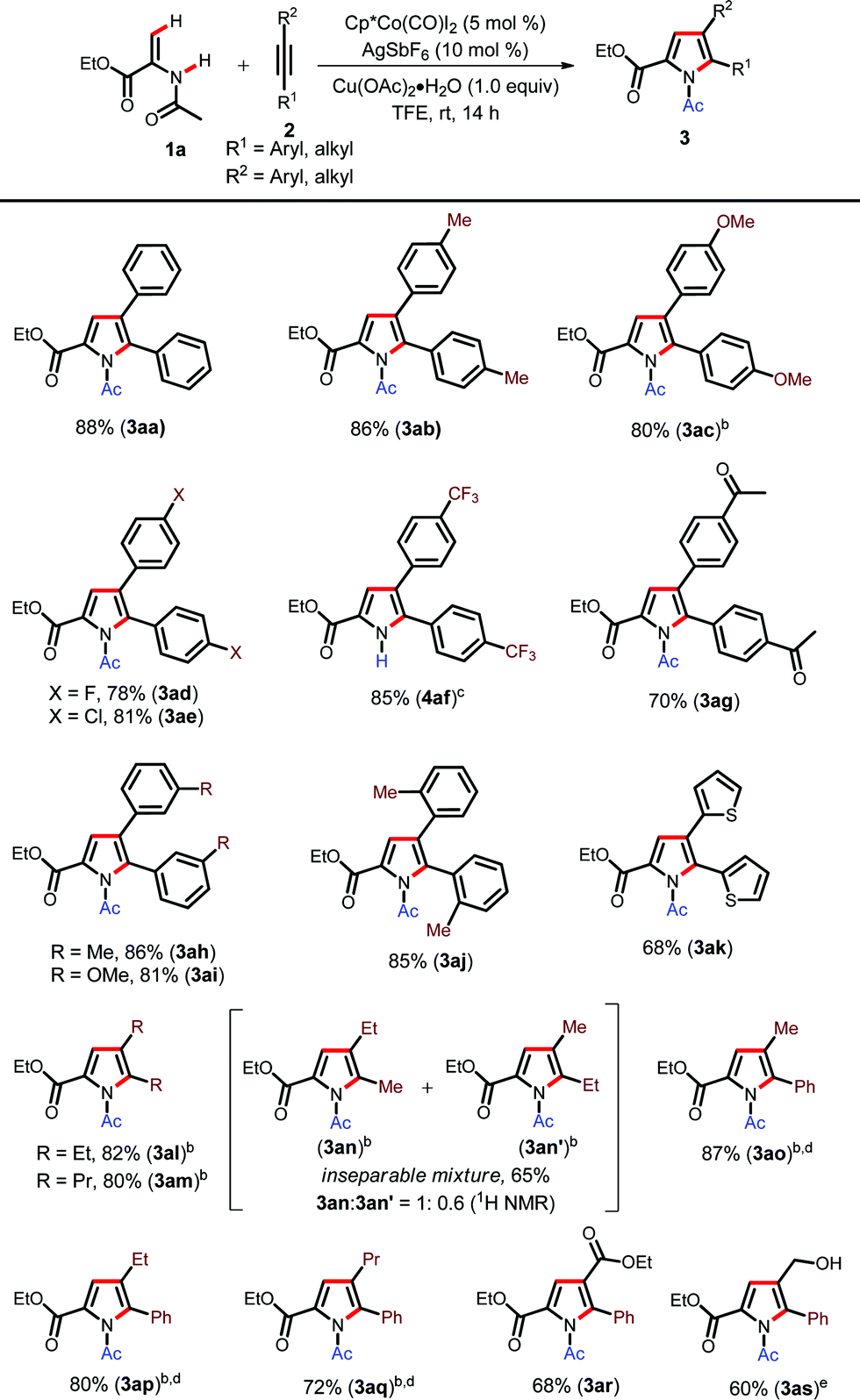
With the optimized conditions in hand, we initially investigated the Cp*Co(III)-catalyzed synthesis of various N-acetyl pyrroles using different internal alkynes (Table 2).20 The alkynes with electron donating groups such as Me and OMe furnished the corresponding pyrroles in excellent yields (3ab–3ac). Halogen containing alkynes were also found to be compatible, furnishing annulated pyrroles in high yields (3ad–3ae). Electron withdrawing groups such as CF3 and acetyl did not inhibit the reaction and produced pyrroles in good yields (4af and 3ag). Interestingly, in the case of the CF3 group we observed N-deacetylation during column chromatography (4af). The meta-substituted alkynes also underwent reaction to furnish the corresponding products in high yields (3ah–3ai). The ortho-methyl group also did not deteriorate product formation (3aj). Heteroaryl alkynes such as di(2-thiophenyl)ethylene (2k) underwent cyclization in good yields (3ak). Dialkyl substituted alkynes such as 3-hexyne and 4-octyne also furnished the products in excellent yields at slightly elevated temperatures (3al–3am). The unsymmetrical dialkyl alkyne, 2-pentyne (2l), produced an inseparable mixture of regioisomers (3an–3an′) with a 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.6 ratio in 65% yield wherein the product that has a methyl group adjacent to the pyrrole nitrogen is the major product.21 Then, we turned our attention towards unsymmetrical aryl–alkyl alkynes such as 1-phenyl-1-propyne (2o), 1-phenyl-1-butyne (2p) and 1-phenyl-1-pentyne (2q) for regioselective annulation. These alkynes smoothly underwent annulation at 50 °C with good regioselectivity (3ao–3aq). Ethyl phenylpropiolate (2r) also furnished the corresponding pyrrole in moderate yield (3ar). The alkyne with a free hydroxyl group was also tolerated under the present reaction conditions (3as); thus showcasing the functional group tolerance of the present protocol. However, our attempt to employ terminal alkynes such as phenylacteylene as a coupling partner was unsuccessful.
:0.6 ratio in 65% yield wherein the product that has a methyl group adjacent to the pyrrole nitrogen is the major product.21 Then, we turned our attention towards unsymmetrical aryl–alkyl alkynes such as 1-phenyl-1-propyne (2o), 1-phenyl-1-butyne (2p) and 1-phenyl-1-pentyne (2q) for regioselective annulation. These alkynes smoothly underwent annulation at 50 °C with good regioselectivity (3ao–3aq). Ethyl phenylpropiolate (2r) also furnished the corresponding pyrrole in moderate yield (3ar). The alkyne with a free hydroxyl group was also tolerated under the present reaction conditions (3as); thus showcasing the functional group tolerance of the present protocol. However, our attempt to employ terminal alkynes such as phenylacteylene as a coupling partner was unsuccessful.
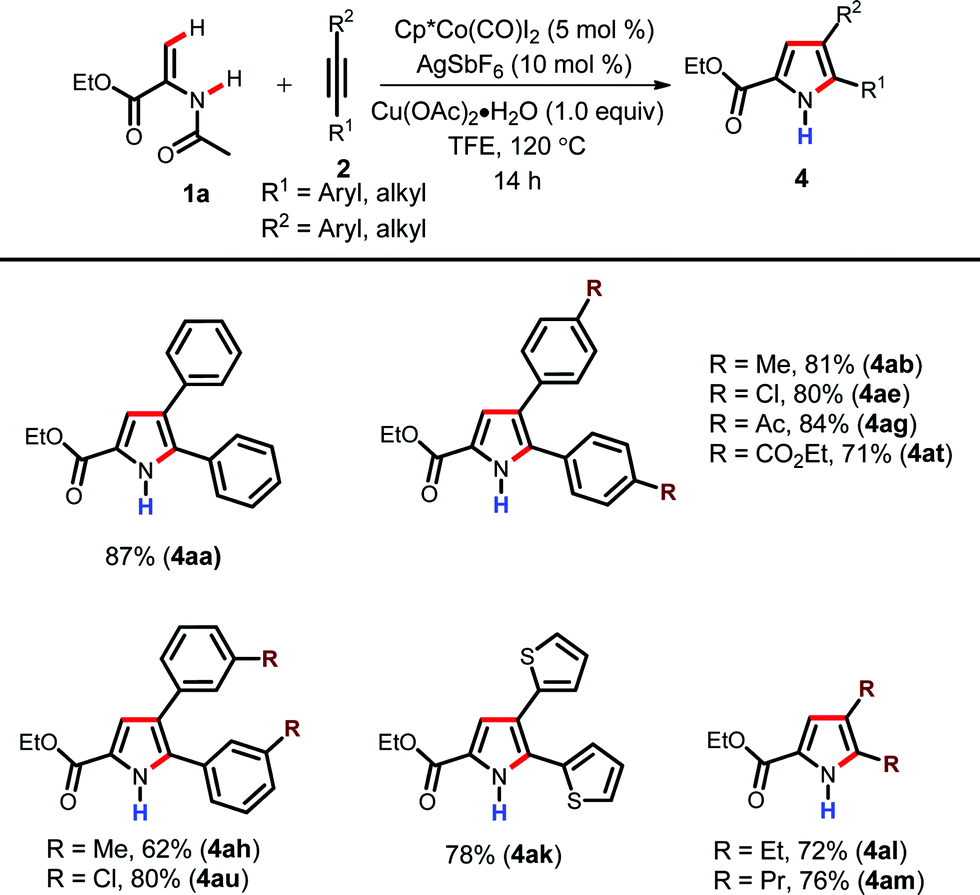
After successfully synthesizing diverse N-acetyl pyrroles, we were interested in expanding the scope of the cobalt catalytic system for the synthesis of synthetically useful N-H pyrroles (Table 3). As described in the optimization table, N-H pyrroles can be obtained by simply heating the reaction mixture at a higher temperature (Table 1, entry 13). Initially, the functional group tolerance of the presented reaction was tested by employing alkynes with various functional groups such as Me, Cl, Ac and ester. Gratifyingly, they gave the corresponding N-H pyrroles in high yields (4ab, 4ae, 4ag and 4at). The meta-substituted alkynes were also coupled efficiently (4ah & 4au). The alkyne with heteroaryl substituents was also well tolerated under the present reaction conditions furnishing the corresponding N-H pyrrole in good yield (4ak). This protocol was further applied to dialkyl alkynes without any difficulties (4al–4am). Li et al. in their Ru-catalyzed pyrrole synthesis have also reported the formation of N-H pyrrole.18d However, they had to employ a mixed solvent system in order to generate these products. However, the present reaction works well in TFE as a single solvent; which makes this procedure operationally friendly.
| a Reaction conditions: 1a (0.20 mmol), 2 (1.2 equiv.), Cp*Co(CO)I2 (5 mol%), AgSbF6 (10 mol%), and Cu(OAc)2·H2O (1.0 equiv.) in TFE (1.2 mL) at 120 °C for 14 h; isolated yields are given. |
|---|
|
In order to check the practical applicability of the present protocol, a gram scale synthesis of pyrrole was carried out at room temperature (Scheme 2). We were pleased to see that when 1.0 g of enamide 1a was treated with diphenylacetylene 2a under Cp*Co(III) catalysis, it delivered the corresponding pyrrole 3aa in 72% yield.
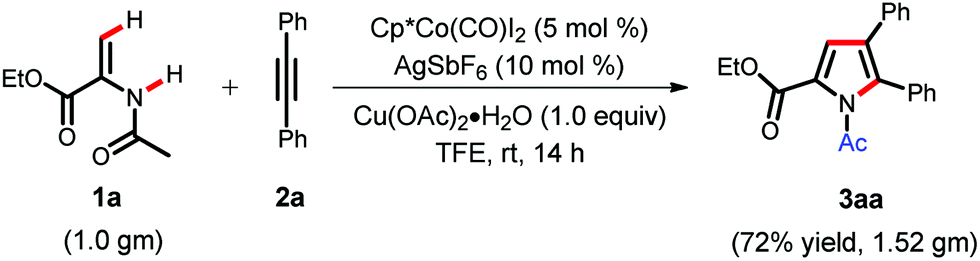 | ||
| Scheme 2 Large scale pyrrole synthesis. | ||
In order to gain insight into the mechanism, some control experiments were carried out (Scheme 3).21 Initially, when enamide 1a was subjected to standard reaction conditions in the absence of Cu(OAc)2·H2O for 14 h at room temperature, it resulted in the formation of 3aa in <5% NMR yield. Afterwards, the addition of 1.0 equiv. of Cu(OAc)2·H2O to the reaction mixture and further stirring of the reaction mixture at room temperature for another 14 h led to the formation of 3aa in 54% yield (Scheme 3). This result clearly indicate that Cu(II) does not play any role in product formation and it acts as a oxidant.18b,d However, at present we cannot rule out the possibility of carboxylate assistance by Cu(OAc)2. An intermolecular competitive experiment with differently functionalized alkynes (2b and 2e) revealed that the more electron rich alkyne 2b reacted solely with respect to the alkyne 2e with an electron withdrawing group.
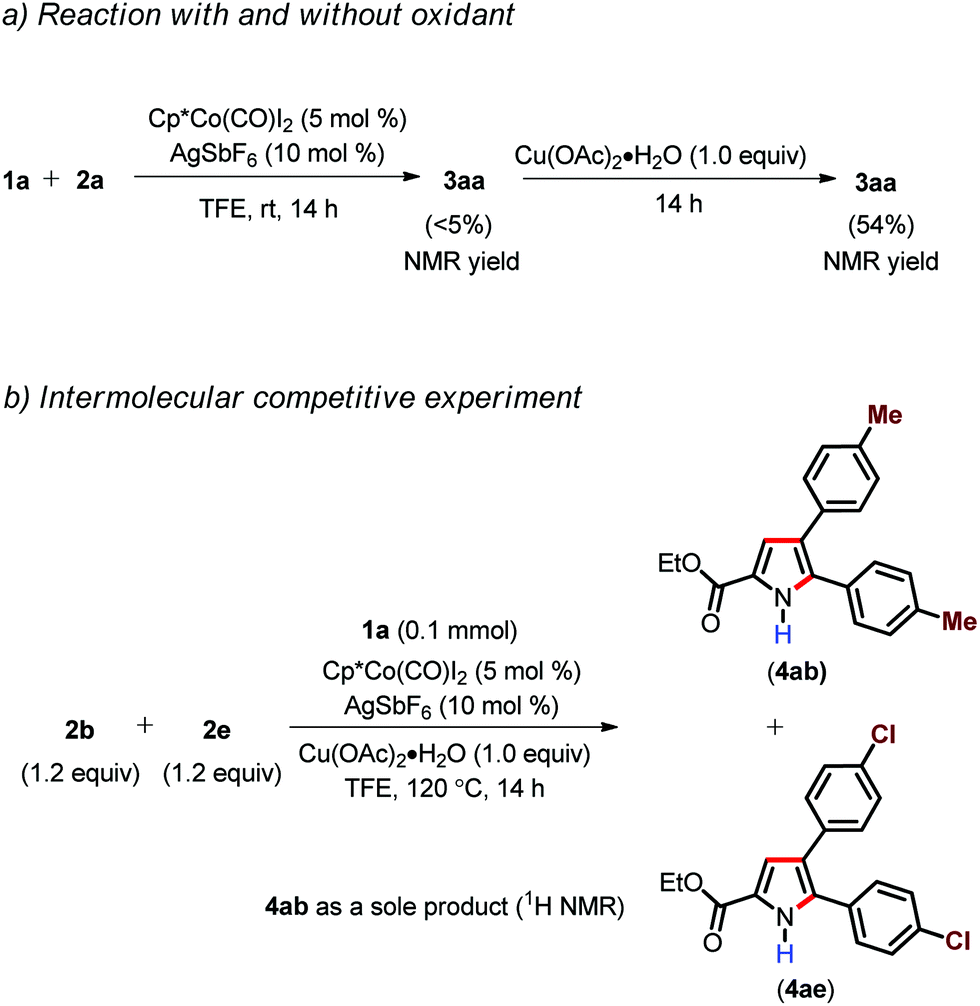 | ||
| Scheme 3 Control experiments. | ||
Based on the control experiments and previous cobalt(III)-catalyzed annulation reactions;11–14 a plausible mechanism for the present study is depicted in Scheme 4. Initially Cp*Co(CO)I2 reacts with AgSbF6 in the presence of Cu(OAc)2·H2O to generate the catalytically active species A; which undergoes cyclometalation via vinylic C–H bond activation to form cobaltacycle B. This is followed by migratory insertion of alkyne into B to form the six-membered cobaltacyclic intermediate C, which undergoes reductive elimination to furnish the pyrrole and cobalt(I) species D. The Co(I) in species D is oxidized to catalytically active Co(III) species A by Cu(OAc)2. The N-acetyl group may be deprotected in the presence of AgSbF6 at elevated temperature to produce N-H pyrroles.18d
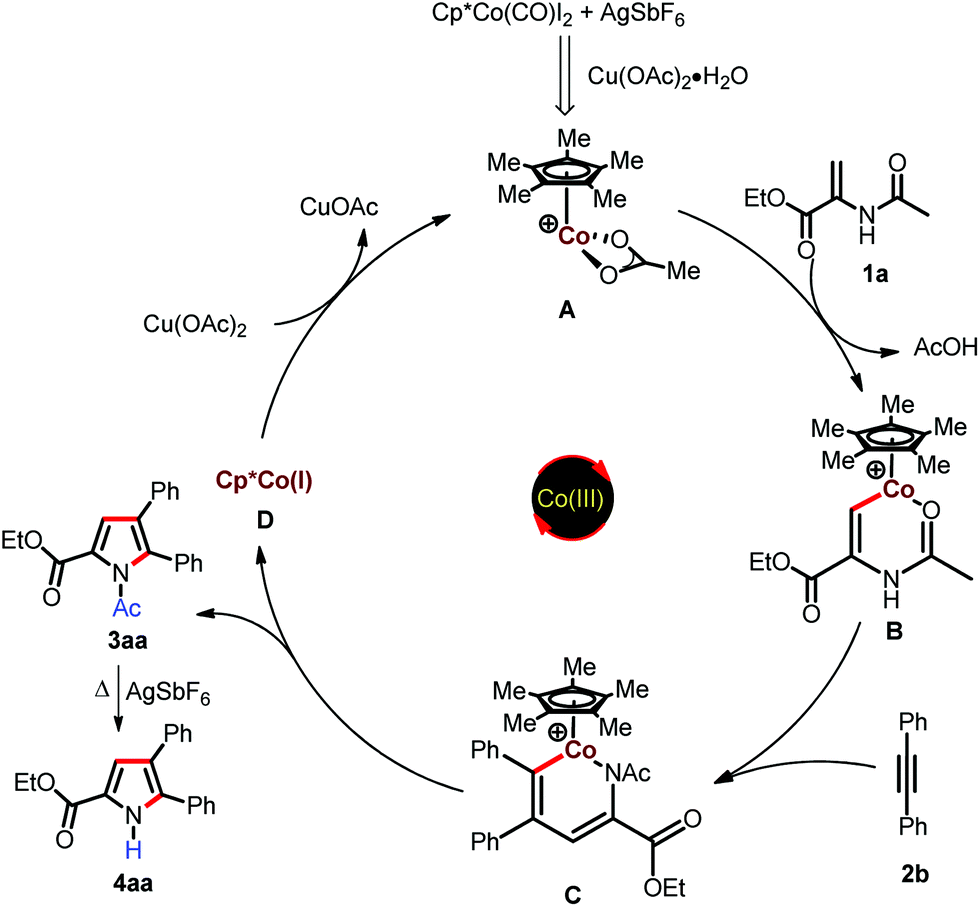 | ||
| Scheme 4 Plausible mechanism. | ||
Conclusions
In conclusion, we have developed a Co(III)-catalyzed (3 + 2) annulation of enamides and alkynes via vinylic C–H bond activation. Most of the N-acetyl pyrroles were formed at room temperature, thus showcasing the power of Cp*Co(III) catalysis. The reaction shows broad scope and excellent functional group tolerance with high yields of the pyrroles. The simple tuning of temperature led to the formation of synthetically useful N-H pyrroles under an identical catalytic system. The practicability of the present protocol is shown by a gram scale synthesis of the pyrrole.Acknowledgements
A. B. P thanks the Department of Science and Technology (DST), New Delhi, India for the INSPIRE Faculty Award (CH-157, GAP 0520). D. M. L thanks CSIR for the senior research fellowship. We thank Dr S. Chandrasekhar (Director, CSIR-IICT) for his support and encouragement.Notes and references
- For recent reviews on C–H bond activation, see: (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (b) T. W. Lyons and M. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (d) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (e) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (f) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- For reviews on Co-catalyzed C–H activation, see: (a) N. Yoshikai, Synlett, 2011, 1047 CrossRef CAS; (b) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed; (c) N. Yoshikai, Bull. Chem. Soc. Jpn., 2014, 87, 843 CrossRef CAS; (d) L. Ackermann, J. Org. Chem., 2014, 79, 8948 CrossRef CAS PubMed; (e) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2015, 6, 498 CrossRef.
- (a) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207 CrossRef CAS PubMed; (b) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Chem. – Eur. J., 2013, 19, 9142 CrossRef CAS PubMed; (c) B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Adv. Synth. Catal., 2014, 356, 1491 CrossRef CAS; (d) H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424 CrossRef CAS PubMed; (e) B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Chem. Commun., 2015, 51, 4659 RSC; (f) Y. Suzuki, B. Sun, T. Yoshino, M. Kanai and S. Matsunaga, Tetrahedron, 2015, 71, 4552 CrossRef CAS; (g) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944 CrossRef CAS PubMed.
- (a) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 3635 CrossRef CAS PubMed; (b) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 8551 CrossRef CAS PubMed; (c) M. Moselage, N. Sauermann, J. Koeller, W. Liu, D. Gelman and L. Ackermann, Synlett, 2015, 1596 CAS; (d) N. Sauermann, M. J. Gonzalez and L. Ackermann, Org. Lett., 2015, 17, 5316 CrossRef CAS PubMed; (e) W. Ma and L. Ackermann, ACS Catal., 2015, 5, 2822 CrossRef CAS; (f) R. Mei, J. Loup and L. Ackermann, ACS Catal., 2016, 6, 793 CrossRef CAS.
- (a) D.-G. Yu, T. Gensch, T. de Azambuja, S. Vasquez-Cespedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722 CrossRef CAS PubMed; (b) D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508 CrossRef CAS PubMed; (c) T. Gensch, S. Vasquez-Cespedes, D.-G. Yu and F. Glorius, Org. Lett., 2015, 17, 3714 CrossRef CAS PubMed.
- (a) A. B. Pawar and S. Chang, Org. Lett., 2015, 17, 660 CrossRef CAS PubMed; (b) P. Patel and S. Chang, ACS Catal., 2015, 5, 853 CrossRef CAS; (c) J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103 CrossRef CAS PubMed.
- (a) J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490 CrossRef CAS PubMed; (b) J. R. Hummel and J. A. Ellman, Org. Lett., 2015, 17, 2400 CrossRef CAS PubMed.
- (a) X.-G. Liu, S.-S. Zhang, J.-Q. Wu, Q. Li and H. Wang, Tetrahedron Lett., 2015, 56, 4093 CrossRef CAS; (b) X.-G. Liu, S.-S. Zhang, C.-Y. Jiang, J.-Q. Wu, Q. Li and H. Wang, Org. Lett., 2015, 17, 5404 CrossRef CAS PubMed; (c) Z.-Z. Zhang, B. Liu, C.-Y. Wang and B.-F. Shi, Org. Lett., 2015, 17, 4094 CrossRef CAS PubMed; (d) Y. Liang, Y.-F. Liang, C. Tang, Y. Yuan and N. Jiao, Chem. – Eur. J., 2015, 21, 16395 CrossRef CAS PubMed; (e) S. Wang, J.-T. Hou, M.-L. Feng, X.-Z. Zhang, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2016, 52, 2709 RSC; (f) X.-K. Guo, L.-B. Zhang, D. Wei and J.-L. Niu, Chem. Sci., 2015, 6, 7059 RSC; (g) E. Ozkal, B. Cacherat and B. Morandi, ACS Catal., 2015, 5, 6458 CrossRef CAS; (h) F. Wang, H. Wang, Q. Wang, S. Yu and X. Li, Org. Lett., 2016, 18, 1306 CrossRef CAS PubMed; (i) Z.-Z. Zhang, B. Liu, J.-W. Xu, S.-Y. Yan and B.-F. Shi, Org. Lett., 2016, 18, 1776 CrossRef CAS PubMed; (j) Q. Yan, Z. Chen, Z. Liu and Y. Zhang, Org. Chem. Front. 10.1039/C6QO00059B.
- For recent reviews on the first row transition metal-catalyzed C–H bond functionalization, see: (a) A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087 CAS; (b) Y. Nakao, Chem. Rec., 2011, 11, 242 CrossRef CAS PubMed; (c) J. Miao and H. Ge, Eur. J. Org. Chem., 2015, 7859 CrossRef CAS.
- (a) E. C. Taylor and J. E. Saxton, The Chemistry of Heterocyclic Compounds, Wiley-Interscience, New York, 1983/1994 Search PubMed; (b) J. Joule and K. Mills, Heterocyclic Chemistry, Blackwell Science, Oxford, 2000 Search PubMed; (c) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, Wiley-VCH Verlag GmbH & Co, Weinheim, 2nd edn, 2003 CrossRef; (d) N. Saracoglu, Top. Heterocycl. Chem., 2007, 11, 145 CrossRef.
- (a) B. Sun, T. Yoshino, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2015, 54, 12968 CrossRef CAS PubMed; (b) H. Wang, J. Koeller, W. Liu and L. Ackermann, Chem. – Eur. J., 2015, 21, 15525 CrossRef CAS PubMed; (c) M. Sen, D. Kalsi and B. Sundararaju, Chem. – Eur. J., 2015, 21, 15529 CrossRef CAS PubMed; (d) K. Muralirajan, R. Kuppusamy, S. Prakash and C.-H. Cheng, Adv. Synth. Catal., 2016, 358, 774 CrossRef CAS.
- S. Prakash, K. Muralirajan and C.-H. Cheng, Angew. Chem., Int. Ed., 2016, 55, 1844 CrossRef CAS PubMed.
- (a) A. Lerchen, S. Vasquez-Cespedes and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 3208 CrossRef CAS PubMed; (b) Q. Lu, S. Vasquez-Cespedes, T. Gensch and F. Glorius, ACS Catal., 2016, 6, 2352 CrossRef CAS; (c) Y. Liang and N. Jiao, Angew. Chem., Int. Ed., 2016, 55, 4035 CrossRef CAS PubMed; (d) H. Wang, M. Moselage, M. Gonzalez and L. Ackermann, ACS Catal., 2016, 6, 2705 CrossRef CAS.
- L. Kong, S. Yu, X. Zhou and X. Li, Org. Lett., 2016, 18, 588 CrossRef CAS PubMed.
- G. Sivakumar, A. Vijeta and M. Jeganmohan, Chem. – Eur. J., 2016, 22, 5899 CrossRef CAS PubMed.
- During preparation of this manuscript, Zhang et al. reported Co-catalyzed pyrrole synthesis. However, the reaction requires very high temperature (100 °C) with higher catalyst and oxidant loading. In addition, they did not observe any N-H pyrrole formation like in our case. see: W. Yu, W. Zhang, L. Yue, Y. Zhou, Z. Liu and Y. Zhang, RSC Adv., 2016, 6, 24768 RSC.
- (a) M. Dipakranjan, S. Brateen and K. D. Bidyut, Pyrrole and Its Derivatives, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 2011, p. 187 Search PubMed; (b) H. Fan, J. N. Peng, M. T. Hamann and J. F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS PubMed.
- (a) D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326 CrossRef CAS PubMed; (b) S. Rakshit, F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585 CrossRef CAS PubMed; (c) T. Besset, N. Kuhl, F. W. Patureau and F. Glorius, Chem. – Eur. J., 2011, 17, 7167 CrossRef CAS PubMed; (d) B. Li, N. Wang, Y. Liang, S. Xu and B. Wang, Org. Lett., 2013, 15, 136 CrossRef CAS PubMed; (e) L. Wang and L. Ackermann, Org. Lett., 2013, 15, 176 CrossRef CAS PubMed; (f) K. Murugan and S. T. Liu, Tetrahedron Lett., 2013, 54, 2608 CrossRef CAS; (g) Y. H. Xu, T. He, Q. C. Zhang and T. P. Loh, Chem. Commun., 2014, 50, 2784 RSC; (h) M. N. Zhao, Z. H. Ren, Y. Y. Wang and Z. H. Guan, Org. Lett., 2014, 16, 608 CrossRef CAS PubMed.
- A. B. Pawar and D. M. Lade, Org. Biomol. Chem., 2016, 14, 3275 CAS.
- Traces of N-deacetylation was observed during silica gel column chromatography.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00108d |
| This journal is © the Partner Organisations 2016 |