
Copper-catalyzed tandem trifluoromethylation–cyclization of olefinic carbonyls: synthesis of trifluoromethylated 2,3-dihydrofurans and 3,4-dihydropyrans†
Xiaohui
Bai
,
Leiyang
Lv
and
Zhiping
Li
*
Department of Chemistry, Renmin University of China, Beijing 100872, China. E-mail: zhipingli@ruc.edu.cn
First published on 26th April 2016
Abstract
A copper-catalyzed trifluoromethylation–cyclization of olefinic carbonyls was developed. With this method, a variety of 2,3-dihydrofuran and 3,4-dihydropyran derivatives containing a CF3 group were selectively obtained in moderate to good yields. This chemistry represents a rare example of direct construction of trifluoromethylated oxygen-containing heterocycles via the combination of radical trifluoromethylation with endo-type radical addition to carbonyls.
Introduction
The carbonyl group is one of the most abundant and versatile functional groups in nature. Theoretically, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond possesses two reactive sites, thus the carbon-centered radical addition to carbonyl could occur in endo- or exo-type reactions.1 Due to the strong electronegativity of the oxygen atom, this kind of radical reaction predominantly occurred at the electropositive carbon position, so the desired alcohol or ether derivatives were selectively obtained (Scheme 1, left). The exo-mode radical addition is typical and conventional, which dominated this field in the past few decades.2 In contrast, the endo-type radical addition to carbonyl was unfavorable and has been less explored by chemists (Scheme 1, right).3,4
O bond possesses two reactive sites, thus the carbon-centered radical addition to carbonyl could occur in endo- or exo-type reactions.1 Due to the strong electronegativity of the oxygen atom, this kind of radical reaction predominantly occurred at the electropositive carbon position, so the desired alcohol or ether derivatives were selectively obtained (Scheme 1, left). The exo-mode radical addition is typical and conventional, which dominated this field in the past few decades.2 In contrast, the endo-type radical addition to carbonyl was unfavorable and has been less explored by chemists (Scheme 1, right).3,4
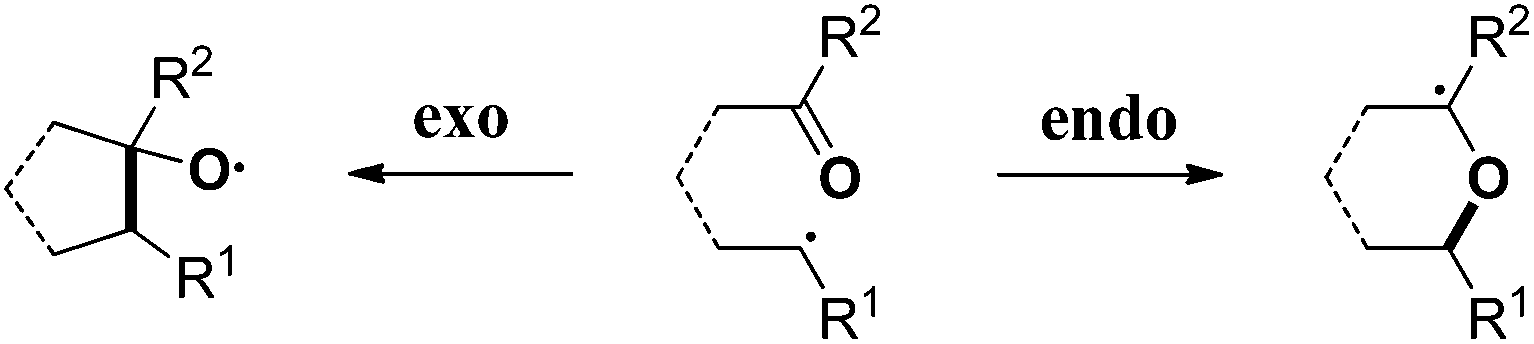 | ||
| Scheme 1 Two types of carbon-centered radical addition to CO bonds. | ||
In 2014, Lei and coworkers reported a novel nickel-catalyzed alkyl radical addition to OC of amides or esters for the preparation of imino-lactones or lactones (Scheme 2, eqn (1)).5 The EPR investigation and DFT calculation revealed that the alkyl radical endo-type addition to the carbonyl is a feasible and key step of this transformation. In the meantime, our group also reported an iron-catalyzed acylation–oxygenation of terminal alkenes for the synthesis of 2,3-dihydrofurans (Scheme 2, eqn (2)).6 The mechanistic studies suggested that the coordination of the carbonyl to the iron catalyst changed the FMOs of the carbonyl, and led to a decrease of the HOMO energy, thus matching with the SOMO of the electron-poor radical, which made the endo-type radical addition to carbonyl come true.
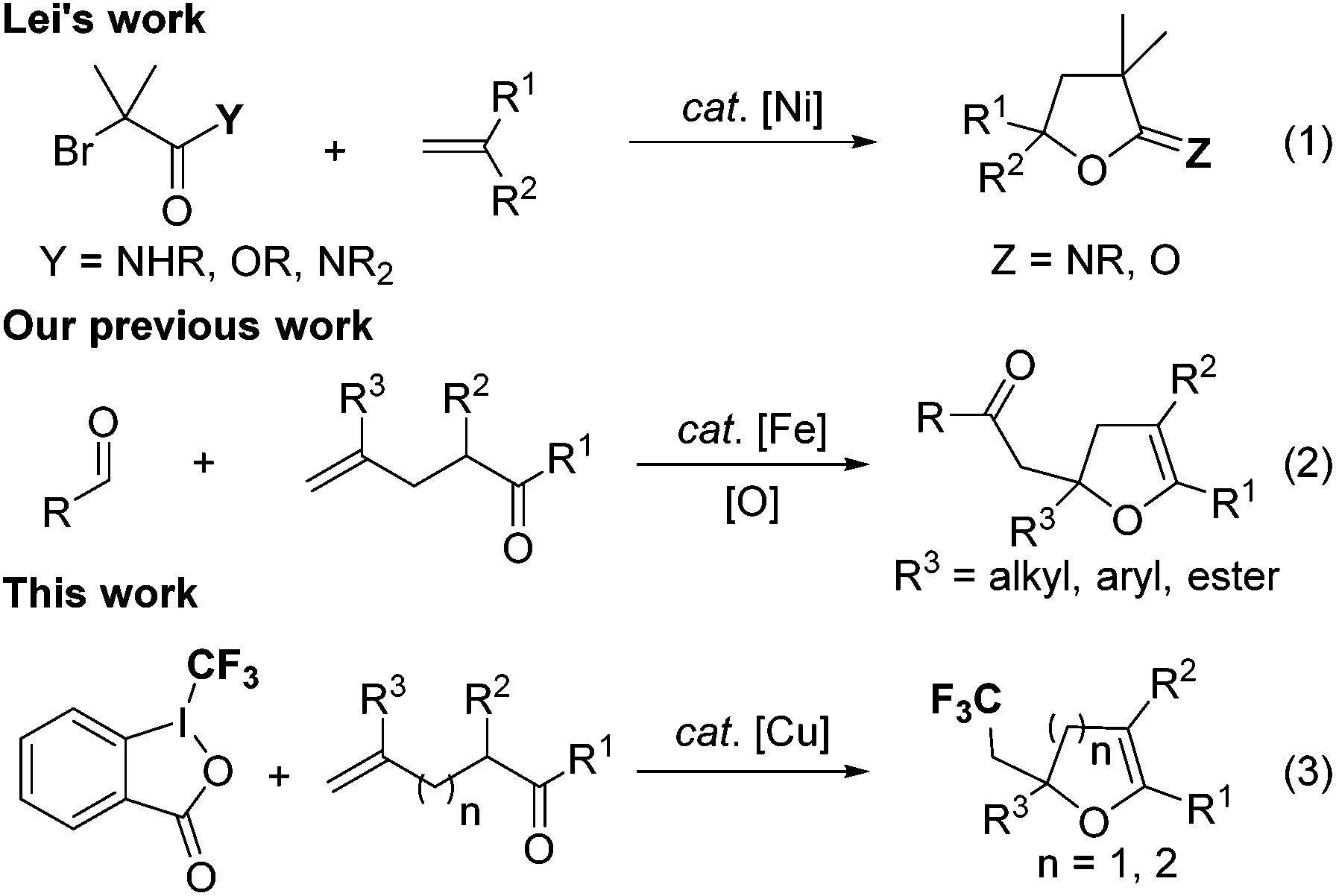 | ||
| Scheme 2 Representative examples of carbon-centered radical addition to the CO bond. | ||
Dihydrofurans, dihydropyrans as well as their derivatives are important structural motifs in a wide range of natural products and bioactive molecules.7 Their wide occurrence and significant drug activity have stimulated considerable interest in developing efficient methods for the synthesis of such oxygen-containing heterocyclic compounds.8 It is well known that the incorporation of a CF3 group into heterocycles can significantly improve their membrane permeability, metabolic stability and bioavailability. Therefore, the development of more simple and efficient approaches to introduce the CF3 motif into heterocyclic skeletons has attracted much attention in the past few decades.9 Generally, trifluoromethylation can be divided into three categories: electrophilic, nucleophilic, and radical processes. However, the use of these protocols to synthesize trifluoromethylated oxygen-containing heterocycles remains less studied.10 On the basis of the successful examples of endo-type radical addition to carbonyl and our ongoing interest in the exploration of functionalized oxygen-containing heterocycles,11 we wish to report a simple and practical Cu(I)-catalyzed trifluoromethylation–cyclization of alkenes, which allowed for the efficient preparation of various trifluoromethylated 2,3-dihydrofurans and 3,4-dihydropyrans (Scheme 2, eqn (3)).
Results and discussion
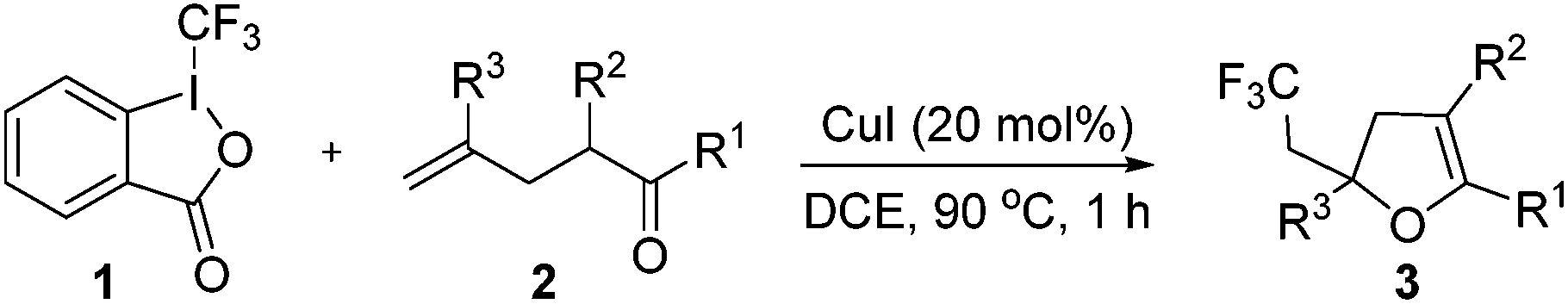
We first investigated the reaction of γ-keto olefin 2a with Togni's reagent to probe the optimal reaction conditions (Table 1). To our delight, the trifluoromethylated 2,3-dihydrofuran 3a was obtained in 42% yield when the reaction was carried out in 1,2-dichloroethane with 20 mol% of CuCl as the catalyst at 90 °C (entry 1). The screening of other copper catalysts (entries 2–9) revealed that CuI showed the highest catalytic efficacy and gave 3a in 80% yield (entry 4).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
|||
|---|---|---|---|
| Entry | [Cu] | Solvent | 3a (Yield %)b |
| a Conditions: 1 (0.35 mmol), 2a (0.2 mmol), catalyst (20 mol%), solvent (1.0 mL), 90 °C, 1 h, under N2. b Reported yields were based on 2a and determined by 1H NMR using CH2Br2 as an internal standard. c CF3SO2Na was used instead of Togni's reagent. d TMSCF3 was used instead of Togni's reagent. | |||
| 1 | CuCl | DCE | 42 |
| 2 | CuBr | DCE | 52 |
| 3 | CuBr2 | DCE | 23 |
| 4 | CuI | DCE | 80(75) |
| 5 | Cu(OAc)2 | DCE | 31 |
| 6 | Cu(OTf)2 | DCE | 40 |
| 7 | CuCl2 | DCE | 28 |
| 8 | Cu2O | DCE | 3 |
| 9 | Cu | DCE | 69 |
| 10 | — | DCE | N.D. |
| 11 | CuI | EtOAc | 62 |
| 12 | CuI | MeCN | 17 |
| 13 | CuI | PhMe | 15 |
| 14 | CuI | DMSO | 27 |
| 15 | CuI | DCE | N.D.c |
| 16 | CuI | DCE | N.D.d |
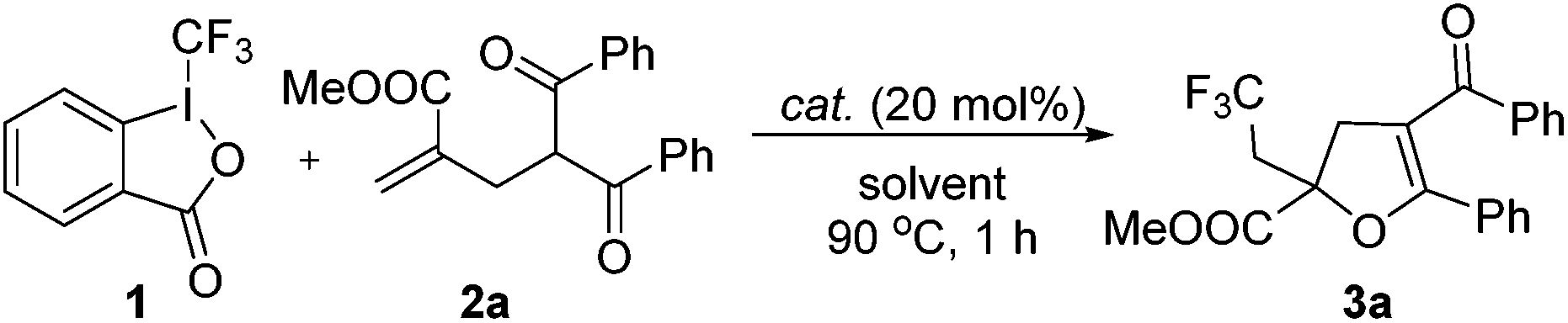
It is worth noting that 3a was not observed in the absence of a catalyst (entry 10), which indicated that copper was crucial for this trifluoromethylation–cyclization reaction. Next, other solvents, such as EtOAc, MeCN, PhMe and DMSO, were also tested and turned out no better than DCE (entries 11–14). Unfortunately, the CF3 group could not be incorporated into the dihydrofuran skeleton when either TMSCF3 or CF3SO2Na was employed (entries 15 and 16).
With the optimized reaction conditions in hand, we started to explore the scope of this transformation. The results are shown in Table 2. γ-Keto olefins 2b–d reacted smoothly with Togni's reagent to give the corresponding trifluoromethylated dihydrofurans 3b–d in good yields. When asymmetric γ-keto olefin 2e was tested, the product 3e was obtained as a major isomer, which indicated a higher reactivity of the aromatic carbonyl group than the corresponding alkyl. Subsequently, the γ-keto ester olefins were synthesized and applied under the standard conditions. The effects of the substituent group R1 were obvious. The results revealed that the substrates with electron withdrawing groups showed better efficiency (3f–i). The reaction of Togni's reagent with 2j bearing a NHPh group was sluggish, yet the reaction of 2k with an attached SO2Ph group underwent smoothly and gave the desired 3k in 54% yield. In addition, the benzyl ester and phenyl group at the CC moiety (R3) were also found to be suitable for this trifluoromethylation–oxygenation cyclization, thus providing the corresponding products 3l and 3m in good yields.
a
|
|
||
|---|---|---|
| Entry | 2 | 3 Yieldb (%) |
| a Conditions: 1 (0.35 mmol), 2 (0.2 mmol), CuI (20 mol%), DCE (1.0 mL), 90 °C, 1 h, under N2. b Reported yields were based on 2 and determined by 1H NMR using CH2Br2 as an internal standard; the isolated yields were given in parentheses. c The ratio of the isomers was determined by 1H NMR analysis. | ||
|
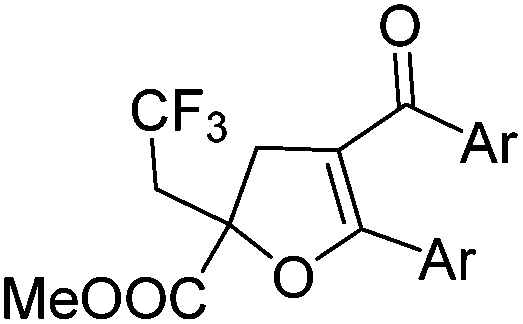
|
|
| 1 | 2b, Ar = 4-Me-C6H4 | 3b, 67(64) |
| 2 | 2c, Ar = 4-OMe-C6H4 | 3c, 69(61) |
| 3 | 2d, Ar = 4-Cl-C6H4 | 3d, 76(70) |
| 4 |
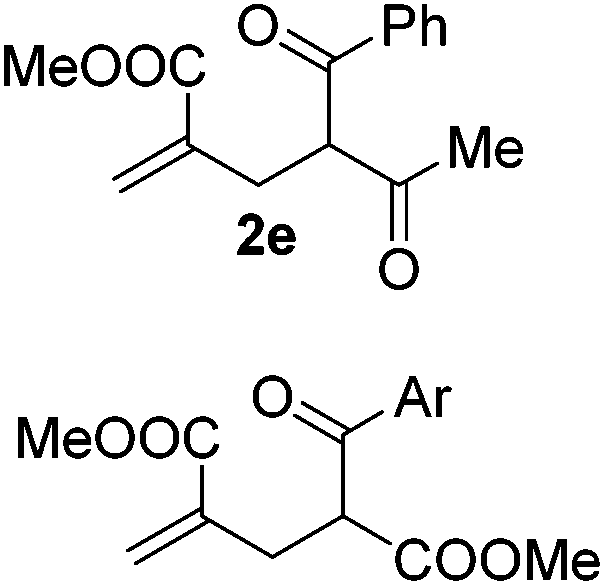
|
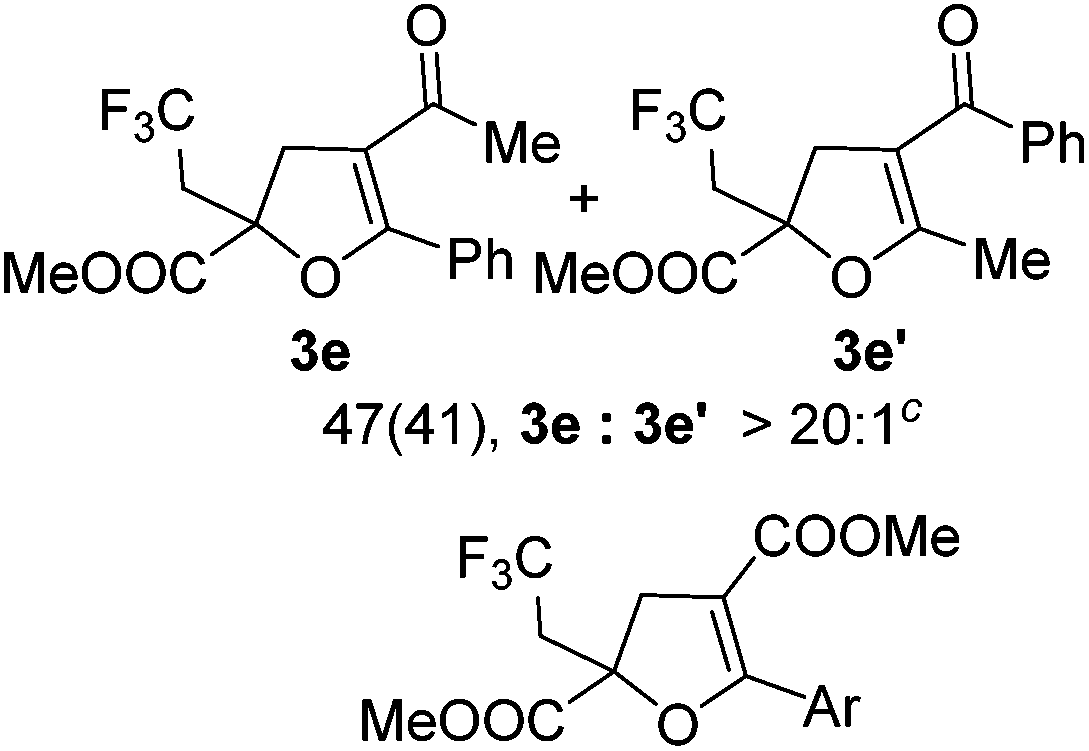
|
| 5 | 2f, Ar = Ph | 3f, 71(65) |
| 6 | 2g, Ar = 4-Me-C6H4 | 3g, 65(63) |
| 7 | 2h, Ar = 4-OMe-C6H4 | 3h, 58(48) |
| 8 | 2i, Ar = 4-Cl-C6H4 | 3i, 75(68) |
| 9 |
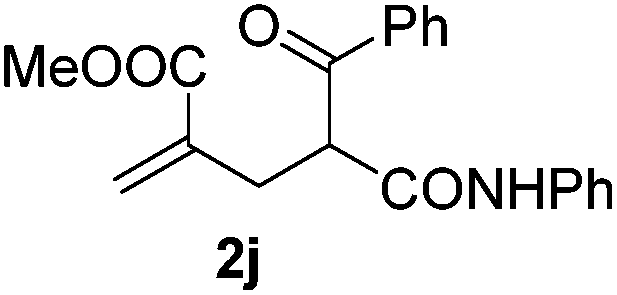
|
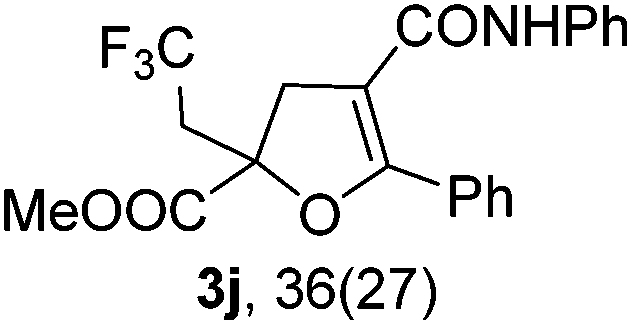
|
| 10 |
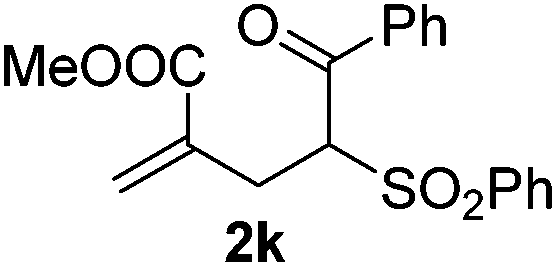
|
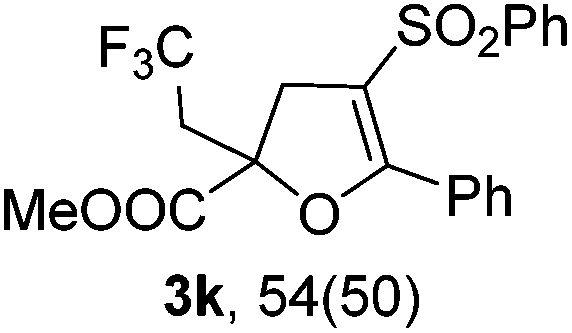
|
| 11 |
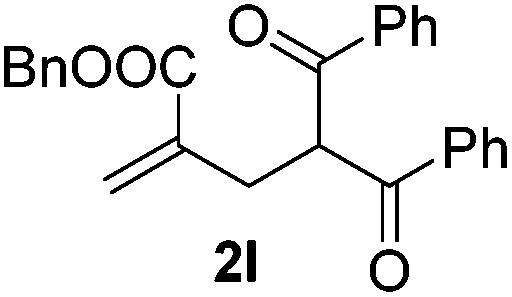
|
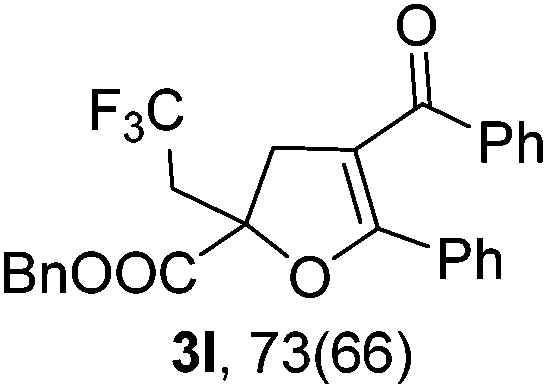
|
| 12 |
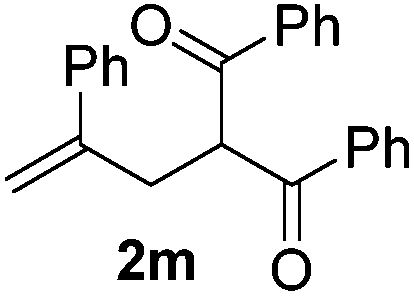
|
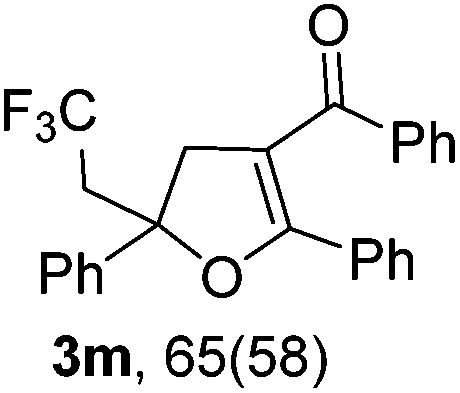
|
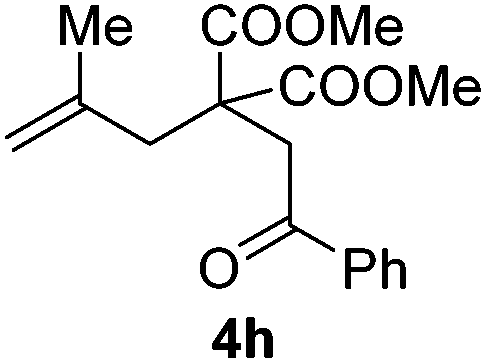
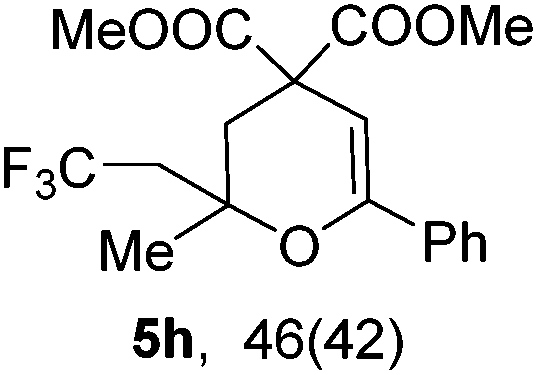
Encouraged by the successful triumph to construct trifluoromethylated dihydrofurans, we attempted to expand this strategy to synthesize six-membered oxygen-containing heterocycles (Table 3). To our delight, the 3,4-dihydropyran 5a was obtained in 70% yield when Togni's reagent and δ-keto olefin 4a were reacted under the optimal reaction conditions. In general, δ-keto olefin 4 containing electron-donating groups, such as OMe and Me, reacted sluggishly with Togni's reagent, thus giving the trifluoromethylated dihydropyrans 5b and 5c in only moderate yields. Instead, the electron-poor substrates 4 with the halogen atoms reacted smoothly to afford the desired products 5d–f in good yields. Gratifyingly, substrates bearing a methyl group at the CC moiety (R3) were also applicable for the present transformation, and delivered the corresponding products in 46% yield.
a
|
|
||
|---|---|---|
| Entry | 4 | 5 Yieldb (%) |
| a Conditions: 1 (0.35 mmol), 4 (0.2 mmol), CuI (20 mol%), DCE (1.0 mL), 90 °C, 1 h, under N2. b Reported yields were based on 4 and determined by 1H NMR using CH2Br2 as an internal standard; the isolated yields were given in parentheses. | ||
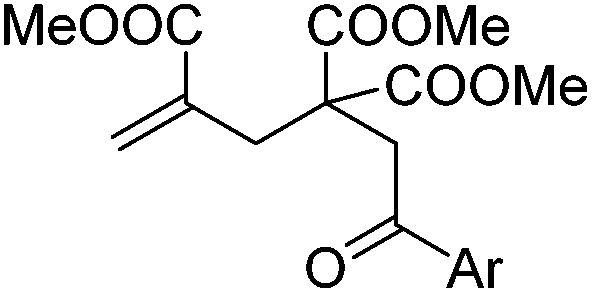
|
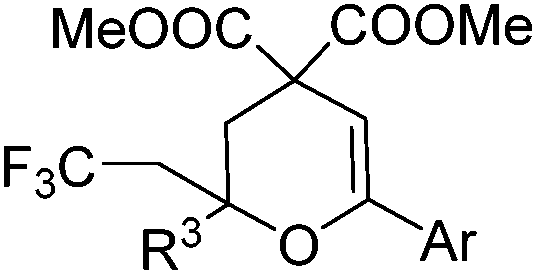
|
|
| 1 | Ar = Ph, 4a | 5a, 70(67) |
| 2 | Ar = 4-Me-C6H4, 4b | 5b, 63(55) |
| 3 | Ar = 4-OMe-C6H4, 4c | 5c, 49(40) |
| 4 | Ar = 4-F-C6H4, 4d | 5d, 69(60) |
| 5 | Ar = 4-Cl-C6H4, 4e | 5e, 73(72) |
| 6 | Ar = 4-Br-C6H4, 4f | 5f, 83(79) |
| 7 | Ar = 2-naphthyl, 4g | 5g, 41(32) |
| 8 |
|
|
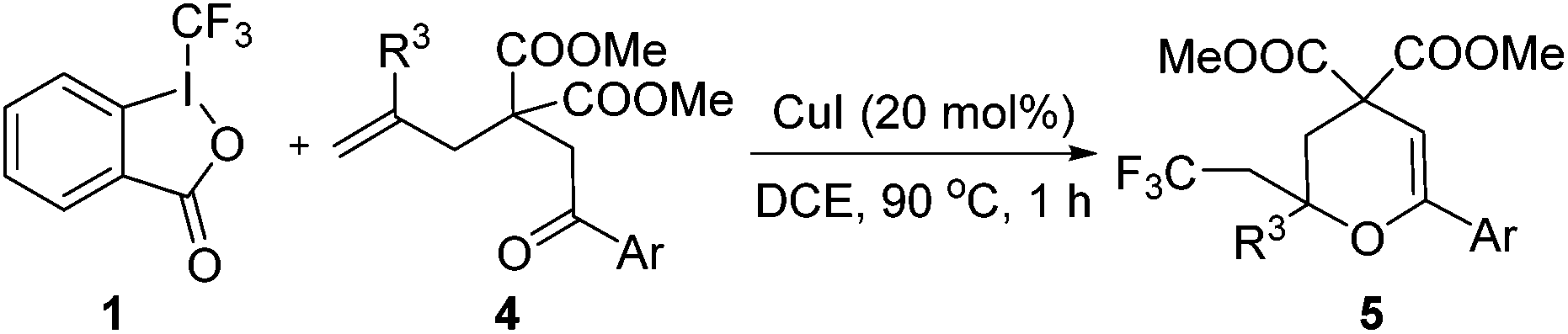
To our delight, the obtained product 5a could be converted into γ-butyrolactone 6 efficiently under acid conditions (Scheme 3). It is worth mentioning that γ-butyrolactones are valuable structural units found in a large number of natural products and biologically active secondary metabolites.12
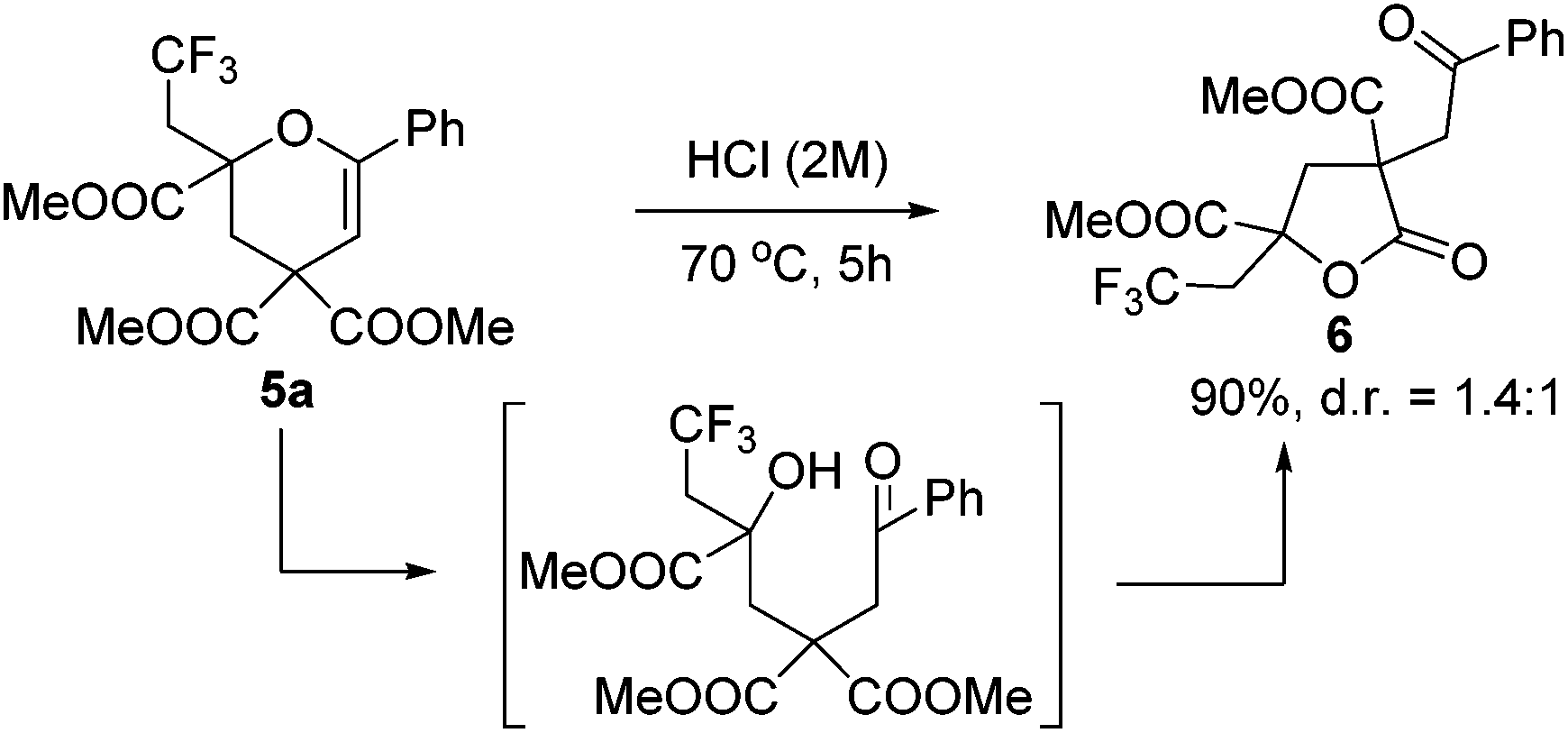 | ||
| Scheme 3 Hydrolysis under acid conditions for the synthesis of γ-butyrolactone. | ||
The control experiments were carried out to probe the possible reaction mechanism (Scheme 4). When the standard reaction was performed in the presence of BHT (butylated hydroxytoluene), 3a was not detected. Moreover, a TEMPO–CF3 adduct was obtained in 22% yield when TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) was introduced, which suggested that a CF3 radical may be involved in the process.
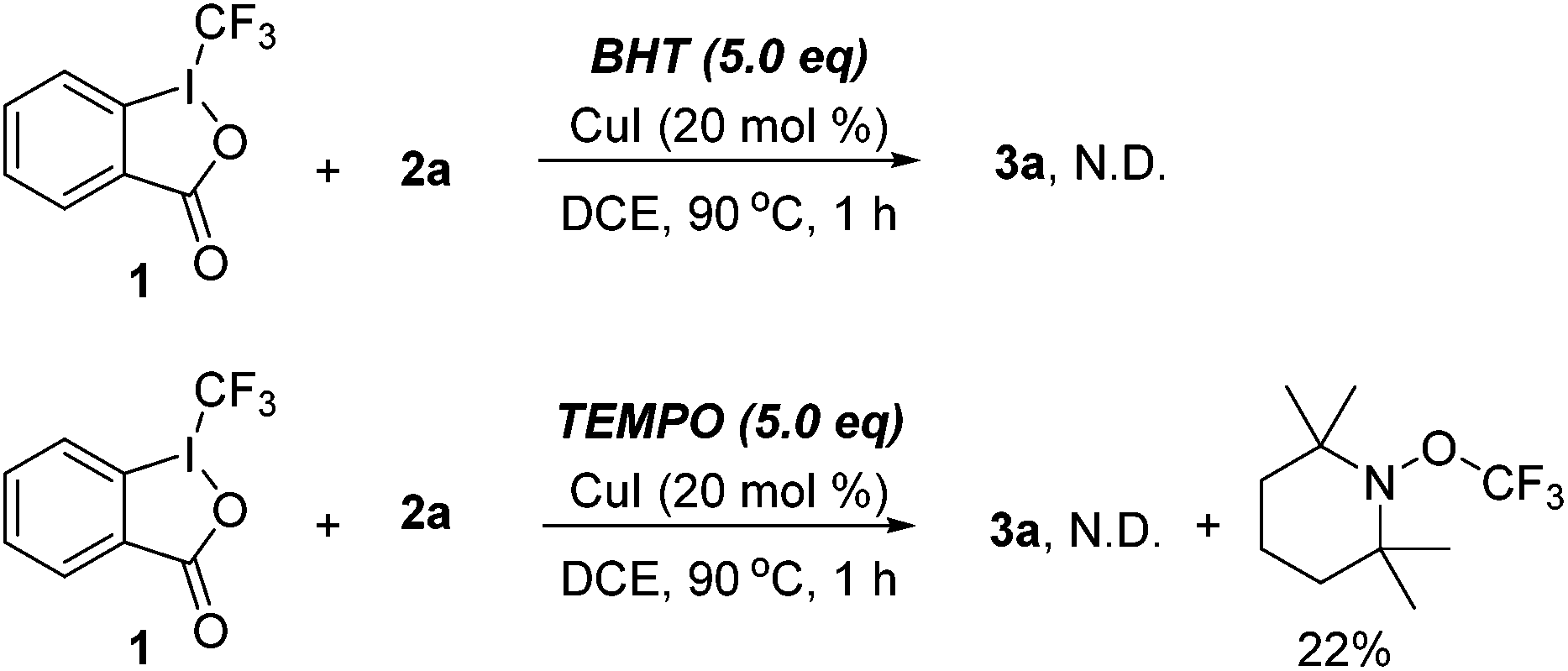 | ||
| Scheme 4 Mechanistic study. | ||
Based on the control experiments and previous related reports,9,11 a plausible mechanism is depicted in Scheme 5. The reaction of CuI with Togni's reagent generates a CF3 radical, which is trapped by the olefinic carbonyl 2 or 4 to give the persistent radical intermediate A (here R3 is an electron-withdrawing ester group). Then A undergoes the key endo-trig radical addition to the carbonyl to generate the cyclic intermediate C, which is oxidized by Cu(II) to give oxonium cation D. During the preparation of this manuscript, Liu and coworkers reported an organic base-catalyzed oxytrifluoromethylation of alkenes with Togni's reagent to synthesize trifluoromethylated dihydrofurans.13 Mechanistically, they proposed that the intermediate A (there R3 is an electron-donating aryl or alkyl group) was firstly oxidized with Togni's reagent or a base to generate a carbocation, which was readily attacked by the corresponding enol. Recent DFT calculations carried out by Lei's group indicated that the endo-trig radical addition to the carbonyl oxygen was a facile step for C–O bond formation, as the carbocation was unstable in the presence of an adjacent electron-withdrawing group.5,14 Finally, the deprotonation is caused by the carboxylic acid anion, thus delivering the final product.
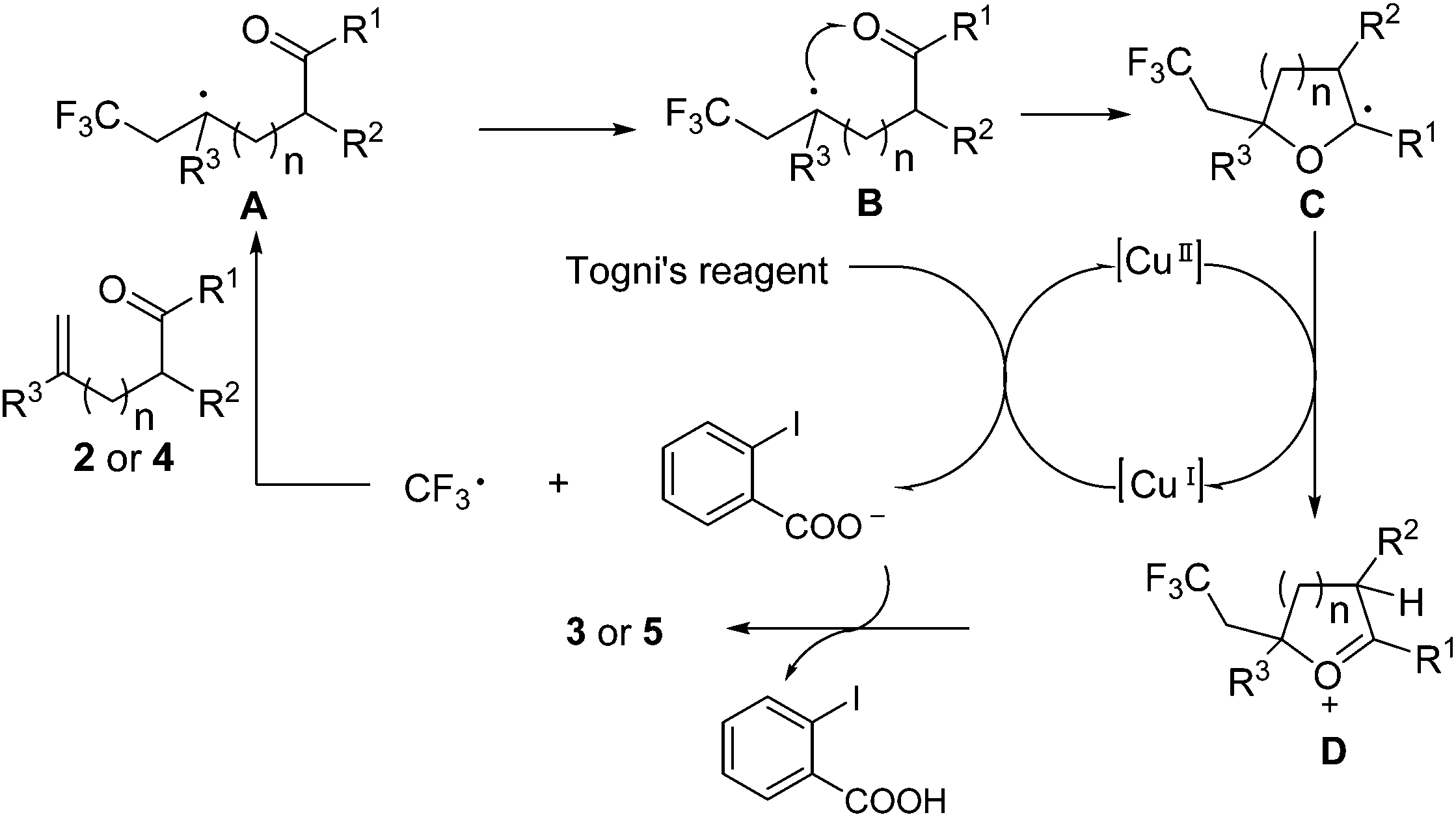 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusion
In conclusion, we have developed a general and practical copper-catalyzed trifluoromethylation–cyclization of olefinic carbonyls. With this method, a variety of trifluoromethylated 2,3-dihydrofuran and 3,4-dihydropyran derivatives were selectively obtained in moderate to good yields. Preliminary mechanistic studies indicated that the transformation proceeded via a CF3 radical addition to the terminal alkene, followed by intramolecular oxidation cyclization to give the target product. Further applications of this protocol to the synthesis of biologically active molecules are underway in our laboratory.Experimental procedure
To a mixture of olefin 2 or 4 (0.2 mmol), Togni reagent 1 (114 mg, 0.35 mmol) and CuI (7.6 mg, 20 mol%), DCE (1.0 mL) was added under nitrogen at room temperature. The resulting mixture was stirred at 90 °C for 1 h. After the mixture was cooled to room temperature, saturated aqueous NaHCO3 (5.0 mL) was added. The resulting mixture was extracted with EtOAc three times, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (ethyl acetate/petroleum ether) to give the product 3 or 5.Acknowledgements
Financial support from the National Science Foundation of China (21272267), the State Key Laboratory of Heavy Oil Processing (SKLOP201401001), and Beijing National Laboratory for Molecular Sciences (BNLMS) is acknowledged.Notes and references
- (a) D. Liu, C. Liu and A. Lei, Chem. – Asian J., 2015, 10, 2040 CrossRef CAS PubMed; (b) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed.
- (a) S.-H. Ueng, M.-J. Chen, S.-F. Chu, Y.-F. Shao, G.-T. Fan, S.-Y. Chang and Y.-M. Tsai, J. Org. Chem., 2006, 71, 1502 CrossRef CAS PubMed; (b) A. Fernández-Mateos, E. M. Nava, G. P. Coca, A. R. Silvo and R. R. González, Org. Lett., 1999, 1, 607 CrossRef; (c) D. P. Curran, U. Diederichsen and M. Palovich, J. Am. Chem. Soc., 1997, 119, 4797 CrossRef CAS; (d) A. L. J. Beckwith and B. P. Hay, J. Am. Chem. Soc., 1989, 111, 2674 CrossRef CAS.
- (a) G. D. Mendenhall, J. D. Protasiewicz, C. E. Brown, K. U. Ingold and J. Lusztyk, J. Am. Chem. Soc., 1994, 116, 1718 CrossRef CAS; (b) D. A. Harrison, R. N. Schwartz and J. Kagan, J. Am. Chem. Soc., 1970, 92, 5793 CrossRef CAS; (c) F. F. Rust, F. H. Seubold and W. E. Vaughan, J. Am. Chem. Soc., 1948, 70, 3258 CrossRef CAS PubMed.
- For recent examples, see: (a) X. Cui, X. Xu, L. Wojtas, M. M. Kim and X. P. Zhang, J. Am. Chem. Soc., 2012, 134, 19981 CrossRef CAS PubMed; (b) H. Jiang, Y. Cheng, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 4884 CrossRef CAS PubMed; (c) J. Yu, H. Yang and H. Fu, Adv. Synth. Catal., 2014, 356, 3669 CrossRef CAS.
- D. Liu, S. Tang, H. Yi, C. Liu, X. Qi, Y. Lan and A. Lei, Chem. – Eur. J., 2014, 20, 15605 CrossRef CAS PubMed.
- L. Lv, S. Lu, Q. Guo, B. Shen and Z. Li, J. Org. Chem., 2015, 80, 698 CrossRef CAS PubMed.
- For selected reviews, see: (a) D. C. M. Albanese and N. Gaggero, Eur. J. Org. Chem., 2014, 5631 CrossRef CAS; (b) A. Goel and V. J. Ram, Tetrahedron, 2009, 65, 7865 CrossRef CAS; (c) A. A. L. Gunatilaka, J. Nat. Prod., 2006, 69, 509 CrossRef CAS PubMed; (d) T. G. Kilroy, T. P. O'Sullivan and P. J. Guiry, Eur. J. Org. Chem., 2005, 4929 CrossRef CAS; (e) M. M. Faul and B. E. Huff, Chem. Rev., 2000, 100, 2407 CrossRef CAS PubMed.
- For selected recent examples, see: (a) R. B. Watson, A. N. Golonka and C. S. Schindler, Org. Lett., 2016, 18, 1310 CrossRef CAS PubMed; (b) S. Tang, K. Liu, Y. Long, X. Gao, M. Gao and A. Lei, Org. Lett., 2015, 17, 2404 CrossRef CAS PubMed; (c) L.-N. Guo, S. Wang, X.-H. Duan and S.-L. Zhou, Chem. Commun., 2015, 51, 4803 RSC; (d) S. Guo, L. Lu, J. Gong, Z. Zhu, F. Xu, Z. Wei and H. Cai, Org. Biomol. Chem., 2015, 13, 4426 RSC; (e) T. Nakano, K. Miyazazaki and A. Kamimura, J. Org. Chem., 2014, 79, 8103 CrossRef CAS PubMed; (f) Y. Zhao, X. Jiang and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2013, 52, 8597 CrossRef CAS PubMed; (g) X. Zheng, S. Lu and Z. Li, Org. Lett., 2013, 15, 5432 CrossRef CAS PubMed; (h) X. Guo, R. Yu, H. Li and Z. Li, J. Am. Chem. Soc., 2009, 131, 17387 CrossRef CAS PubMed; (i) K. A. Jørgensen, Angew. Chem., Int. Ed., 2000, 39, 3558 CrossRef.
- For selected recent reviews, see: (a) A. Prieto, O. Baudoin, D. Bouyssi and N. Monteiro, Chem. Commun., 2016, 52, 869 RSC; (b) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed; (c) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (d) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed. For selected recent examples, see: (e) K. Shen and Q. Wang, Org. Chem. Front., 2016, 3, 222 RSC; (f) K. Liu, S. Chen, X. G. Li and P. N. Liu, J. Org. Chem., 2016, 81, 265 CrossRef CAS PubMed; (g) L.-Z. Yu, Q. Xu, X.-Y. Tang and M. Shi, ACS Catal., 2016, 6, 526 CrossRef CAS; (h) Y. Wang, M. Jiang and J.-T. Liu, Org. Chem. Front., 2015, 2, 542 RSC; (i) L. Huang, S.-C. Zheng, B. Tan and X.-Y. Liu, Chem. – Eur. J., 2015, 21, 6718 CrossRef CAS PubMed; (j) Y.-L. Ji, J.-H. Lin, J.-C. Xiao and Y.-C. Gu, Org. Chem. Front., 2014, 1, 1280 RSC; (k) J.-S. Lin, Y.-P. Xiong, C.-L. Ma, L.-J. Zhao, B. Tan and X.-Y. Liu, Chem. – Eur. J., 2014, 20, 1332 CrossRef CAS PubMed; (l) F. Wang, D. Wang, X. Mu, P. Chen and G. Liu, J. Am. Chem. Soc., 2014, 136, 10202 CrossRef CAS PubMed; (m) W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480 CrossRef CAS PubMed; (n) W. Kong, M. Casimiro, N. Fuentes, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2013, 52, 13086 CrossRef CAS PubMed.
- (a) X. Bao, D. A. Hrovat and W. T. Borden, Chem. – Eur. J., 2013, 19, 5687 CrossRef CAS PubMed; (b) P. Cao, X.-B. Yan, T. Tao, F. Yang, T. He, X.-R. Song, X.-Y. Liu and Y.-M. Liang, Chem. – Eur. J., 2013, 19, 14420 CrossRef PubMed; (c) Y. Zheng, J. Cleaveland, D. Richardson and Y. Yuan, Org. Lett., 2015, 17, 4240 CrossRef CAS PubMed; (d) Q. Wei, J.-R. Chen, X.-Q. Hu, X.-C. Yang, B. Lu and W.-J. Xiao, Org. Lett., 2015, 17, 4464 CrossRef CAS PubMed; (e) B. Wang, D.-C. Xiong and X.-S. Ye, Org. Lett., 2015, 17, 5698 CrossRef CAS PubMed; (f) R. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 12655 CrossRef CAS PubMed; (g) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 12462 CrossRef CAS PubMed.
- (a) L. Lv, H. Xi, X. Bai and Z. Li, Org. Lett., 2015, 17, 4324 CrossRef CAS PubMed; (b) L. Lv, L. Qi, Q. Guo, B. Shen and Z. Li, J. Org. Chem., 2015, 80, 12562 CrossRef CAS PubMed.
- (a) M. W. Pertino, C. Theoduloz, J. A. Rodríguez, T. Yáñez, V. Lazo and G. Schmeda-Hirschmann, J. Nat. Prod., 2010, 73, 639 CrossRef CAS PubMed; (b) C. Ito, M. Itoigawa, K. Aizawa, K. Yoshida, N. Ruangrungsi and H. Furukawa, J. Nat. Prod., 2009, 72, 1202 CrossRef CAS PubMed; (c) W. Zhang, K. Krohn, J. Ding, Z.-H. Miao, X.-H. Zhou, S.-H. Chen, G. Pescitelli, P. Salvadori, T. Kurtan and Y.-W. Guo, J. Nat. Prod., 2008, 71, 961 CrossRef CAS PubMed; (d) J.-H. Jang, K. Kanoh, K. Adachi and Y. Shizuri, J. Nat. Prod., 2006, 69, 1358 CrossRef CAS PubMed.
- N.-Y. Yang, Z.-L. Li, L. Ye, B. Tan and X.-Y. Liu, Chem. Commun., 2016 10.1039/C6CC00364H.
- (a) S. Tang, K. Liu, Y. Long, X. Gao, M. Gao and A. Lei, Org. Lett., 2015, 17, 2404 CrossRef CAS PubMed; (b) S. Tang, K. Liu, Y. Long, X. Qi, Y. Lan and A. Lei, Chem. Commun., 2015, 51, 8769 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR, 13C NMR and 19F NMR spectra for new compounds. See DOI: 10.1039/c6qo00137h |
| This journal is © the Partner Organisations 2016 |