
Cu-catalyzed one-pot synthesis of fused oxazepinone derivatives via sp2 C–H and O–H cross-dehydrogenative coupling†
Zeyuan
Zhang
a,
Zhen
Dai
a,
Xinkun
Ma
a,
Yihan
Liu
a,
Xiaojun
Ma
a,
Wanli
Li
a and
Chen
Ma
*ab
aSchool of Chemistry and Chemical Engineering, Shandong University, Jinan, 250100, P R China
bState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing, 100191, P R China
First published on 19th April 2016
Abstract
An efficient Cu-catalyzed cascade reaction protocol was developed for the synthesis of fused oxazepinone derivatives via sp2 C–H and O–H cross-dehydrogenative coupling. A readily available catalyst, Cu2O, was used in this modular and convergent approach. An unusual Smiles rearrangement reaction was involved in this synthesis. Various reaction parameters were evaluated and optimized, and the target compounds were obtained in good-to-excellent yields.
Fused oxazepinone derivatives have attracted considerable attention due to their many useful biological properties, including anticancer,1 anti-HIV,2 and antitumor3 activities. These compounds also display a wide range of pharmacological and neurochemical activities, and thus they are being investigated as potential central nervous system agents,4 potential atypical antipsychotics,5 β-secretase inhibitors,6 histone deacetylase inhibitors,7 and H4R agonist.8 In particular, Sintamil(I)5a and its derivatives have been reported as antidepressants. Another derivative, Loxapine,5b has been reported to show clozapine-like properties (Scheme 1).
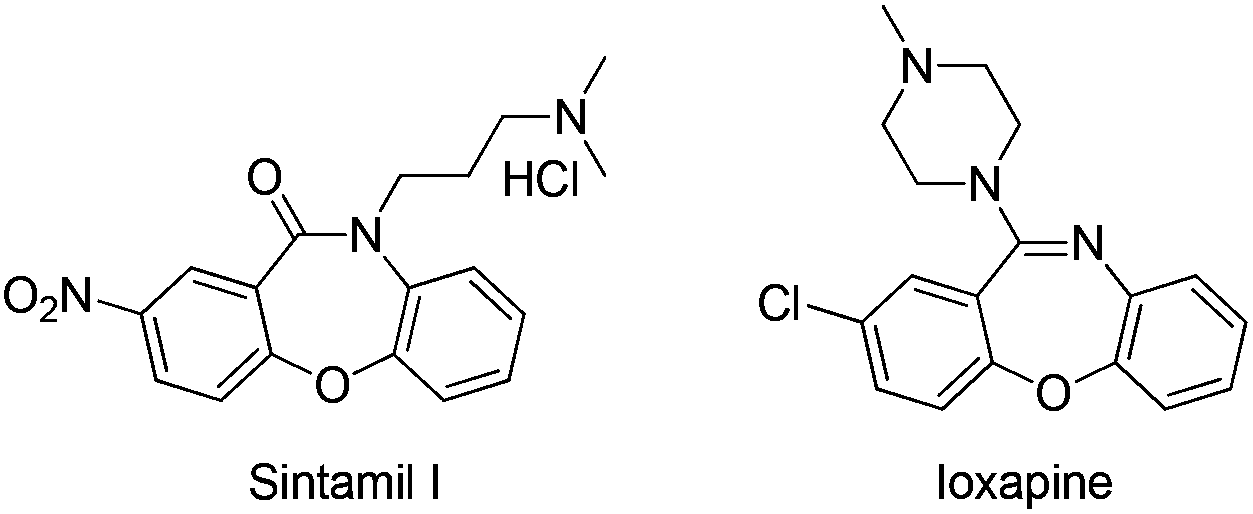 | ||
| Scheme 1 Sintamil I and ioxapine. | ||
The functionalization of carbon–hydrogen bonds by bidentate auxiliary-directed cross-coupling approaches has become increasingly popular in recent decades.9–13 While these coupling approaches have been demonstrated to be effective in single-bond constructions, there is much interest in improving their efficiency by using one-pot procedures that can bring about multiple transformations in a cascade manner. Success in this area has come from a few impressive examples10d,e,j,12g,13a,c of exploitation of established multicomponent reactions as far as we know.
The Smiles rearrangement (SR) is an intramolecular nucleophilic aromatic substitution reaction which can be easily included in cascade reactions.14–18 Based on our previous work14 and experience with the Smiles rearrangement and construction of fused oxazepinone derivatives, we envisaged that a copper-catalyzed one-pot annulation between N-(quinolin-8-yl)benzamides 1 and 2-bromophenols 2 would allow access to dibenzoxazepinones 3 by a cross-dehydrogenative coupling (C–O construction)/Smiles rearrangement (SR, C–N construction)/O-arylation (C–O construction) domino approach (Scheme 2).
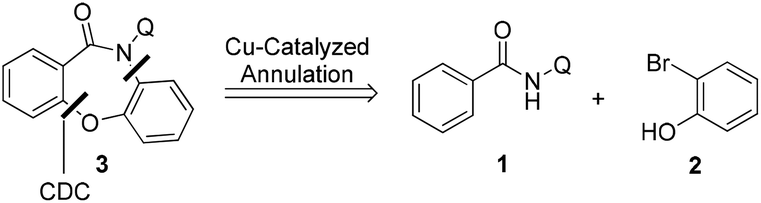 | ||
| Scheme 2 Initial retrosynthetic analysis of dibenzoxazepinones 3. | ||
To confirm the feasibility of this approach, it was tested using N-(quinolin-8-yl)benzamide 1a and 2-bromophenol 2a as the model substrates, with Cu(OAc)2 as the catalyst, K2CO3 or Na2CO3 as the base, and DMF as the solvent. Surprisingly, we found that the SR product 4a was obtained when K2CO3 was used as the base, while Na2CO3 as the base gave a direct coupling product 3a (Scheme 3c). This result was contrary to our expectations, as the SR product is often obtained by a weaker base-mediated process, due to the slightly lower potential energy of the SR metastable intermediate.
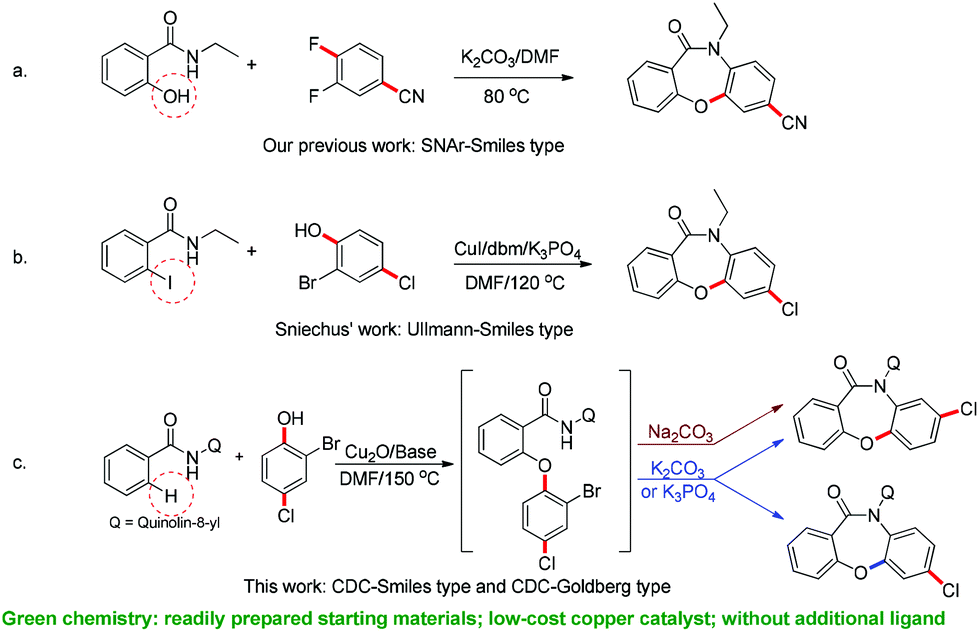 | ||
| Scheme 3 Several kinds of cascade reactions for construction of dibenzoxazepinones. | ||
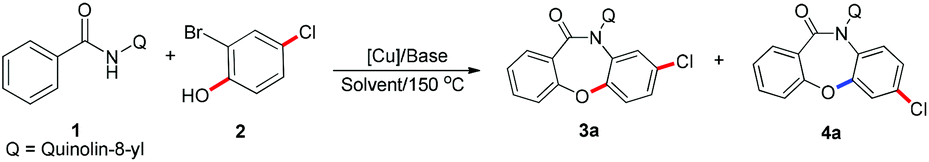
In this work, we report the development of an efficient and selective copper-catalyzed domino synthesis of dibenzoxazepinones. This domino synthesis includes a C–N coupling reaction of secondary benzamides via an unusual Smiles rearrangement reaction.19 Several methods for the synthesis of fused oxazepinone scaffolds have been reported.14i,17a,20 In 2011, we developed a metal-free methodology for the synthesis of fused oxazepinone derivatives via the Smiles rearrangement by the reaction of N-substituted salicylamides with substituted benzenes or pyridines (Scheme 3a).14i In 2012, Sniechus reported a highly regioselective CuI-initiated domino reaction which provides a general route to dibenzoxazepinones from 2-iodobenzamides and 2-bromophenols using dbm as the ligand (Scheme 3b).17a This method provides an efficient, scalable, and modular approach for the rapid construction of dibenzoxazepinones. However, these methods are not ideal because they either involve multiple steps or use 2-functionalized benzamide as the substrate. To overcome these drawbacks, the synthesis reported here is a modular and convergent approach using commercially available or readily prepared starting materials. Moreover, this method involves a directed ortho-metalation strategy, which enables the rapid formation of fused oxazepinone derivatives.
The reaction conditions were optimized using the model reaction between N-(quinolin-8-yl)benzamide 1a and 2-bromo-4-chlorophenol 2a, and the results are summarized in Table 1. First, several bases were investigated under atmospheric conditions, and we found that only 3a was obtained when Na2CO3 was used (Table 1, entry 2). As the strength of the base increased, the proportion of product 4a increased but the total yield gradually decreased (Table 1, entries 1 and 3–5). To obtain the single compound 3a, different reaction conditions were optimized using Na2CO3 as the base. Then, several different copper catalysts such as CuBr, Cu(OH)2·CuCO3, Cu(OTf)2, CuO, CuI, and Cu2O were screened. The yield catalyzed by 20% Cu(OTf)2 (91%) was little higher than that realized by 10% Cu2O (90%), while no desired product was obtained when CuO was used (Table 1, entries 6–11 and 20). Given that Cu2O was much cheaper than Cu(OTf)2 (see the ESI†), Cu2O was selected for further experiments, and the effects of Cu2O loading were also investigated. It was found that addition of 10 mol% Cu2O resulted in 90% yield, while higher catalyst loadings did not increase the yield any further (Table 1, entries 6 and 11–15). Furthermore, several different reaction solvents were investigated, and there was no increase in the yield upon changing the solvent (Table 1, entries 16–19). The other relevant reaction conditions are summarized in the ESI.†
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Base | Solvent | Yieldb (%) | |
| 3a | 4a | ||||
| DMF = N,N-dimethylformamide, DMA = N,N-dimethylacetamide, NMP = N-methylpyrrolidone, DMSO = dimethyl sulfoxide.a Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), catalyst (10 mol%), base (3.5 equiv.), solvent (2 mL), in a sealed tube at 150 °C under air for 6 h.b Yield of isolated products. | |||||
| 1 | Cu(OAc)2 (10) | K2CO3 | DMF | 35 | 22 |
| 2 | Cu(OAc)2 (10) | Na2CO3 | DMF | 77 | Trace |
| 3 | Cu(OAc)2 (10) | K3PO4·H2O | DMF | 25 | 30 |
| 4 | Cu(OAc)2 (10) | Cs2CO3 | DMF | 16 | 30 |
| 5 | Cu(OAc)2 (10) | NaOH | DMF | Trace | Trace |
| 6 | CuBr (10) | Na2CO3 | DMF | 71 | Trace |
| 7 | Cu(OH)2CuCO3 (10) | Na2CO3 | DMF | 69 | Trace |
| 8 | Cu(OTf)2 (10) | Na2CO3 | DMF | 85 | Trace |
| 9 | CuO (10) | Na2CO3 | DMF | N.D. | Trace |
| 10 | CuI (10) | Na2CO3 | DMF | 69 | Trace |
| 11 | Cu 2 O (10) | Na 2 CO 3 | DMF | 90 | Trace |
| 12 | Cu2O (5) | Na2CO3 | DMF | 72 | Trace |
| 13 | Cu2O (7) | Na2CO3 | DMF | 81 | Trace |
| 14 | Cu2O (12) | Na2CO3 | DMF | 88 | Trace |
| 15 | Cu2O (15) | Na2CO3 | DMF | 86 | Trace |
| 16 | Cu2O (10) | Na2CO3 | DMSO | 86 | Trace |
| 17 | Cu2O (10) | Na2CO3 | DMA | 82 | Trace |
| 18 | Cu2O (10) | Na2CO3 | NMP | 75 | Trace |
| 19 | Cu2O (10) | Na2CO3 | Toluene | 40 | Trace |
| 20 | Cu(OTf) 2 (20) | Na 2 CO 3 | DMF | 91 | Trace |
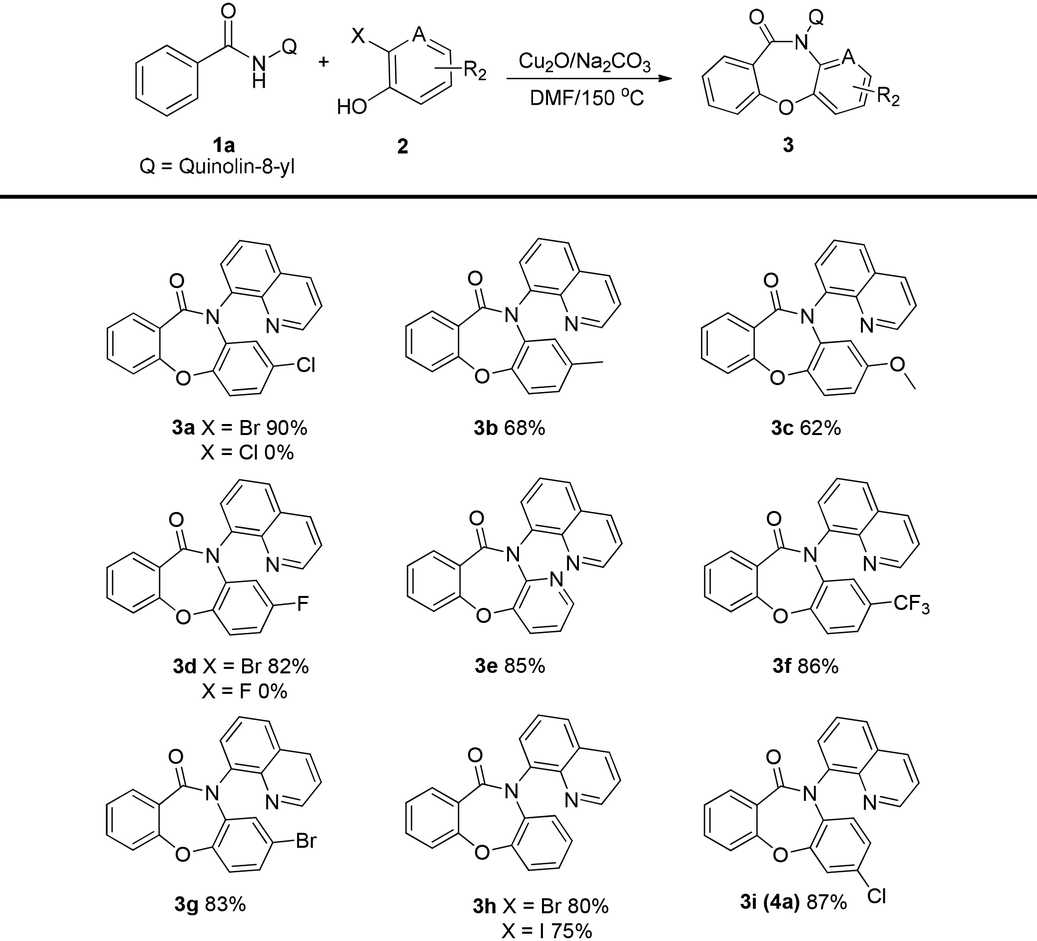
With the optimized reaction conditions, the substrate scope of this method was investigated. Firstly, a variety of 2-halogenophenols 2a–l were investigated in the reaction with N-(quinolin-8-yl)benzamide 1a (Table 2). The corresponding products were obtained in good-to-excellent yields, and the yields were found to improve in the presence of electron-withdrawing substituents R2 on the aryl ring. Low yields were obtained when electron-donating substituents R2 were present on the aryl ring. Notably, having Br and I as the 2-substituent in the 2-halogenophenols gave good yields, while no desired product was obtained when F and Cl were the 2-substituents.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| a Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), catalyst (10 mol%), base (3.5 equiv.), solvent (2 mL), in a sealed tube at 150 °C under air for 6 h. |
|---|
|
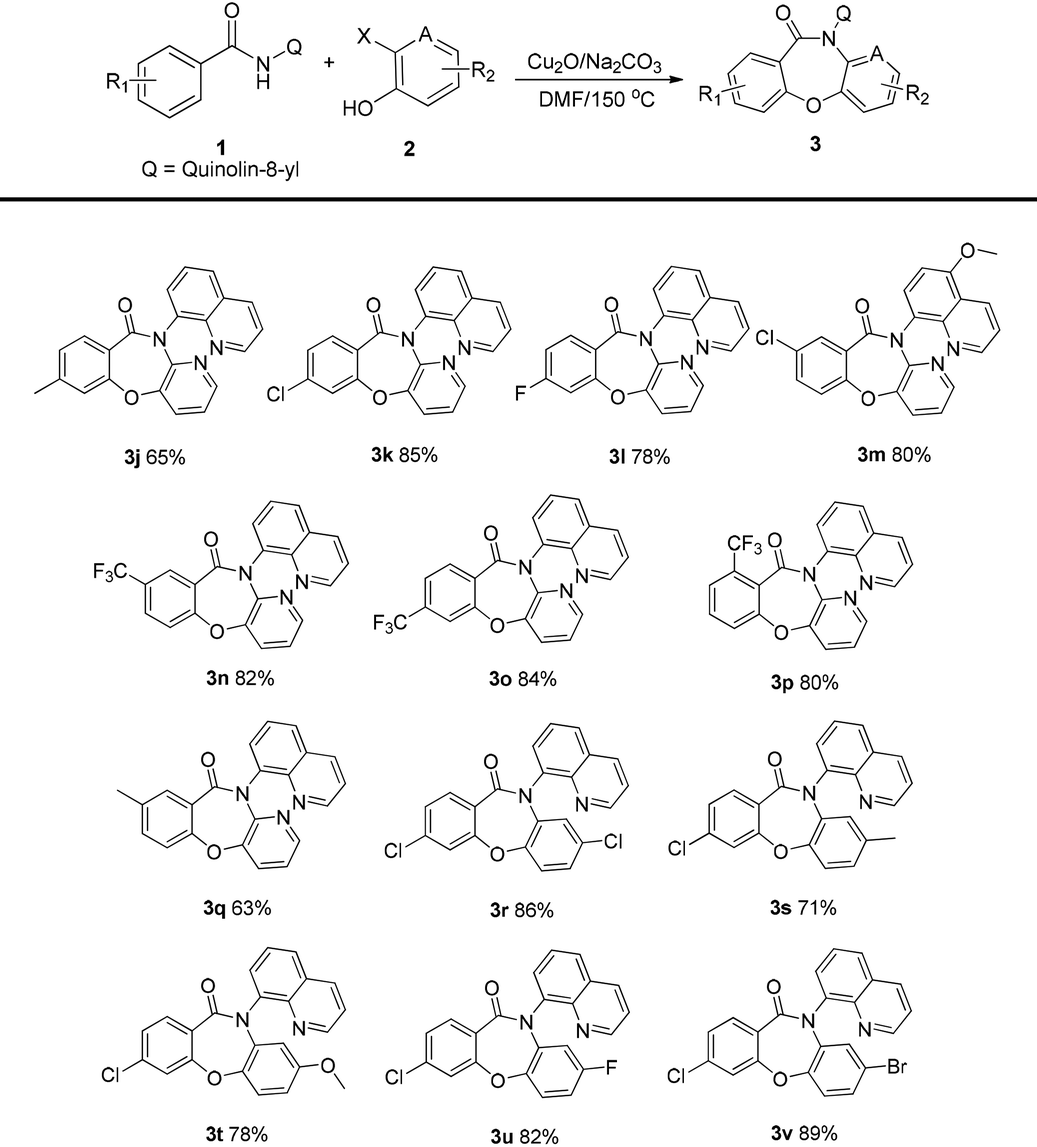
Next, several different quinolinyl benzamide derivatives, N-(quinolin-8-yl)benzamides 1b–j, were evaluated in the reaction and the results are shown in Table 3. The target compounds were obtained in good yields. The presence of electron-withdrawing groups (–CF3, –F, and Cl) on the substrate slightly increased the yields of the target products 3k–3p and 3r–3v compared to the yields of the derivatives 3g and 3q with an election-donating group (–Me). The halide (fluoride and chloride) derivatives 3k–3m and 3r–3v were also obtained in good yields. With regard to the meta-substituted benzamides, cleavage of the C–H bond occurred para to the substituent to yield single regioisomers 3n and 3q.
a
| a Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), catalyst (10 mol%), base (3.5 equiv.), solvent (2 mL), in a sealed tube at 150 °C under air for 6 h. |
|---|
|
To elucidate the mechanism of the reaction, several control experiments were performed (Scheme 4). The ortho-blocked benzamide 1l failed to generate the Goldberg amidation product (Scheme 4a), supporting the hypothesis that the cross-dehydrogenative coupling (CDC) should take place first in this reaction. To allow the reaction to remain in the first step, the amount of the base was reduced to 2.0 equiv. and the temperature was lowered to 75 °C. After the amide starting material was fully consumed, the mixture was cooled to room temperature. Then, 1.5 equiv. strong base t-BuOK was added to the mixture. Finally, the desired product 4a was obtained in 65% yield (Scheme 4b). It was further evidenced that the SR step requires higher energy to overcome the resistance from sterically hindered ligands.
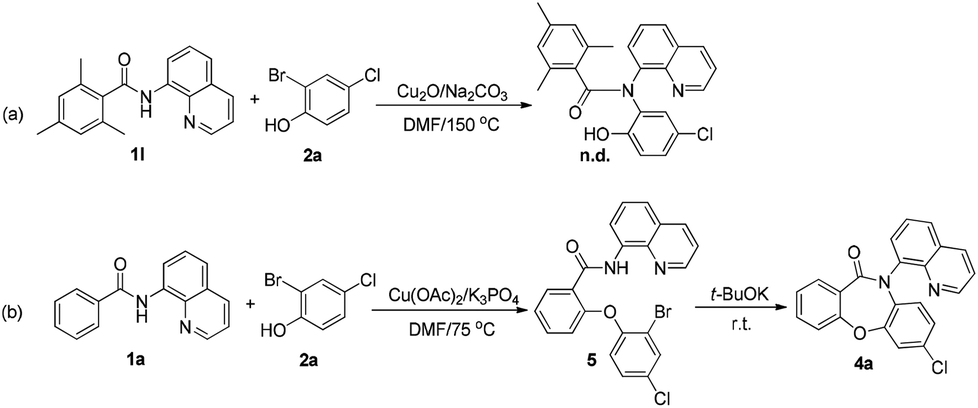 | ||
| Scheme 4 Control experiments. | ||
Based on the above control experiments and our previous work on Smiles rearrangement chemistry, a possible mechanism for this reaction has been proposed, as shown in Scheme 5. Firstly, the reaction of N-(quinolin-8-yl)benzamide 1a and 2-bromo-4-chlorophenol 2a affords the intermediate 5. Then, a carboxamide anion 6 is formed. Subsequently, intermediate 6 can react via two pathways (a and b). Pathway a leads to 3a by direct intramolecular cyclization. In contrast, pathway b forms the intermediate 8via the Smiles rearrangement, which then undergoes another intramolecular cyclization, leading to the corresponding product 4a.
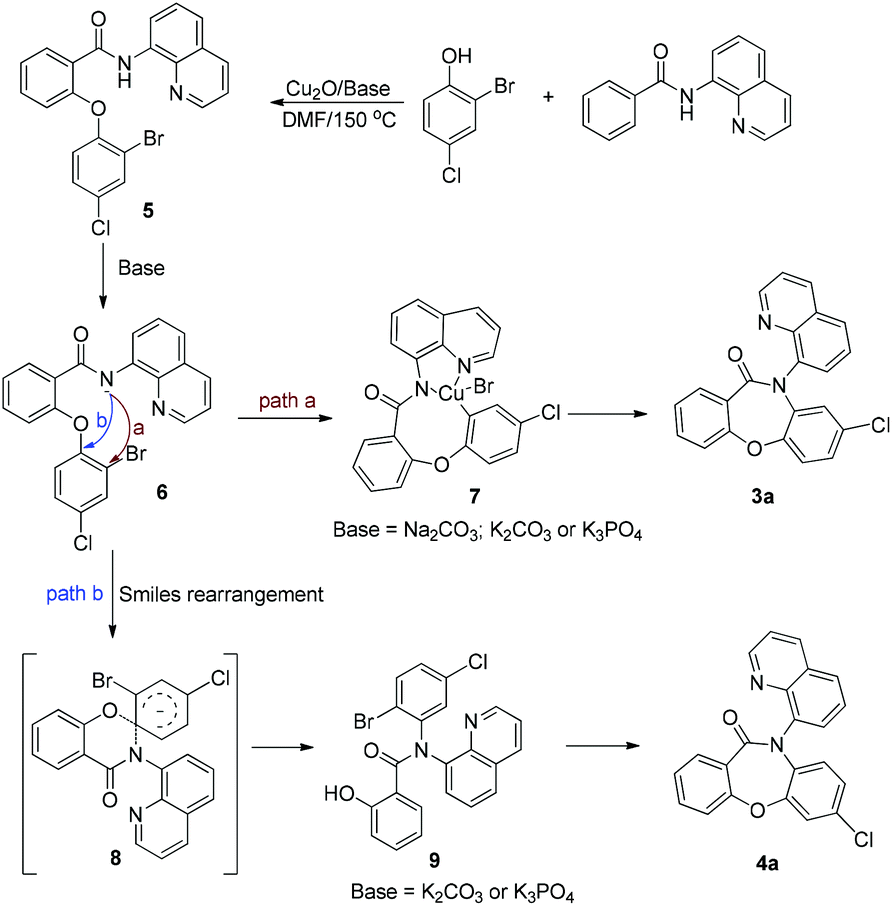 | ||
| Scheme 5 Proposed mechanism. | ||
In summary, an efficient method has been developed for the synthesis of fused oxazepinone derivatives via sp2 C–H and O–H cross-dehydrogenative coupling, catalysed by the readily available Cu2O. This method is applicable for a wide variety of substrates, and the target compounds were obtained in good-to-excellent yields. Further studies on the application of this method towards the synthesis of pharmaceutical compounds are in progress.
Acknowledgements
We are grateful to the National Science Foundation of China (no. 21572117) and the State Key Laboratory of Natural and Biomimetic Drugs of Peking University (no. K20140208) for financial support of this research.Notes and references
- H. A. Soliman, A. Y. Mubarak, A. El-Mekabaty, H. M. Awad and S. S. Elmorsy, Monatsh. Chem., 2016, 147, 809 CrossRef CAS.
- (a) S. Vilar, L. Santana and E. Uriarte, J. Med. Chem., 2006, 49, 1118 CrossRef CAS PubMed; (b) J. M. Klunder, K. D. Hargrave, M. West, E. Cullen, K. Pal, M. L. Behnke, S. R. Kapadia, D. W. McNeil, J. C. Wu and G. C. Chow, J. Med. Chem., 1992, 35, 1887 CrossRef CAS PubMed; (c) K. D. Hargrave, J. R. Proudfoot, K. G. Grozinger, E. Cullen, S. R. Kapadia, U. R. Patel, V. U. Fuchs, S. C. Mauldin and J. Vitous, J. Med. Chem., 1991, 34, 2231 CrossRef CAS PubMed.
- M. Binaschi, A. Boldetti, M. Gianni, C. A. Maggi, M. Gensini, M. Bigioni, M. Parlani, A. Giolitti, M. Fratelli, C. Valli, M. Terao and E. Garattini, ACS Med. Chem. Lett., 2010, 1, 411 CrossRef CAS PubMed.
- J. F. F. Liegeois, F. A. Rogister, J. Bruhwyler, J. Damas, T. P. Nguyen, M. O. Inarejos, E. M. G. Chleide, M. G. A. Mercier and J. E. Delarge, J. Med. Chem., 1994, 37, 519 CrossRef CAS PubMed.
- (a) R. Jain, J. A. Rather and A. Dwivedi, Mater. Sci. Eng. C, 2011, 31, 230 CrossRef CAS; (b) Y. Liao, B. J. Venhuis, N. Rodenhuis, W. Timmerman and H. Wikström, J. Med. Chem., 1999, 42, 2235 CrossRef CAS PubMed.
- T. H. Al-Tel, R. A. Al-Qawasmeh, M. F. Schmidt, A. Al-Aboudi, S. N. Rao, S. S. Sabri and W. Voelter, J. Med. Chem., 2009, 52, 6484 CrossRef CAS PubMed.
- M. Binaschi, A. Boldetti, M. Gianni, C. A. Maggi, M. Gensini, M. Bigioni, M. Parlani, A. Giolitti, M. Fratelli, C. Valli, M. Terao and E. Garattini, ACS Med. Chem. Lett., 2010, 1, 411 CrossRef CAS PubMed.
- R. A. Smits, H. D. Lim, B. Stegink, R. A. Bakker, I. J. P. de Esch and R. Leurs, J. Med. Chem., 2006, 49, 4512 CrossRef CAS PubMed.
- For selected reviews, see: (a) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (b) M. Corbet and F. De Campo, Angew. Chem., Int. Ed., 2013, 52, 9896 CrossRef CAS PubMed; (c) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed.
- For selected work, see Daugulis’ work: (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed; (c) L. Grigorjeva and O. Daugulis, Org. Lett., 2015, 17, 1204 CrossRef CAS PubMed; (d) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4684–4687 CrossRef CAS PubMed; (e) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4688 CrossRef CAS PubMed; (f) J. Roane and O. Daugulis, Org. Lett., 2013, 15, 5842–5845 CrossRef CAS PubMed; (g) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7 CrossRef CAS PubMed; (h) L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237 CrossRef CAS PubMed; (i) T. Truong, K. Klimovica and O. Daugulis, J. Am. Chem. Soc., 2013, 135, 9342 CrossRef CAS PubMed; (j) T. T. Nguyen, L. Grigorjeva and O. Daugulis, ACS Catal., 2015, 551 Search PubMed; (k) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, J. Org. Chem., 2013, 78, 9689 CrossRef CAS PubMed; (l) L. D. Tran, J. Roane and O. Daugulis, Angew. Chem., Int. Ed., 2013, 52, 6043 CrossRef CAS PubMed.
- Yu and Dai's work: (a) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 3354 CrossRef CAS PubMed; (b) M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590 CrossRef CAS PubMed; (c) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, Org. Lett., 2014, 16, 5666 CrossRef CAS PubMed; (d) M. Shang, S.-Z. Sun, H.-L. Wang, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 10439 CrossRef CAS PubMed.
- Shi's work: (a) K. Chen, X. Li, S.-Q. Zhang and B.-F. Shi, Chem. Commun., 2016, 52, 1915–1918 RSC; (b) K. Chen, X. Li, S.-Q. Zhang and B.-F. Shi, Org. Chem. Front., 2016, 3, 204–208 RSC; (c) P.-X. Ling, S.-L. Fang, X.-S. Yin, K. Chen, B.-Z. Sun and B.-F. Shi, Chem. – Eur. J., 2015, 21, 17503 CrossRef CAS PubMed; (d) S. Zhao, J. Yuan, Y.-C. Li and B.-F. Shi, Chem. Commun., 2015, 51, 12823 RSC; (e) W.-H. Rao and B.-F. Shi, Org. Lett., 2015, 17, 2784 CrossRef CAS PubMed; (f) S. Zhao, Y.-J. Liu, S.-Y. Yan, F.-J. Chen, Z.-Z. Zhang and B.-F. Shi, Org. Lett., 2015, 17, 3338 CrossRef CAS PubMed; (g) F.-J. Chen, G. Liao, X. Li, J. Wu and B.-F. Shi, Org. Lett., 2014, 16, 5644 CrossRef CAS PubMed; (h) Q. Zhang, X.-S. Yin, K. Chen, S.-Q. Zhang and B.-F. Shi, J. Am. Chem. Soc., 2015, 137, 8219 CrossRef CAS PubMed; (i) W.-H. Rao, B.-B. Zhan, K. Chen, P.-X. Ling, Z.-Z. Zhang and B.-F. Shi, Org. Lett., 2015, 17, 3552 CrossRef CAS PubMed; (j) X. Li, Y.-H. Liu, W.-J. Gu, B. Li, F.-J. Chen and B.-F. Shi, Org. Lett., 2014, 16, 3904 CrossRef CAS PubMed; (k) W.-H. Rao, B.-B. Zhan, K. Chen, P.-X. Ling, Z.-Z. Zhang and B.-F. Shi, Org. Lett., 2015, 17, 3552 CrossRef CAS PubMed; (l) K. Chen, F. Hu, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 3906 RSC; (m) F.-J. Chen, S. Zhao, F. Hu, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 4187 RSC; (n) Q. Zhang, K. Chen, W. Rao, Y. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588 CrossRef CAS PubMed; (o) Q. Zhang, X.-S. Yin, S. Zhao, S.-L. Fang and B.-F. Shi, Chem. Commun., 2014, 50, 8353 RSC; (p) K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950 CrossRef CAS PubMed; (q) X.-S. Yin, Y.-C. Li, J. Yuan, W.-J. Gu and B.-F. Shi, Org. Chem. Front., 2015, 2, 119 RSC.
- Other reports: (a) W. Zhu, D. Zhang, N. Yang and H. Liu, Chem. Commun., 2014, 50, 10634 RSC; (b) M. Nishino, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 4457 CrossRef CAS PubMed; (c) H.-W. Liang, W. Ding, K. Jiang, L. Shuai, Y. Yuan, Y. Wei and Y.-C. Chen, Org. Lett., 2015, 17, 2764 CrossRef CAS PubMed.
- For selected work, see SNAr–Smiles type (our work): (a) Y.-M. Zhao, Y.-M. Wu, J. Jia, D.-J. Zhang and C. Ma, J. Org. Chem., 2012, 77, 8501 CrossRef CAS PubMed; (b) B.-C. Yang, X.-C. Tan, R.-Y. Guo, S.-W. Chen, Z.-Y. Zhang, X.-L. Chu, C.-X. Xie, D.-J. Zhang and C. Ma, J. Org. Chem., 2014, 79, 8040 CrossRef CAS PubMed; (c) C.-J. Zhan, J. Jia, B.-C. Yang, A.-P. Huang, Y.-L. Liu and C. Ma, RSC Adv., 2012, 2, 7506 RSC; (d) X.-Y. Niu, B.-C. Yang, Y.-Q. Li, S. Fang, Z.-X. Huang, C.-X. Xie and C. Ma, Org. Biomol. Chem., 2013, 11, 4102 RSC; (e) Y.-M. Zhao, Q.-L. Dai, Z. Chen, Q.-H. Zhang, Y.-C. Bai and C. Ma, ACS Comb. Sci., 2013, 15, 130 CrossRef CAS PubMed; (f) Y.-M. Zhao, Y.-C. Bai, Q.-H. Zhang, Z. Chen, Q.-L. Dai and C. Ma, Tetrahedron Lett., 2013, 54, 3253 CrossRef CAS; (g) Y.-L. Liu, X.-H. Zhang, Y. Ma and C. Ma, Tetrahedron Lett., 2013, 54, 402 CrossRef CAS; (h) Y.-L. Liu, Y. Ma, C.-J. Zhan, A.-P. Huang and C. Ma, Synlett, 2012, 255 Search PubMed; (i) Y.-L. Liu, C. Chu, A.-P. Huang, C.-J. Zhan, Y. Ma and C. Ma, ACS Comb. Sci., 2011, 13, 547 CrossRef CAS PubMed.
- Ugi–Smiles type: (a) L. El Kaim and L. Grimaud, Tetrahedron, 2009, 65, 2153 CrossRef CAS; (b) L. El Kaim, L. Grimaud and J. Oble, Angew. Chem., Int. Ed., 2005, 44, 7961 CrossRef CAS PubMed; (c) L. El Kaim, L. Grimaud and J. Oble, J. Org. Chem., 2007, 72, 5835 CrossRef PubMed; (d) L. El Kaim, M. Gizolme, L. Grimaud and J. Oble, J. Org. Chem., 2007, 72, 4169 CrossRef PubMed; (e) L. El Kaim, M. Gizolme, L. Grimaud and J. Oble, Org. Lett., 2006, 8, 4019 CrossRef CAS PubMed; (f) D. Coffinier, L. El Kaim and L. Grimaud, Org. Lett., 2009, 11, 995 CrossRef CAS PubMed; (g) A. Barthelon, L. El Kaim, M. Gizolme and L. Grimaud, Eur. J. Org. Chem., 2008, 5974 CrossRef CAS; (h) A. Barthelon, X. F. Legoff, L. El Kaim and L. Grimaud, Synlett, 2010, 153 CAS; (i) L. El Kaim and L. Grimaud, Mol. Diversity, 2010, 14, 855 CrossRef CAS PubMed.
- Julia–Smiles type: J. B. Baudin, G. Hareau, S. A. Julia and O. Ruel, Tetrahedron Lett., 1991, 32, 1175 CrossRef CAS.
- Ullmann–Smiles type: (a) M. O. Kitching, T. E. Hurst and V. Sniechus, Angew. Chem., Int. Ed., 2012, 51, 2925 CrossRef CAS PubMed; (b) P. Sang, M. Yu, H. Tu, J. Zou and Y. Zhang, Chem. Commun., 2013, 49, 701 RSC; (c) P. Mondal, P. Mitra and N. C. Ganguly, RSC Adv., 2014, 4, 55640 RSC.
- Radical-Smiles type: (a) M. Pudlo, I. Allart-Simon, B. Tinant, S. Gérard and J. Sapi, Chem. Commun., 2012, 48, 2442 RSC; (b) E. Bacqué, M. El Qacemi and S. Z. Zard, Org. Lett., 2005, 7, 3817 CrossRef PubMed.
- While the manuscript was under preparation, a Cu-catalyzed synthesis of oxazepinone with a controllable Smiles rearrangement was reported online. See: Y. Zhou, J. Zhu, B. Li, Y. Zhang, J. Feng, A. Hall, J. Shi and W. Zhu, Org. Lett., 2016, 18, 380–383 CrossRef CAS PubMed.
- (a) A. V. Samet, V. N. Marshalkin, K. A. Kislyi, N. B. Chernysheva, Y. A. Strelenko and V. V. Semenov, J. Org. Chem., 2005, 70, 9371 CrossRef CAS PubMed; (b) I. Ott, B. Kircher, G. Heinisch and B. Matuszczak, J. Med. Chem., 2004, 47, 4628 CrossRef PubMed; (c) B. Stegink, R. A. Bakker, I. J. P. de Esch and R. Leurs, J. Med. Chem., 2006, 49, 4513 Search PubMed; (d) Q. Yang, H. Cao, A. Robertson and H. Alper, J. Org. Chem., 2010, 75, 6297 CrossRef CAS PubMed; (e) K. Nagarajan, Indian J. Chem., 1974, 12, 252 CAS; (f) S.-M. Lu and H. Alper, J. Am. Chem. Soc., 2005, 127, 14776 CrossRef CAS PubMed; (g) N. C. Ganguly, P. Mondal, S. Roy and P. Mitra, RSC Adv., 2014, 4, 55640 RSC; (h) M. O. Kitching, T. E. Hurst and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 2925 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1449079 and 1449087. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00040a |
| This journal is © the Partner Organisations 2016 |