
One-carbon homologation of arylboronic acids: a convenient approach to the synthesis of pinacol benzylboronates†
Chaoqiang
Wu
a,
Guojiao
Wu
a,
Yan
Zhang
a and
Jianbo
Wang
*ab
aBeijing National Laboratory of Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: wangjb@pku.edu.cn; Fax: (+86)10-6275-7248; Tel: (+86)10-6275-1708
bState Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 3rd May 2016
Abstract
A transition-metal-free method for the synthesis of benzylic boronate esters with arylboronic acids and trimethylsilyldiazomethane (TMSCHN2) has been developed. This transformation is a straightforward homologation of arylboronic acids, which represents a unique approach toward the preparation of pinacol benzylboronates. The reaction has a wide substrate scope and good functional-group tolerance, and it can be scaled up easily.
Introduction
Organoboron compounds are important intermediates in organic synthesis, and are widely used in transition-metal-catalyzed cross-coupling reactions.1 They are also essential building blocks for the synthesis of medicines, agrochemicals and materials.2 Thus, great progress has been achieved for the synthesis of arylboron compounds in recent years, many of which are now commercially available and the methods for their synthesis are also well-established. In contrast, the commercial availability of the corresponding alkylboron compounds is relatively limited, especially benzyl boronates.Benzyl boronates are versatile synthetic intermediates as they can be converted into relevant diarylmethanes, benzyl alcohols and benzyl amides, however the methods for their preparation are limited. The classical method for the synthesis of benzyl boronates involves the reaction of highly reactive benzyl lithium3 and Grignard4 reagents with suitable boron compounds, such as B(OR)3 or BX3 (Scheme 1a). This method suffers from poor functional group tolerance because of the high reactivity of organometallic reagents. Rh- and Ir-catalyzed hydroboration of alkenes5 is another method to access benzyl boronates (Scheme 1b). The regioselectivity is not satisfactory in some cases and primary benzyl boronates cannot be accessed by this method. In recent years, transition metal (such as Pd, Cu, Fe)-catalyzed cross-coupling reactions have established as effective methods to directly convert benzyl halides to benzyl boronates, although expensive metal catalysts and ligands either in a catalytic or stoichiometric amount are indispensable (Scheme 1c).6–9 Ir-, Rh-, and Pd-catalyzed borylation of benzylic C–H bonds is an alternative method for the preparation of benzyl boronates (Scheme 1d)10,11 Recently, Ir- and Co-catalyzed borylation of primary benzylic C–H bonds without a directing group has also been reported.12 Aside from the methods mentioned above, direct borylation of arylmethanols with B2pin2 through palladium-catalyzed C–O bond activation has also been developed by Shi and co-workers very recently.13 In addition, we have reported a transition-metal-free borylation of N-tosylhydrazones with B2pin2 to access benzyl boronates.14
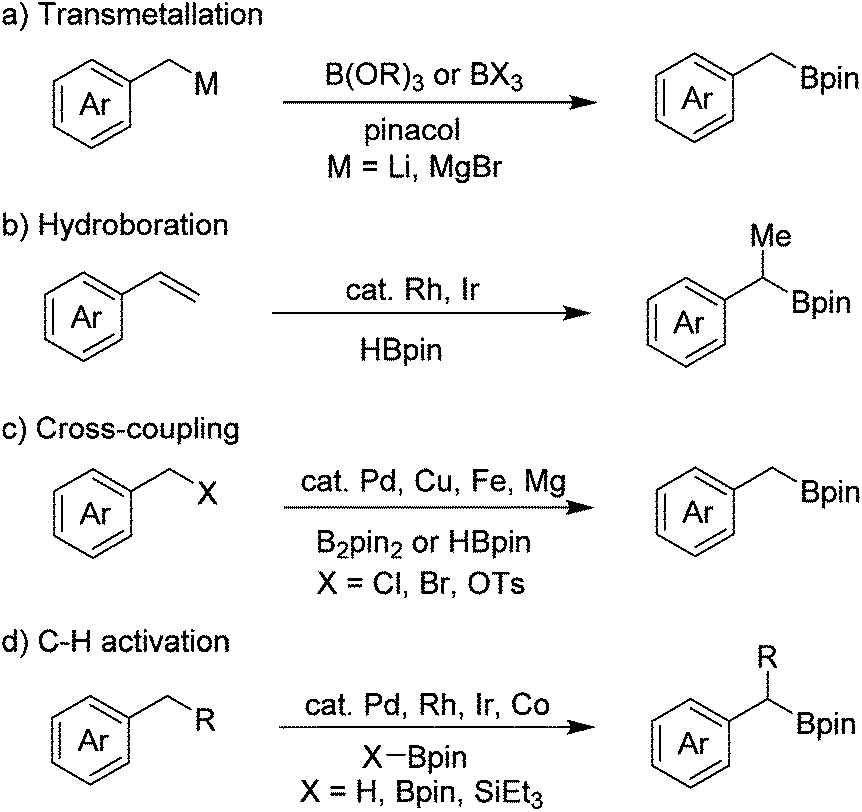 | ||
| Scheme 1 Methods for the synthesis of benzyl boronates. | ||
The reactions of diazo compounds with organoboron compounds have been previously established as a unique method to form C–C bonds under transition-metal-free conditions. Our group and others have previously developed the transition-metal-free reaction of diazo compounds with boronic acids.14–16 As the continuation of our interest in both diazo compounds and alkylboron compounds, we report herein a transition-metal-free method for the preparation of primary benzyl boronates from the corresponding arylboronic acids and TMSCHN2 in one pot. As shown in Scheme 2, this type of transformation follows a simple process involving the coordination of the electron-rich diazo carbon atom to the electron-deficient boron centre, which is followed by a 1,2-shift of the aryl group to form a carbon–carbon bond. Upon subsequent esterification and protodesilylation, benzyl boronates can be obtained from the corresponding arylboronic acids in one pot.
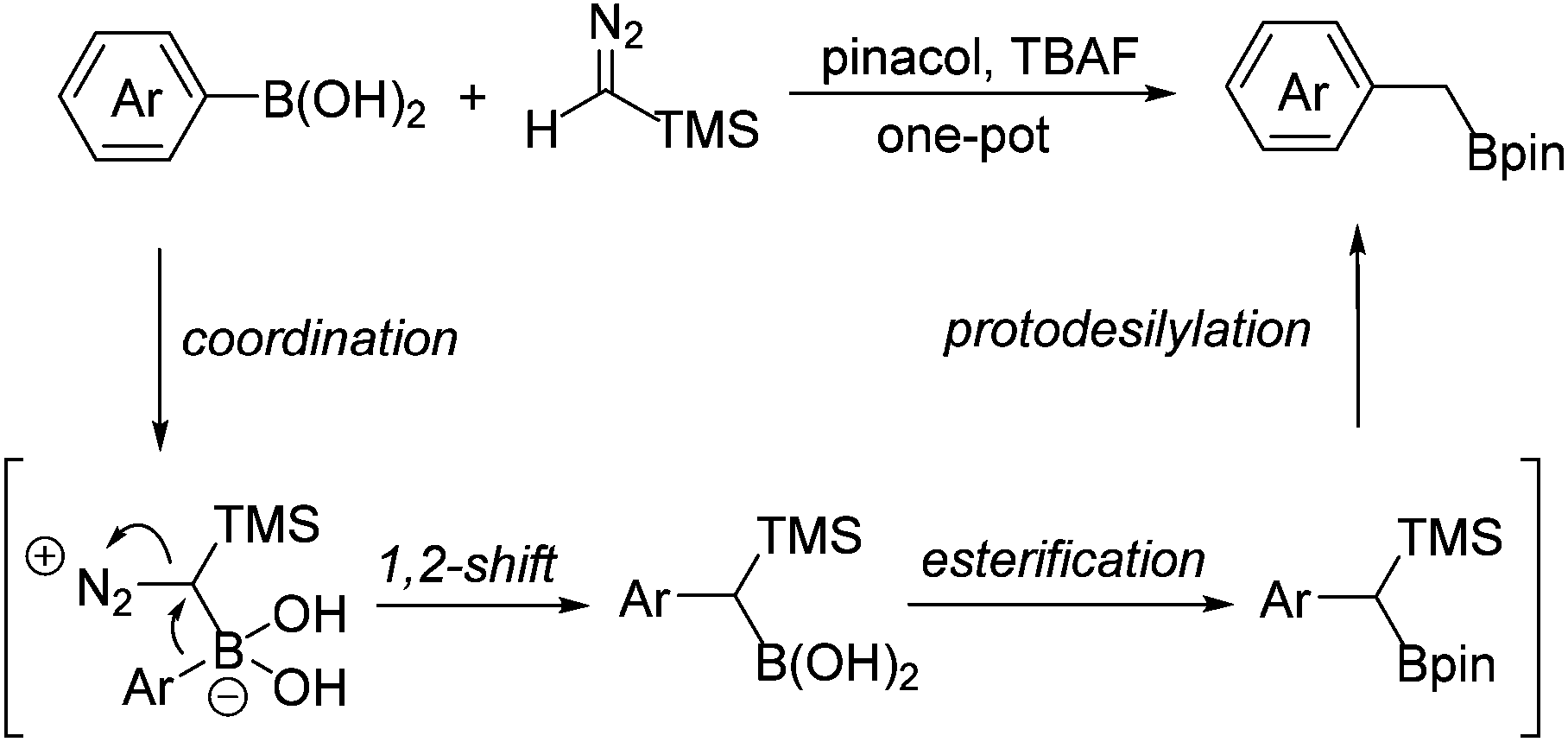 | ||
| Scheme 2 Transition-metal-free process for the synthesis of benzyl boronates. | ||
Results and discussion
At the outset of this study, we chose phenylboronic acid (1a) as the substrate for the optimization of the reaction conditions (Table 1). With toluene as the solvent, the desired product 3a could be obtained as detected by GC-MS, albeit in low yield (entry 1). Then a series of solvents were screened and the reaction was slightly improved with THF and DCE as the solvent (entries 2 and 3). When 1,4-dioxane was used as the solvent, the expected product 3a was isolated with 61% yield (entry 4). Since the proton source in the reaction could be beneficial to the protodesilylation, H2O was then chosen as the additive and the reaction was indeed improved to 76% yield (entry 5). The effect of temperature was also investigated (entries 6 and 7). The yields diminished at either lower or higher temperature, and 50 °C offered the optimal result. A diminished yield was also observed when the equivalent of pinacol was reduced (entry 8). However, no desired product 3a was observed when Ph-B(OH)2 (1a) was replaced with Ph-Bpin.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | H2Ob (mL) | T (°C) | Yieldc (%) |
| a The reaction was carried out with 1a and 2 in 1 mL solvent (0.4 mmol, 0.4 M) for 4 h and then pinacol (dissolved in 1 mL solvent) and TBAF were added for another 4 h. b Water was added in the last step. c All the yields refer to the isolated products. d 1.3 equiv. pinacol was added instead of 1.5 equiv. pinacol. | ||||
| 1 | Toluene | — | 50 | <10 |
| 2 | THF | — | 50 | 24 |
| 3 | DCE | — | 50 | 29 |
| 4 | Dioxane | — | 50 | 61 |
| 5 | Dioxane | 0.2 | 50 | 76 |
| 6 | Dioxane | 0.2 | 40 | 50 |
| 7 | Dioxane | 0.2 | 60 | 64 |
| 8d | Dioxane | 0.2 | 50 | 56 |
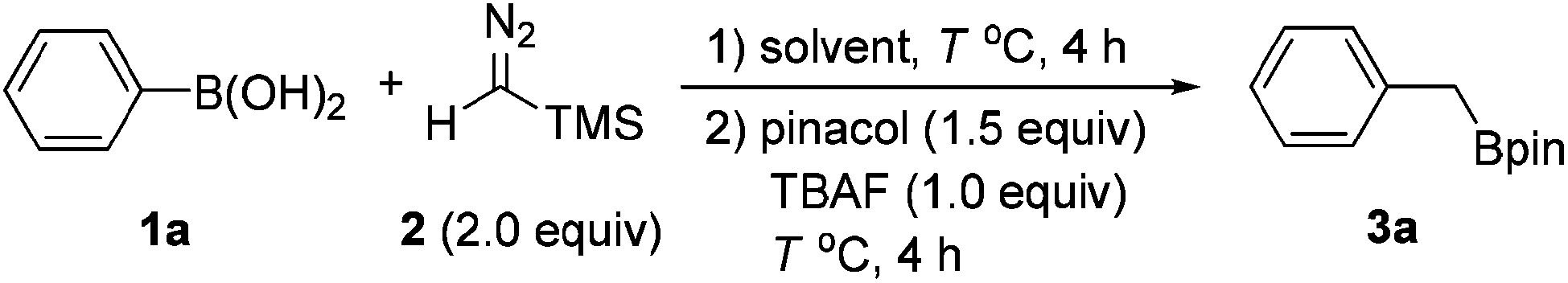
With the optimized reaction conditions in hand, we next proceeded to investigate the scope of the reaction with various arylboronic acids. As shown in Scheme 3, the reaction worked well with a series of arylboronic acids (1a–w), thus affording the corresponding products (3a–w) in moderate to good yields with good functional group tolerance. Arylboronic acids bearing electron-donating groups such as alkyl (3b, 3c, 3l, 3r), vinyl (3k), methoxy (3d, 3m), trifluoromethoxy (3n) and methylthio (3e), all can be converted into the corresponding benzyl boronates successfully. Notably, the reaction tolerates halogen substituents on the aromatic rings, including fluoro (3f, 3s), chloro (3g, 3o, 3t), bromo (3h, 3p) and iodo (3i), which provides the possibility for additional transformations through transition-metal-catalyzed coupling reactions. Arylboronic acids with strong electron-withdrawing groups, such as the nitro group, on the aromatic rings failed to afford the desired products. However, para-phenyl substituted boronic acid (3j) and meta-trifluoromethyl substituted boronic acid (3q) afforded the desired product in moderate to good yields. In addition, the steric effect was observed for ortho-substituted arylboronic acids (3r, 3s, 3t) and diminished yields were obtained with bulkier substituents. Naphthylboronic acids (3u, 3v) could also afford the desired products albeit with relatively low yields as there were some byproducts generated through protodeboronation instead of protodesilylation. Furthermore, poly-substituted arylboronic acid (3w) could also work under the reaction conditions to afford the corresponding products.
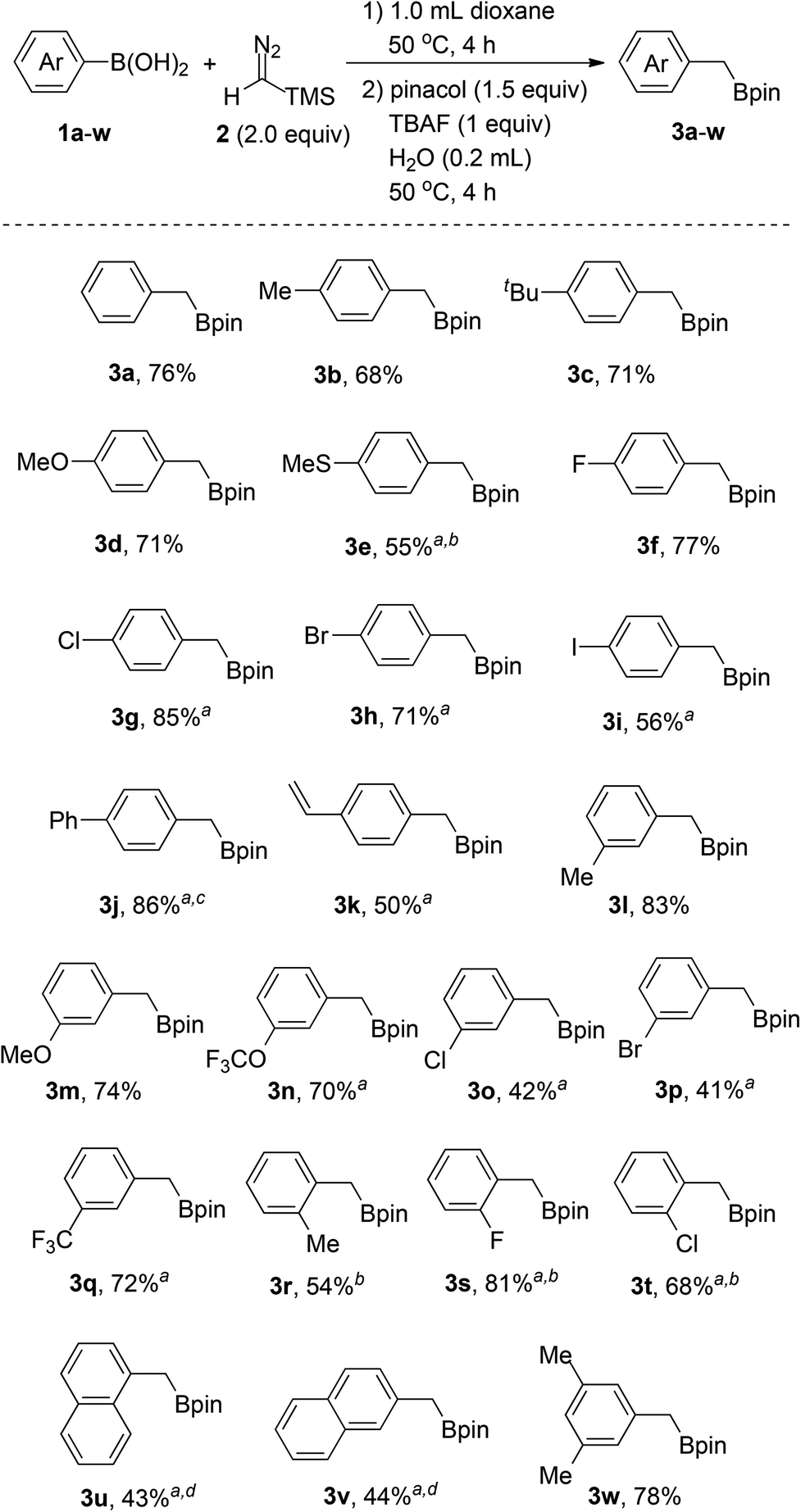 | ||
| Scheme 3 Substrate scope of arylboronic acids for the synthesis of benzyl boronates. The reaction was carried out with 1 and 2 in 1 mL solvent (0.4 mmol, 0.4 M) at 50 °C for 4 h and then pinacol (dissolved in 1 mL solvent), TBAF and water were added for another 4 h. All the yields refer to isolated yields with silica gel column chromatography if not otherwise noted. aThe reaction was carried out in 1.0 mL PhMe as the solvent instead of dioxane in step one. bThe reaction was carried out with 3.0 equiv. TMSCHN2. cThe reaction was carried out with 1.5 mL dioxane in step two. dThe reaction was carried out with 2.5 equiv. TMSCHN2. | ||
To demonstrate the practical usefulness of this reaction, a gram-scale experiment was performed with 4-chlorophenylboronic acid (1g). As shown in Scheme 4, the reaction proceeded to afford the benzyl boronate (3g) in good yield, which is similar to a small scale experiment.
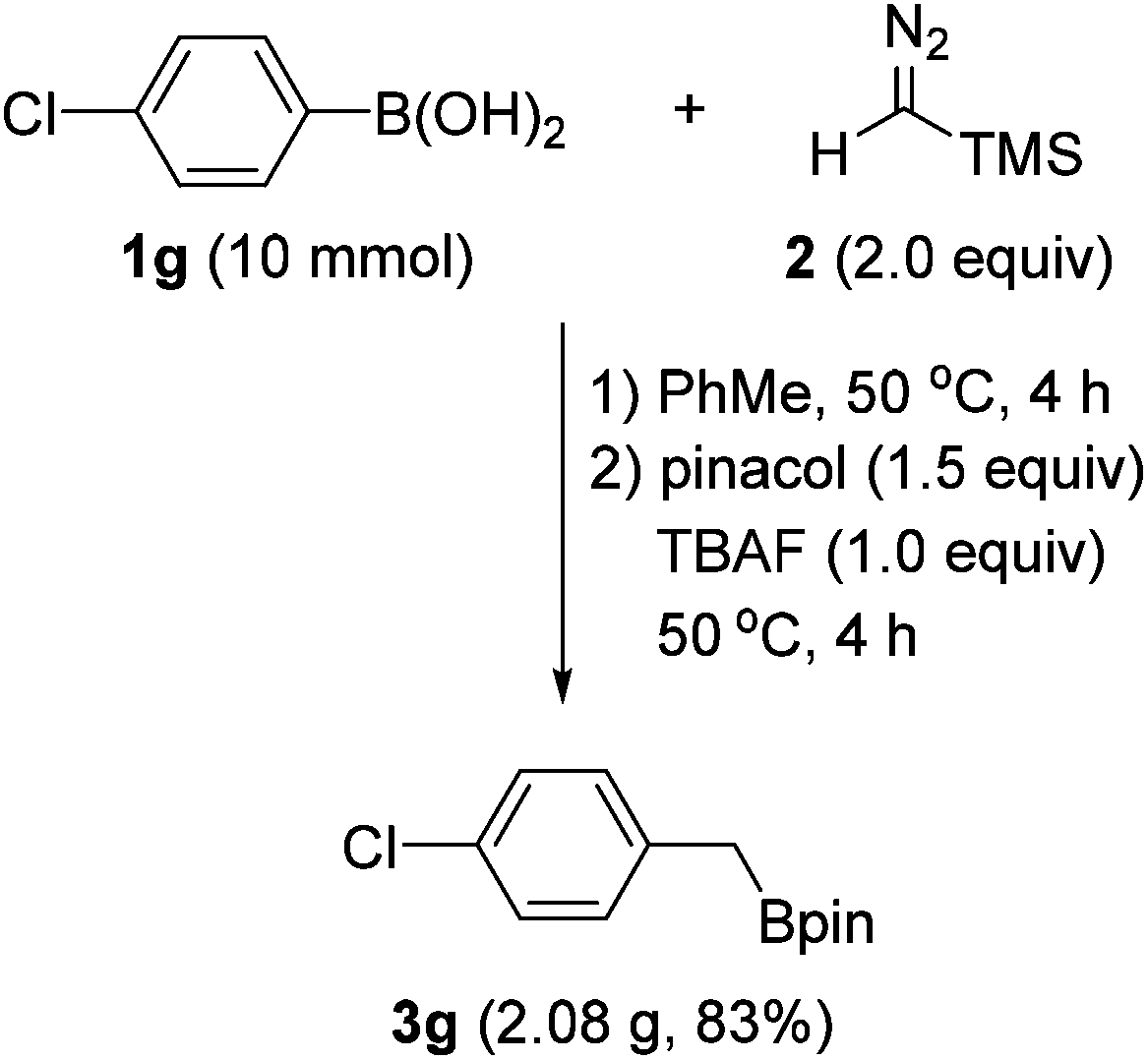 | ||
| Scheme 4 Gram-scale experiment. | ||
Having established an efficient entry to benzyl boronates, we then tried to explore their synthetic utility. As shown in Scheme 5, with hydrogen peroxide as the oxidant, benzylic alcohol could be accessed from the corresponding arylboronic acid in good yield by a two-step one-pot procedure.
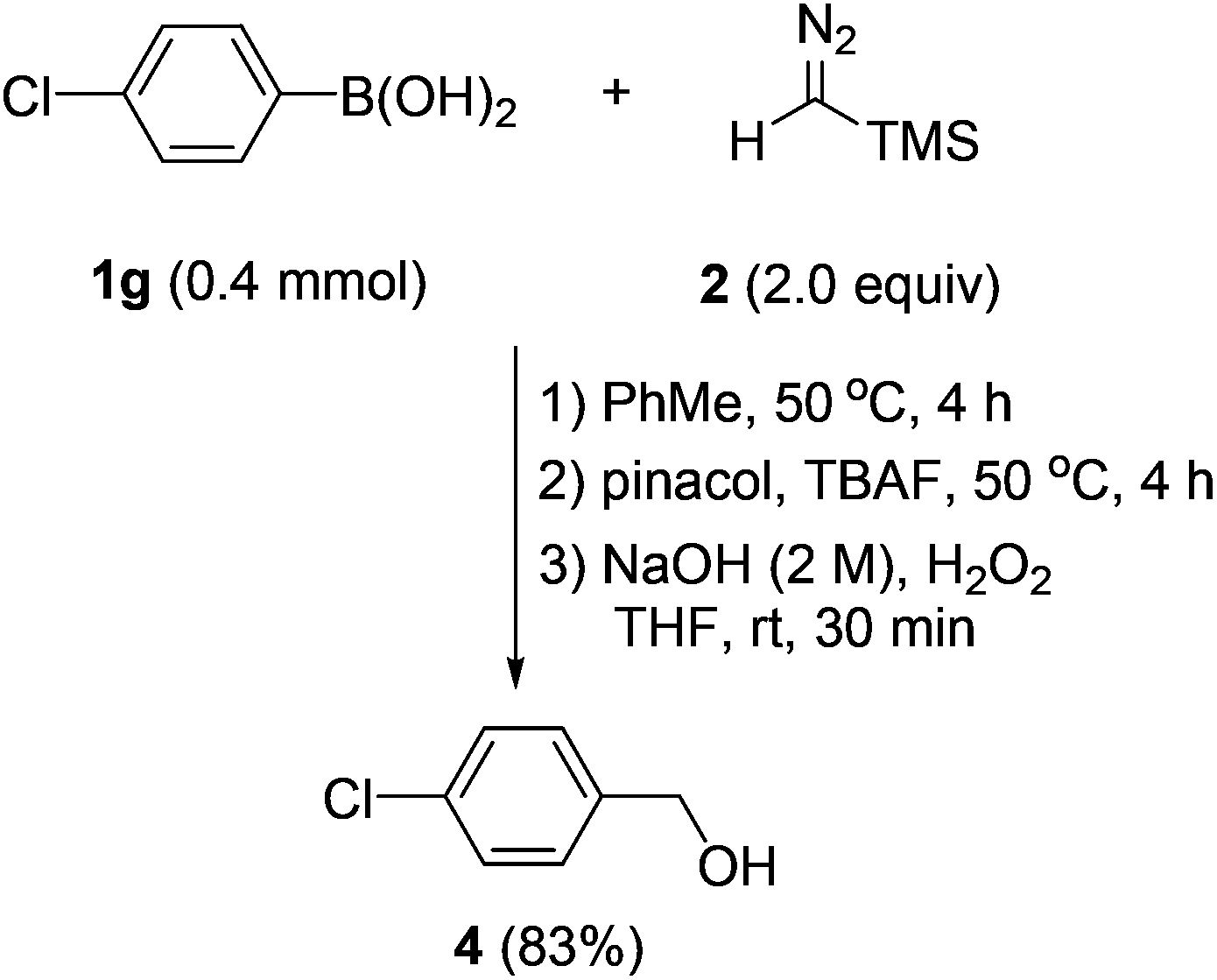 | ||
| Scheme 5 Conversion of arylboronic acid to benzylic alcohol in one-pot reaction. | ||
In summary, we have developed a transition-metal-free one-carbon homologation of arylboronic acids to synthesize benzyl boronates. This method is featured by mild reaction conditions, simple operation and commercially available reactants. The reaction has a wide substrate scope and good functional group tolerance, and it can be scaled up easily. With these advantages, it would be an attractive method to access benzyl boronates and find useful applications in organic synthesis.
Experimental section
General
All the reactions were performed under a nitrogen atmosphere in a Schlenk reaction flask. All solvents were distilled under a nitrogen atmosphere prior to use. Toluene, THF and 1,4-dioxane were dried over Na with the benzophenone-ketyl intermediate as the indicator. DCE was dried over CaH2. The boiling point of petroleum ether was between 60 and 70 °C. For chromatography, 200–300 mesh silica gel was employed. Chemical shifts for 1H NMR (400 MHz) and 13C{1H} NMR spectra are reported relative to the chemical shift of tetramethylsilane (TMS): chemical shifts (δ) were reported in ppm, and coupling constants (J) are in Hertz (Hz). The resonances for carbon atoms directly attached to boron were not observed due to quadrupolar relaxation. IR spectra are reported in wavenumbers, cm−1. For HRMS measurements, the mass analyzer is FT-ICR. PE: petroleum ether; EA: ethyl acetate. Unless otherwise noted, materials obtained from commercial suppliers were used without further purification.2-Benzyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3a)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 40:1), the product was isolated as a colorless oil (66 mg, 76%). 1H NMR (400 MHz, CDCl3) δ 7.26–7.20 (m, 2H), 7.18 (d, J = 6.9 Hz, 2H), 7.11 (t, J = 7.1 Hz, 1H), 2.29 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 138.7, 129.0, 128.3, 124.8, 83.4, 24.7.
:EtOAc = 40:1), the product was isolated as a colorless oil (66 mg, 76%). 1H NMR (400 MHz, CDCl3) δ 7.26–7.20 (m, 2H), 7.18 (d, J = 6.9 Hz, 2H), 7.11 (t, J = 7.1 Hz, 1H), 2.29 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 138.7, 129.0, 128.3, 124.8, 83.4, 24.7.
4,4,5,5-Tetramethyl-2-(4-methylbenzyl)-1,3,2-dioxaborolane (3b)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (63 mg, 68%). 1H NMR (400 MHz, CDCl3) δ 7.07 (d, J = 8.2 Hz, 2H), 7.04 (d, J = 8.2 Hz, 2H), 2.29 (s, 3H), 2.25 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 135.4, 134.1, 129.0, 128.9, 83.4, 24.7, 21.0.
2-(4-tert-Butylbenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3c)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a yellow oil (78 mg, 71%). 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J = 8.1 Hz, 2H), 7.11 (d, J = 8.1 Hz, 2H), 2.26 (s, 2H), 1.29 (s, 9H), 1.24 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 147.5, 135.4, 128.7, 125.2, 83.4, 34.2, 31.4, 24.8.
2-(4-Methoxybenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3d)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (70 mg, 71%). 1H NMR (400 MHz, CDCl3) δ 7.09 (d, J = 8.5 Hz, 2H), 6.79 (d, J = 8.6 Hz, 2H), 3.77 (s, 3H), 2.22 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 157.1, 130.5, 129.8, 113.8, 83.4, 55.2, 24.7.
4,4,5,5-Tetramethyl-2-(4-(methylthio)benzyl)-1,3,2-dioxaborolane (3e)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a pale yellow oil (58 mg, 55%). 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 8.2 Hz, 2H), 7.11 (d, J = 8.2 Hz, 2H), 2.45 (s, 3H), 2.25 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 135.9, 134.0, 129.5, 127.4, 83.5, 24.7, 16.5.
2-(4-Fluorobenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3f)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (73 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 7.14–7.10 (m, 2H), 6.95–6.87 (m, 2H), 2.25 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 160.8 (d, J = 241.9 Hz), 134.1 (d, J = 3.1 Hz), 130.2 (d, J = 7.6 Hz), 114.9 (d, J = 21.2 Hz), 83.5, 24.7.
2-(4-Chlorobenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3g)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (86 mg, 85%). 1H NMR (400 MHz, CDCl3) δ 7.19 (d, J = 8.3 Hz, 2H), 7.10 (d, J = 8.3 Hz, 2H), 2.25 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 137.2, 130.6, 130.3, 128.3, 83.6, 24.7.
2-(4-Bromobenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3h)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (84 mg, 71%). 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 8.3 Hz, 2H), 7.05 (d, J = 8.2 Hz, 2H), 2.23 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 137.7, 131.2, 130.7, 118.6, 83.6, 24.7.
2-(4-Iodobenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3i). The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (77 mg, 56%). 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 8.3 Hz, 2H), 6.93 (d, J = 8.3 Hz, 2H), 2.22 (s, 2H), 1.22 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 138.4, 137.2, 131.2, 89.7, 83.6, 24.7; IR (film) 2973, 1484, 1331, 1166, 1142, 1007, 968, 847 cm−1; HRMS (ESI) calcd for C13H19BIO2 [M + H]+, 345.0520; found, 345.0522.
2-(Biphenyl-4-ylmethyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3j)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a white solid (101 mg, 86%). 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 7.8 Hz, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.41 (t, J = 7.6 Hz, 2H), 7.30 (t, J = 7.7 Hz, 1H), 7.27–7.24 (m, 2H), 2.34 (s, 2H), 1.25 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 141.3, 137.8, 137.8, 129.4, 128.7, 127.0, 126.9, 126.8, 83.5, 24.8.
4,4,5,5-Tetramethyl-2-(4-vinylbenzyl)-1,3,2-dioxaborolane (3k)6b. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (49 mg, 50%). 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.1 Hz, 2H), 7.14 (d, J = 8.0 Hz, 2H), 6.67 (dd, J = 17.6, 10.9 Hz, 1H), 5.67 (d, J = 17.6 Hz, 1H), 5.15 (d, J = 10.9 Hz, 1H), 2.28 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 138.5, 136.8, 134.3, 129.1, 126.2, 112.4, 83.5, 24.7.
4,4,5,5-Tetramethyl-2-(3-methylbenzyl)-1,3,2-dioxaborolane (3l)6b. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (77 mg, 83%). 1H NMR (400 MHz, CDCl3) δ 7.12 (t, J = 7.4 Hz, 1H), 6.98 (d, J = 7.9 Hz, 2H), 6.93 (d, J = 7.4 Hz, 1H), 2.30 (s, 3H), 2.25 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 138.4, 137.6, 129.8, 128.1, 125.9, 125.6, 83.3, 24.7, 21.3.
2-(3-Methoxybenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3m)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (73 mg, 74%). 1H NMR (400 MHz, CDCl3) δ 7.15 (t, J = 7.9 Hz, 1H), 6.79–6.73 (m, 2H), 6.67 (dd, J = 8.2, 2.4 Hz, 1H), 3.78 (s, 3H), 2.27 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 159.5, 140.2, 129.2, 121.5, 114.6, 110.4, 83.5, 55.1, 24.7.
4,4,5,5-Tetramethyl-2-(3-(trifluoromethoxy)benzyl)-1,3,2-dioxaborolane (3n). The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (85 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 7.26–7.21 (m, 1H), 7.12–7.05 (m, 2H), 6.97 (d, J = 8.1 Hz, 1H), 2.31 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 149.2, 141.0, 129.4, 127.5, 121.5, 120.5 (q, J = 256.7 Hz), 117.4, 83.6, 24.7; IR (film) 2979, 1333, 1260, 1217, 1163, 1144, 842 cm−1; HRMS (ESI) calcd for C14H18BF3O3 [M + H]+, 303.1376; found, 303.1379.
2-(3-Chlorobenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3o)6c. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a pale yellow oil (42 mg, 42%). 1H NMR (400 MHz, CDCl3) δ 7.17 (s, 1H), 7.14 (d, J = 7.6 Hz, 1H), 7.09 (d, J = 8.2 Hz, 1H), 7.05 (d, J = 7.4 Hz, 1H), 2.26 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 140.8, 133.9, 129.4, 129.1, 127.2, 125.1, 83.6, 24.7.
2-(3-Bromobenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3p)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (49 mg, 41%). 1H NMR (400 MHz, CDCl3) δ 7.33 (s, 1H), 7.26–7.22 (m, 1H), 7.12–7.07 (m, 2H), 2.26 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 141.1, 132.0, 129.7, 128.0, 127.7, 122.3, 83.6, 24.7.
4,4,5,5-Tetramethyl-2-(3-(trifluoromethyl)benzyl)-1,3,2-dioxaborolane (3q). The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (82 mg, 72%). 1H NMR (400 MHz, CDCl3) δ 7.44 (s, 1H), 7.39–7.31 (m, 3H), 2.35 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 139.7, 132.4, 130.4 (q, J = 31.7 Hz), 128.6, 125.7 (q, J = 3.6 Hz), 124.4 (q, J = 270.0 Hz), 121.8 (q, J = 3.9 Hz), 83.7, 24.7; IR (film) 2976, 1329, 1167, 1143, 1124, 1075, 700 cm−1; HRMS (ESI) calcd for C14H19BF3O2 [M + H]+, 287.1430; found, 287.1429.
4,4,5,5-Tetramethyl-2-(2-methylbenzyl)-1,3,2-dioxaborolane (3r)6a. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (50 mg, 54%). 1H NMR (400 MHz, CDCl3) δ 7.14–7.01 (m, 4H), 2.27 (s, 3H), 2.25 (s, 2H), 1.22 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 137.5, 135.9, 129.7, 129.4, 125.8, 125.1, 83.3, 24.7, 20.1.
2-(2-Fluorobenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3s)6c. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (77 mg, 81%). 1H NMR (400 MHz, CDCl3) δ 7.19 (t, J = 7.5 Hz, 1H), 7.11 (dd, J = 13.7, 6.6 Hz, 1H), 7.04–6.95 (m, 2H), 2.26 (s, 2H), 1.24 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 161.0 (d, J = 243.6 Hz), 131.3 (d, J = 5.0 Hz), 126.6 (d, J = 8.0 Hz), 126.0 (d, J = 16.7 Hz), 123.8 (d, J = 3.6 Hz), 114.9 (d, J = 22.2 Hz), 83.6, 24.7.
2-(2-Chlorobenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3t)6d. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (68 mg, 68%). 1H NMR (400 MHz, CDCl3) δ 7.30 (dd, J = 7.8, 1.4 Hz, 1H), 7.22 (dd, J = 7.5, 1.6 Hz, 1H), 7.14 (td, J = 7.4, 1.4 Hz, 1H), 7.07 (td, J = 7.6, 1.8 Hz, 1H), 2.38 (s, 2H), 1.24 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 137.5, 133.9, 130.8, 129.0, 126.7, 126.5, 83.6, 24.7.
4,4,5,5-Tetramethyl-2-(naphthalen-1-ylmethyl)-1,3,2-dioxaborolane (3u)6a. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (46 mg, 43%). 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 7.8 Hz, 1H), 7.84–7.79 (m, 1H), 7.65 (d, J = 7.5 Hz, 1H), 7.50–7.42 (m, 2H), 7.36 (q, J = 7.1 Hz, 2H), 2.69 (s, 2H), 1.19 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 135.6, 133.8, 132.5, 128.5, 126.5, 125.8, 125.4, 125.3, 124.5, 83.5, 24.7.
4,4,5,5-Tetramethyl-2-(naphthalen-2-ylmethyl)-1,3,2-dioxaborolane (3v)13. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (47 mg, 44%). 1H NMR (400 MHz, CDCl3) δ 7.78–7.71 (m, 3H), 7.61 (s, 1H), 7.43–7.32 (m, 3H), 2.45 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 136.3, 133.8, 131.5, 128.2, 127.7, 127.6, 127.3, 126.6, 125.7, 124.7, 83.5, 24.7.
2-(3,5-Dimethylbenzyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3w)9. The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 40:1), the product was isolated as a colorless oil (77 mg, 78%). 1H NMR (400 MHz, CDCl3) δ 6.80 (s, 2H), 6.76 (s, 1H), 2.26 (s, 6H), 2.21 (s, 2H), 1.23 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 138.4, 137.6, 126.9, 126.6, 83.4, 24.7, 21.3.
(4-Chlorophenyl)methanol (4). The title compound was prepared via the general procedure. After purification by silica gel column chromatography (petroleum ether
:EtOAc = 3:1), the product was isolated as a white solid (48 mg, 83%). 1H NMR (400 MHz, CDCl3) δ 7.30 (q, J = 8.5 Hz, 1H), 4.65 (s, 2H), 1.87 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 139.3, 133.4, 128.7, 128.3, 64.5.
Acknowledgements
The project was supported by the 973 Program (no. 2012CB821600) and the National Nature Science Foundation of China (Grant 21272010 and 21332002).Notes and references
- For selected recent reviews, see:
(a) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed
; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed
-
(a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2009, 110, 890 CrossRef PubMed
-
(a) H. C. Brown and T. E. Cole, Organometallics, 1983, 2, 1316 CrossRef CAS
- H. C. Brown, N. G. Bhat and V. Somayaji, Organometallics, 1983, 2, 1311 CrossRef CAS
-
(a) K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179 CrossRef CAS
-
(a) T. Ishiyama, Z. Oohashi, T. Ahiko and N. Miyaura, Chem. Lett., 2002, 31, 780 CrossRef
- C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528 CrossRef CAS PubMed
- T. C. Atack, R. M. Lecker and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 9521 CrossRef CAS PubMed
- For an Mg-catalyzed cross-coupling reaction, see: C. Pintaric, S. Olivero, Y. Gimbert, P. Y. Chavant and E. Duñach, J. Am. Chem. Soc., 2010, 132, 11825 CrossRef CAS PubMed
-
(a) S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168 CrossRef CAS
- S. H. Cho and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8157 CrossRef CAS PubMed
-
(a) M. A. Larsen, C. V. Wilson and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 8633 CrossRef CAS PubMed
- Z.-C. Cao, F.-X. Luo, W.-J. Shi and Z.-J. Shi, Org. Chem. Front., 2015, 2, 1505 RSC
- H. Li, L. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 2943 CrossRef CAS PubMed
-
(a) C. Peng, W. Zhang, G. Yan and J. Wang, Org. Lett., 2009, 11, 1667 CrossRef CAS PubMed
- For related reactions, see:
(a) J. Barluenga, M. Tomas-Gamasa, F. Aznar and C. Valdes, Nat. Chem., 2009, 1, 494 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of substrates, characterization data, and 1H, 13C NMR, MS and IR spectra. See DOI: 10.1039/c6qo00141f |
| This journal is © the Partner Organisations 2016 |