DOI:
10.1039/C6QO00315J
(Research Article)
Org. Chem. Front., 2016,
3, 1286-1294
Homochiral association of binuclear trans-bis(β-iminoaryloxy)palladium(II) complexes doubly linked with m-xylylene spacers: drastic linker-dependence of the association chirality of chiral clothespin-shaped molecules†
Received
30th June 2016
, Accepted 5th August 2016
First published on 8th August 2016
Abstract
A significant effect of the linker on association chirality has been observed during the interpenetrative dimeric association of clothespin-shaped Pd complexes bearing either rigid or flexible linkers. Optically pure, planar, chiral binuclear trans-bis(β-iminoaryloxy)palladium(II) complexes doubly linked with m-xylylene spacers (1 and 2) were newly prepared by the reaction of Pd salts with the corresponding spacer-linked diimine ligands, and their rigid H-shaped conformation was confirmed by single-crystal XRD analysis. Analysis of the concentration dependence of optically pure and racemic mixtures of 1 in CDCl3 based on 1H NMR revealed that this complex exhibits homochiral association with an equilibrium constant ratio, [KD(homo)/KD(hetero)], of 1.2 × 102. This result is completely opposite to the reported heterochiral association of the pentamethylene-linked analogue 3, which has a KD(homo)/KD(hetero) value of 0.033 under the same conditions. Based on XRD analysis and molecular modelling, the drastic linker-dependence of the association chirality of 1 and 3 has been rationalized by proposing contrasting interpenetrative stacking modes during the dimeric association of these clothespin-shaped molecules. An outward double stacking mode, leading to homochiral association, was determined to be well-suited to the H-shaped, constrained conformation of the rigidly-linked 1, while an inward single stacking mode providing heterochiral association is preferred based on the A-shaped tilted conformation of the flexibly-linked 3.
Introduction
Control of supramolecular chirality in molecular association and aggregation is important with regard to the design and creation of novel chirality-driven molecular functions.1 The molecular association/aggregation of chiral compounds generally proceeds in a homochiral manner in solution,2 gel,2c,3 liquid crystalline,4 and crystalline states,5 because high degrees of symmetry and regularity are required for molecular alignment. For this reason, the heterochiral association/aggregation of chiral molecular units has been recognized as unfavorable with regard to molecular assembly and there have been only a few examples of species that undergo this phenomenon, including [Ru(bpy)(eilatin)](PF6)2,6 bis(2-hydroxyphenyl)diamides7 and norbornene-fused porphyrins.8 Understanding the effects of the structure on chirality control would not only provide significant information regarding the chemistry of supramolecular chirality, but could also contribute to the development of forward-looking technologies based on molecular devices that are highly sensitive to molecular chirality. However, despite the high profile of several chiral self-assembling molecules, the correlation between the molecular structure and association/aggregation chirality is poorly understand, since there have not yet been any systematic studies on this topic.
As part of a program aimed at the development of new functional materials based on the controlled d–π conjugation of flexible d8 transition metal planar platforms,9–12 we have been investigating the association11 and aggregation12 properties of clothespin-shaped binuclear trans-bis(β-iminoaryloxy)palladium(II) complexes doubly linked with pentamethylene spacers, in which two co-facial coordination blades undergo interpenetrative, intermolecular π-stacking stabilization to generate specific association/aggregation properties in conjunction with dynamic molecular mobility. This effect leads to appropriate conformational adjustments with the support of the flexible linkers. One of the most important aspects of this phenomenon is that the interpenetrative association/aggregation of these chiral clothespin-shaped complexes proceeds in a strict heterochiral manner, in which the units undergo regular molecular alignment to afford RS dimers in association,11 and RSRSRS… supramolecular polymers in aggregation.12 This rare case of heterochiral molecular alignment results from dynamic conformational alterations of the clothespin-shaped complexes. The driving force for these alterations is the synergistic effect arising from the flexibility of the coordination blades and the pentamethylene linkers, which in turn results from the weak d–π conjugation of the coordination platforms and the low rotation barrier for the carbon–carbon single bonds of the linkers, respectively. Understanding the effects of the structure on the dynamic molecular behavior of a series of clothespin-shaped complexes would provide significant information that should assist in the creation of functional molecules exhibiting unique association/aggregation properties.
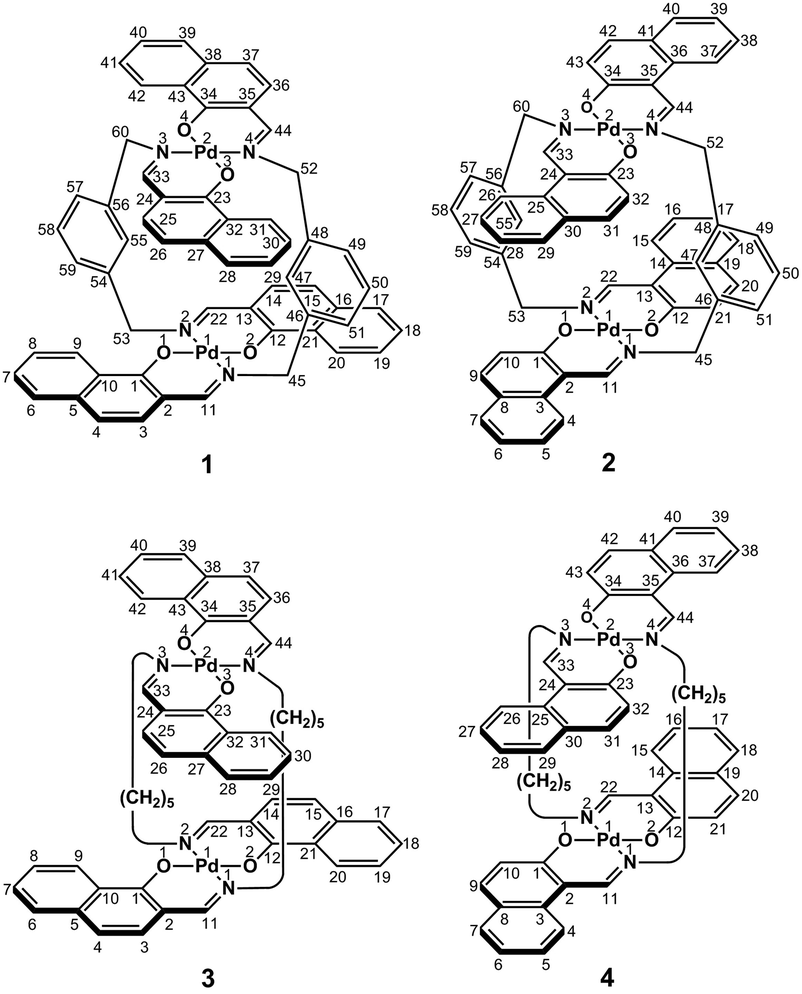
In the present study, we focused on the correlation between the conformational mobility of linker moieties and the association properties of clothespin-shaped palladium complexes. For this purpose, novel binuclear trans-bis(β-iminoaryloxy)palladium(II) complexes (1 and 2) incorporating rigid m-xylylene linkers were newly synthesized, and compared with pentamethylene-linked analogues (3 and 4)11 in terms of molecular structure and association properties in the solution state (Scheme 1). The m-xylylene-linked complex 1, having Z-shaped coordination blades, exhibited homochiral association properties in CDCl3, in contrast to the heterochiral association properties of the pentamethylene-linked analogue 3 under the same conditions. This represents the first reported case of a drastic change in association chirality following a slight molecular alteration of the association units. Single-crystal X-ray diffraction (XRD) studies revealed that the greater degree of molecular constraint in the case of 1 resulting from the conformational rigidity of the m-xylylene linkers is the key factor in obtaining the inversion of the association chirality. This paper describes the synthesis, structure, and homochiral association properties of these m-xylylene-linked palladium complexes, paying special attention to the mechanistic rationale with respect to the linker-dependence of the association chirality of these clothespin-shaped complexes.
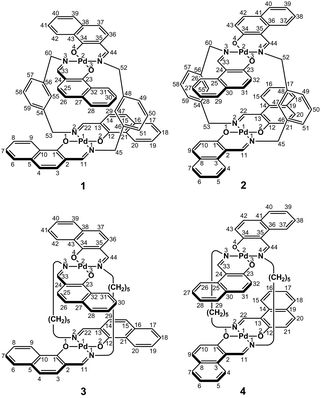 |
| | Scheme 1 Rigid or flexible dinuclear clothespin-shaped molecules 1–4. | |
Results and discussion
Synthesis and the structure of clothespin-shaped binuclear palladium complexes bearing rigid m-xylylene linkers
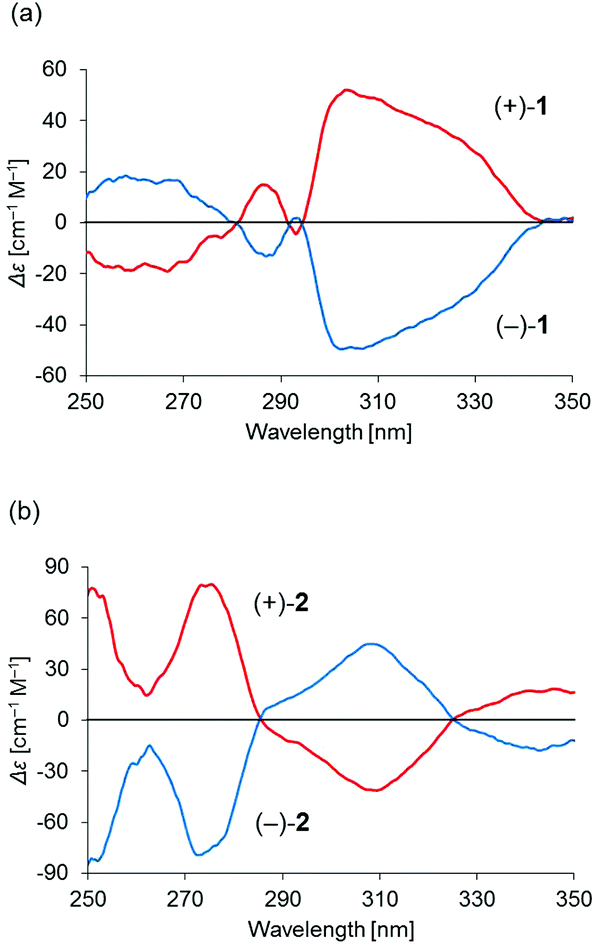
Binuclear palladium complexes 1 and 2 with anti-conformations were prepared via the reaction of Pd(OAc)2 with an equimolar amount of the corresponding N,N′-bis(β-hydroxyarylmethylene)-m-xylylenediamine in the presence of N,N,N′,N′-tetramethylethylenediamine in boiling toluene. The reactions gave anti-forms exclusively without generating any of the corresponding syn-forms. This is in contrast to the synthesis of the pentamethylene-linked analogues 3 and 4 with the corresponding ligands, in which both syn- and anti-forms were produced under similar reaction conditions.11 Complexes 1 and 2 were characterized by 1H and 13C nuclear magnetic resonance spectroscopy (NMR), Fourier-transform infrared spectroscopy (FT-IR), high-resolution mass spectrometry (HRMS), and nuclear Overhauser effect spectroscopy (NOESY). Optically pure (100% ee) complexes of 1 and 2 were obtained from the racemic mixtures by employing a preparative high performance liquid chromatography (HPLC) system with a chiral column. The absolute configurations of the (+)-forms were tentatively assigned as R by analogy to the configurations of other clothespin-shaped binuclear trans-bis(β-iminoaryloxy) transition metal complexes.11,12 The circular dichroism (CD) spectra of optically pure 1 and 2 in CHCl3 are shown in Fig. 1.
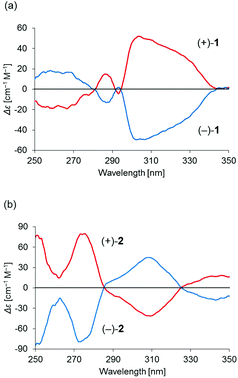 |
| | Fig. 1 CD spectra of 2.00 × 10−5 M solutions of optically pure (a) 1 and (b) 2 in CHCl3. | |
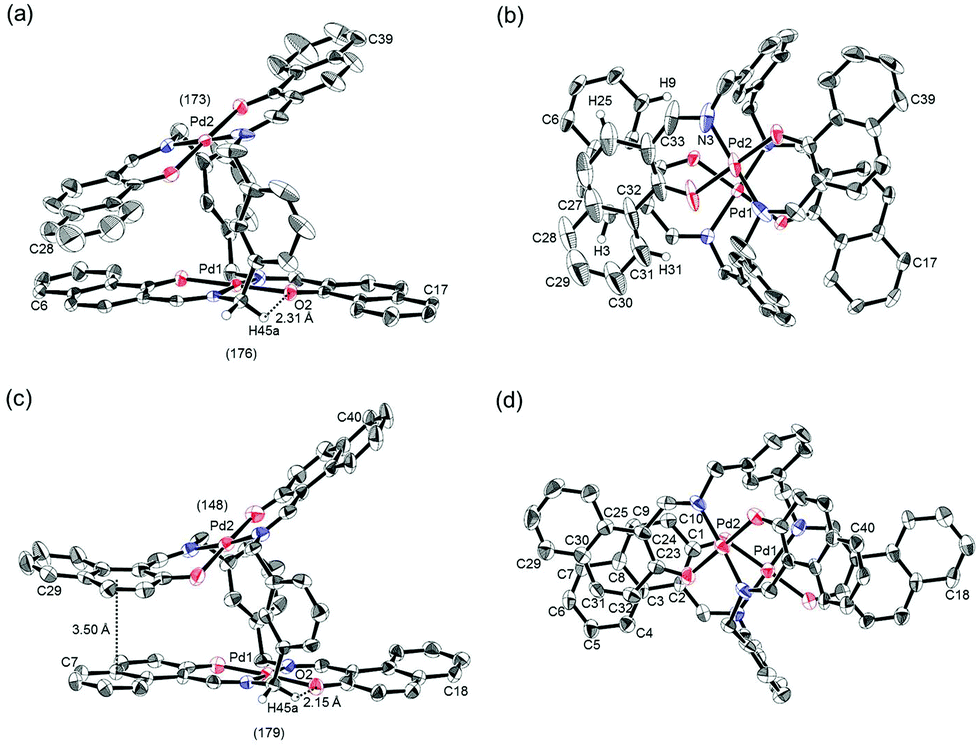
The trans-coordination, anti-conformation, and clothespin-shaped three-dimensional overall structures of complexes 1 and 2 were unequivocally established by XRD analysis of crystals obtained by recrystallization from CHCl3. The crystallographic data and ORTEP drawings of 1 and 2 are summarized in Table 1 and Fig. 2. The side view of 1 indicates that the co-facial naphthalene rings on the upper and lower Z-shaped coordination platforms are situated in a declined manner without intermolecular π-stacking stabilization in the crystalline state (Fig. 2a). The C(6)–Pd(1)–C(17) and C(28)–Pd(2)–C(39) angles for 1, showing a macroscopic view of the planarity of a trans-bis(iminonaphthoxy)Pd(II) coordination platform, are 176° and 173°, respectively (Fig. 2a). This is in contrast to the reported molecular structure of the pentamethylene-linked analogue 3, which is stabilized by significant intermolecular π-stacking interactions between co-facial naphthalene rings as a result of bent platforms and a highly tilted molecular conformation.11 Based on these results, it is apparent that the doubly-linked m-xylylene moieties of 1 strongly inhibit the conformational latitude of the co-facial coordination blades due to their conformational rigidity, impeding firm intermolecular stacking. The specific planarity of the coordination platforms of 1 is the result of compensation for the loss of the stacking stabilization arising from the linker-induced conformational constraint of the clothespin-shaped molecules.
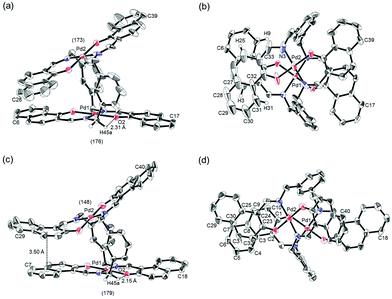 |
| | Fig. 2 ORTEP representations of (a, b) (R)-1 and (c, d) (R)-2 as their racemic crystals, showing (a, c) side and (b, d) overhead views. The angles of (a) C(6)–Pd(1)–C(17)/C(28)–Pd(2)–C(39) and (c) C(7)–Pd(1)–C(18)/C(29)–Pd(2)–C(40) are provided in parentheses. Stacking and hydrogen-bonding distances are indicated for each structure. Thermal ellipsoids are shown at the 50% probability level. | |
Table 1 Crystallographic data and structure refinements for (±)-1 and (±)-2
| Parameter |
(±)-1·CHCl3 |
(±)-2 |
|
R
1 = ∑(|Fo| − |Fc|)/∑(|Fo|).
wR2 = [∑[w(Fo2 − Fc2)2]/∑w(Fo2)2]1/2.
|
| Formula |
C61H45Cl3N4O4Pd2 |
C60H44N4O4Pd2 |
| Formula weight |
1217.21 |
1097.83 |
| Temperature (K) |
113 |
113 |
| Crystal color, habit |
Yellow, block |
Yellow, platelet |
| Crystal size, mm |
0.60 × 0.60 × 0.30 |
0.20 × 0.16 × 0.04 |
| Crystal system |
Monoclinic |
Orthorhombic |
| Space group |
C2/c (#15) |
Pccn (#56) |
|
a, Å |
27.618(2) |
20.2007(6) |
|
b, Å |
13.0551(9) |
25.2676(7) |
|
c, Å |
30.802(2) |
31.3498(8) |
|
α, ° |
90 |
90 |
|
β, ° |
113.650(2) |
90 |
|
γ, ° |
90 |
90 |
|
V, Å3 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 173(2) 173(2) |
16001.7(8) |
|
Z value |
8 |
12 |
|
D
calcd, g cm−3 |
1.589 |
1.367 |
|
μ (MoKα), cm−1 |
9.197 |
7.238 |
|
F(000) |
4912.00 |
6672.00 |
| 2θmax, ° |
55.0 |
54.9 |
| No. of reflections measured |
89431 |
270470 |
| No. of observed reflections |
11605 |
18213 |
| No. of variables |
667 |
946 |
|
R
1 (I > 2σ(I))a |
0.073 |
0.069 |
| wR2 (all reflns)b |
0.17 |
0.20 |
| Goodness of fit |
1.05 |
1.04 |
Complex 2 undergoes significant intermolecular stacking interactions between the co-facial naphthalene rings on the I-shaped coordination platforms (Fig. 2c). The side and overhead views of this complex show that the entire naphthalene ring of the lower blade [C(1)–C(2)–C(3)–C(4)–C(5)–C(6)–C(7)–C(8)–C(9)–C(10)] interacts with the inner benzene ring of the upper blade [C(23)–C(24)–C(25)–C(30)–C(31)–C(32)], with a typical π-stacking distance of 3.50 Å (Fig. 2c and d). In order to obtain significant stacking interactions, the upper blade of 2 is highly bent with a C(29)–Pd(2)–C(40) angle of 148°, whereas the lower blade maintains a high degree of coordination planarity with a C(7)–Pd(1)–C(18) angle of 179° (Fig. 2c). The contrasting stacking scenario in the case of 2 can be ascribed to the I-shaped coordination platforms, which enable stacking stabilization following only a slight conformational change of the rigid structure.
The other relevant interaction in the clothespin-shaped molecules is the intramolecular contact between the α-protons of nitrogen atoms and remote oxygen atoms of the counterpart iminoaryloxy ligands. The H(45a)/H(53a)/H(52a)/H(60a) atoms of 1 interact with the remote O(2)/O(1)/O(3)/O(4) atoms with typical hydrogen-bonding distances of 2.31, 2.21, 2.21, and 2.25 Å, respectively. Complex 2 undergoes similar hydrogen bonding interactions between H(45a)–O(2) (2.15 Å), H(53a)–O(1) (2.28 Å), H(52a)–O(3) (2.23 Å), and H(60a)–O(4) (2.39 Å).
Static molecular mobility in the solution state
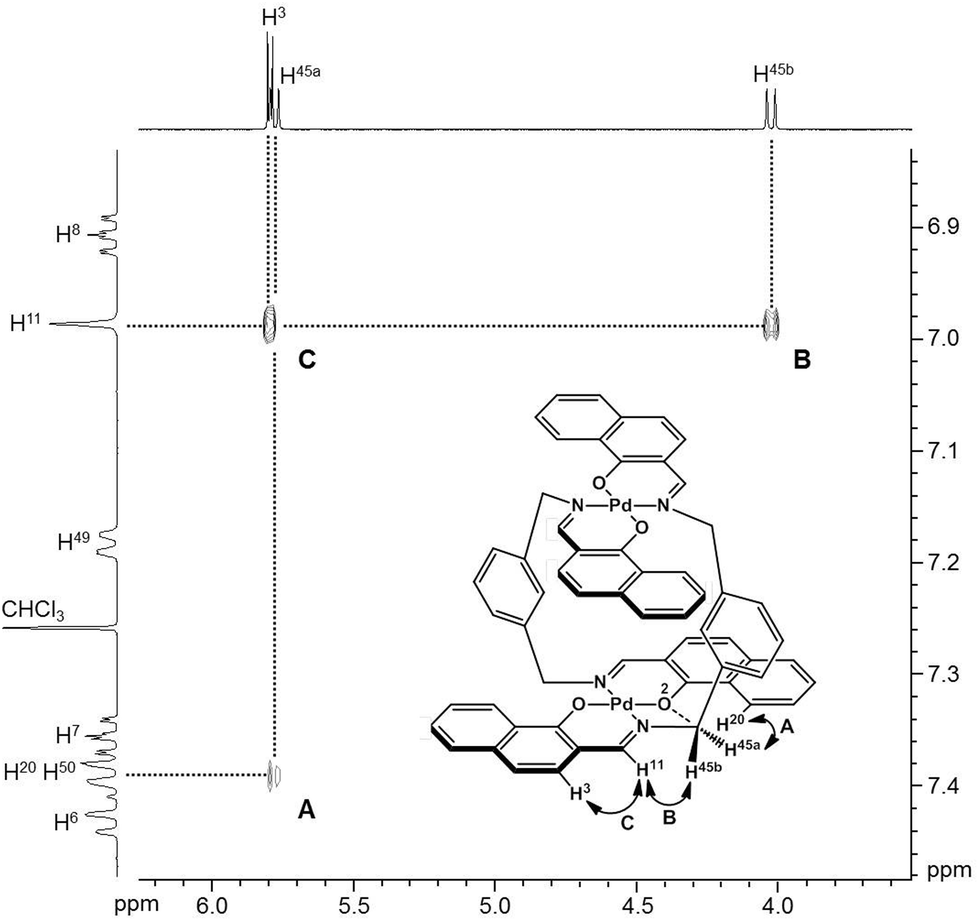
To obtain insight into the dynamic behavior of these m-xylylene-linked clothespin-shaped molecules in the solution state, the molecular mobilities of 1 and 2 in solution were examined by means of 1H NMR. The 1H NMR (500 MHz) spectra of 1 and 2 in CDCl3 exhibit two doublet signals at 5.78 and 4.03 ppm (for 1) or 5.83 and 4.19 ppm (for 2), which are assigned to the geminal protons (H45a/H53a/H52a/H60a and H45b/H53b/H52b/H60b) at the α-position of the nitrogen atoms, as shown in the upper vertical axes in Fig. 3 and S1, ESI.† The NCHaHb protons of 1 were assigned on the basis of the NOESY (500 MHz) spectra (Fig. 3), in which an intense remote correlation is observed between the H45a and H20 signals (correlation A) as well as the expected correlations between H45b–H11 (B) and H11–H3 (C). The clear splitting and extreme down-field shift of the H45a/H53a/H52a/H60a protons indicate that complexes 1 and 2 both undergo multiple intramolecular H-bonding fixation of the coordination platforms with remote oxygen atoms in the solution state, similar to those determined for the crystalline state on the basis of XRD analysis (Fig. 2a and c). This fixation would be expected to suppress the random conformational mobility of the two trans-bis(β-iminoaryloxy)Pd(II) coordination blades, leading to the association properties discussed in the following section.
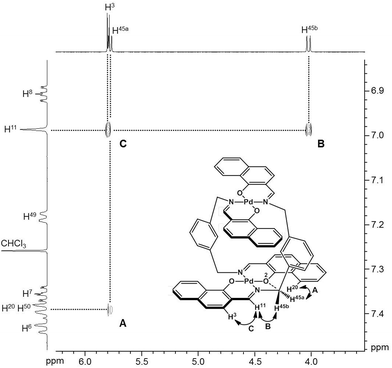 |
| | Fig. 3 NOESY spectrum (500 MHz) of 1 in CDCl3 (298 K, mixing time = 0.700 s). | |
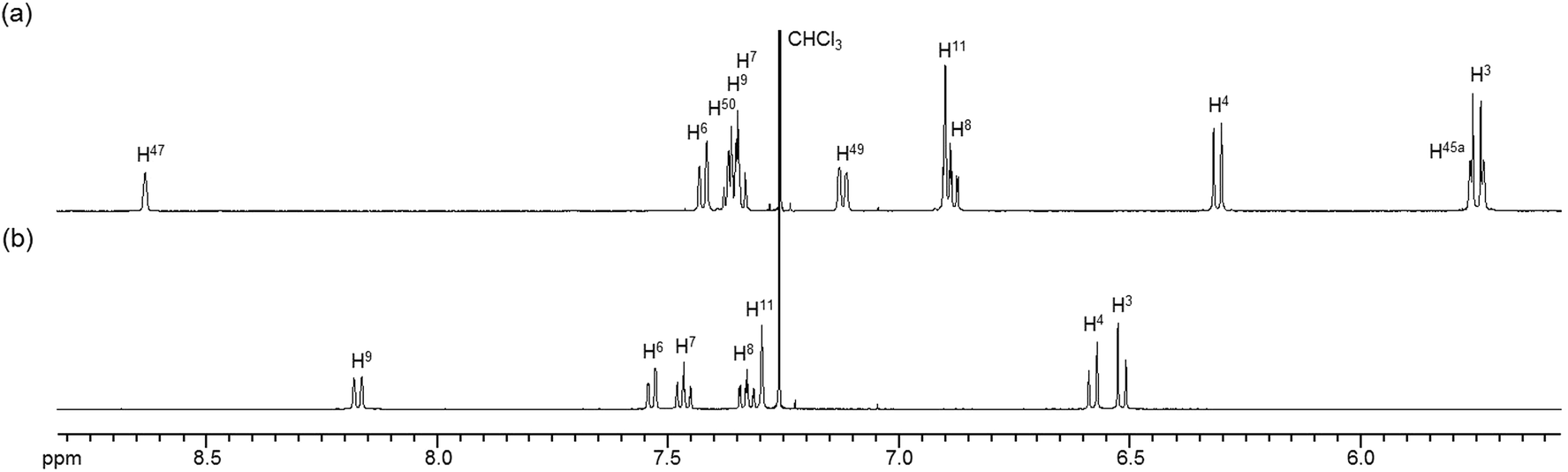
The 1H NMR spectra of the aromatic regions of the m-xylylene-linked complex 1 and the pentamethylene-linked analogue 3, obtained in CDCl3 at 298 K, are compared in Fig. 4. The signals generated by the aromatic protons H3, H4, H6, H7, H8 and H9 on the coordination platforms of 1 undergo a remarkable upfield shift compared to those of 3, which can be attributed to the especially strong shielding effect of the co-facial aromatic ring arising from the rigid conformation of 1. According to the molecular structure determined by XRD analysis (Fig. 2a and b), the extremely high upfield shifts of the H3 and H9 signals of 1 result from the rigid conformation of this complex, as shown in Fig. 5a, such that these two aromatic protons experience strong shielding due to the ring current from the outer benzene ring [C(27)–C(28)–C(29)–C(30)–C(31)–C(32)] in the case of H3 and the imine moiety [C(33)–N(3)] in the case of H9. The relative downfield shifts of the H3 and H9 signals of 3 can also be explained by the XRD data for this complex (Fig. S3a†),11 which show that a much more highly tilted and stacked conformation is formed by intermolecular π-stacking interactions between the co-facial aromatic rings of the coordination platforms due to flexibility of the pentamethylene linkers. As a result of the strong stacking interactions between the outer benzene ring of the upper blade and the entire naphthalene ring of the lower blade, protons H3 and H9 in 3 are not significantly affected by shielding from the ring current of the benzene ring or the imine moiety, as can be seen from Fig. 5b.
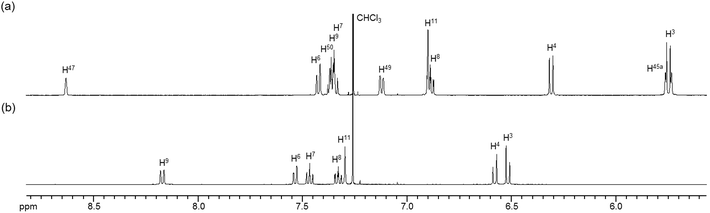 |
| | Fig. 4
1H NMR spectra (500 MHz) of the aromatic region of (a) m-xylylene-linked complex 1 and (b) pentamethylene-linked analogue 3 in CDCl3 (6.0 × 10−3 M) at 298 K. | |
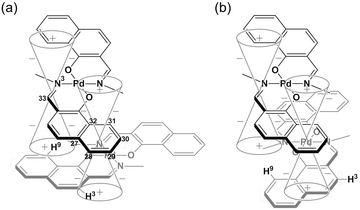 |
| | Fig. 5 Schematic representation of the shielding effects of (a) 1 and (b) 3. | |
In contrast, the chemical shifts of the signals from the aromatic protons H4, H5, H6, H7, H9 and H10 of the m-xylylene-linked complex 2, having an I-shaped coordination platform, are close to those of the pentamethylene-linked analogue 4, as is evident in their respective 1H NMR spectra (Fig. S2†). Based on the XRD analysis of 3 and 4, the contrasting similarity in the chemical shifts of complexes 2 and 4, having I-shaped blades, can be ascribed to the conformational resemblance of these complexes, both of which have significant intramolecular π-stacking interactions between the inner benzene ring in the upper blade and the entire naphthalene ring in the lower blade (Fig. 2c, d and S3c, d†), leading to similar shielding or deshielding patterns in 2 and 4.
Variable-temperature 1H NMR analysis of toluene-d8 solutions demonstrated that the aromatic and aliphatic proton signals of both 1 and 2 are not split and exhibit shifts even at 188 K (Fig. S4†). This result is in contrast to the observation that the aromatic signals of the pentamethylene-linked analogues 3 and 411 as well as the 3-azaheptamethylene-linked analogues10 are clearly split into two broad signals representing the open and closed sides in the A-shaped conformers at 193 and 298 K, respectively. Given the above experimental results obtained by 1H NMR analysis, we can be reasonably certain that the m-xylylene-linked complexes are highly static in the solution state owing to the molecular constraint arising from the rigid linkers. These complexes thus maintain the open-mouth, rigid conformations of the clothespin-shaped structure. Conversely, the flexible pentamethylene-linked analogues reportedly exhibit dynamic flapping and twisting motions based on rapid consecutive intramolecular association/dissociation of aromatic moieties in the co-facial coordination blades enhanced by the significant flexibility of the linkers under similar conditions.
Linker-dependence of the association chirality of clothespin-shaped complexes
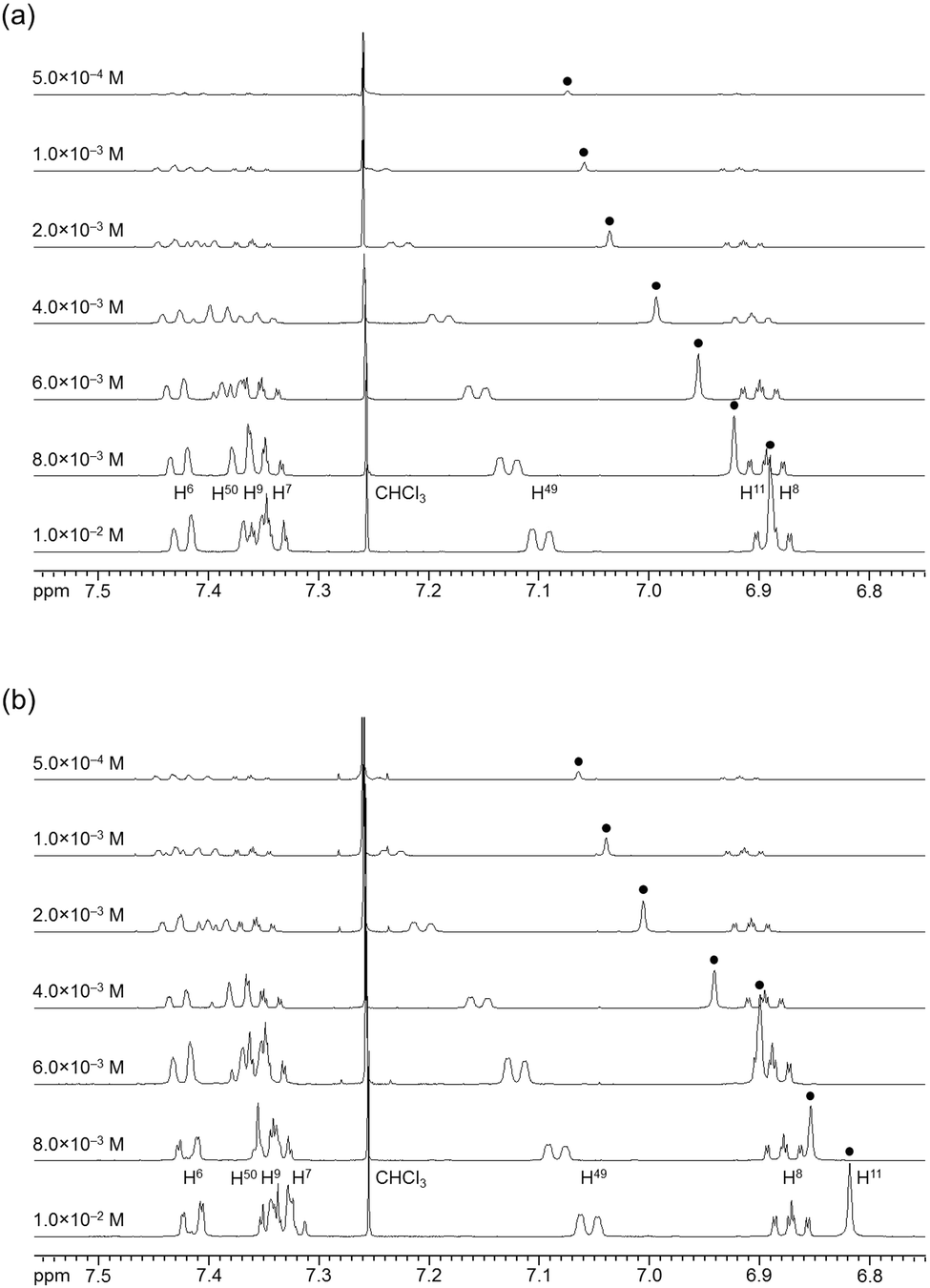
The association behavior of the m-xylylene-linked complex 1, bearing Z-shaped coordination blades, in the solution state was examined by means of 1H NMR analysis. Fig. 6 shows the concentration-dependent changes in the 1H NMR spectra of the aromatic region obtained from solutions of racemic (±)- (0% ee) and optically pure (+)-1 (100% ee) in CDCl3 at 293 K. All signals originating from aromatic protons for the (±)- and (+)-1 complexes undergo a significant upfield shift when the concentration is increased from 5.0 × 10−4 to 1.0 × 10−2 M. Notably, the imine proton (H11/H22/H33/H44) signal undergoes a remarkable change in the chemical shift, from 7.07 to 6.89 ppm in the case of (±)-1, and from 7.06 to 6.82 ppm for (+)-1, over the range of the concentrations examined. The other concentration-sensitive signal of 1 is that of the H49/H51/H57/H59 protons on the benzene rings on the m-xylylene linkers, whose chemical shift varies over the range of 7.10–7.26 ppm for (±)-1 and 7.05–7.26 ppm for (+)-1 under the same conditions. The most important point is that all the signals generated by (+)-1 (Fig. 6b) are shifted upfield to a much greater extent than those of (±)-1 upon increasing the concentration (Fig. 6a). This is exactly opposite to the upfield shift behavior of the aromatic signals of the racemic pentamethylene-linked analogue (±)-3, which are much larger than those of the optically active (+)-3 under the same conditions.11Fig. 7 focuses on the contrasting concentration-dependencies of the 1H NMR chemical shifts of the imine moiety (H11) signal for racemic and optically pure 1 and 3 in CDCl3 at 293 K. These results imply that complex 1 prefers homochiral over heterochiral association, which is in contrast to the reported heterochiral association of the pentamethylene-linked analogue. Complex 2, bearing I-shaped coordination blades, did not exhibit any significant change in the chemical shift of the aromatic protons, most likely due to its reduced tendency to associate.
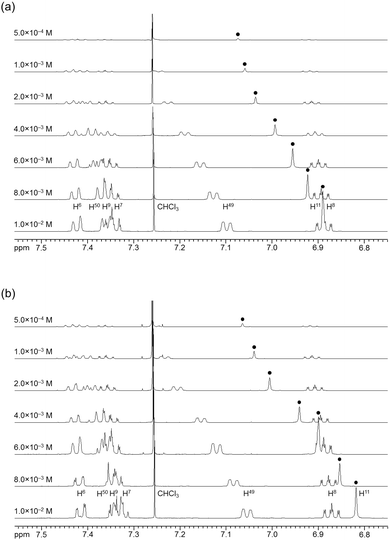 |
| | Fig. 6 Concentration dependence of the 1H NMR spectra (500 MHz) of (a) (±)-1 and (b) (+)-1 in CDCl3 at 293 K. | |
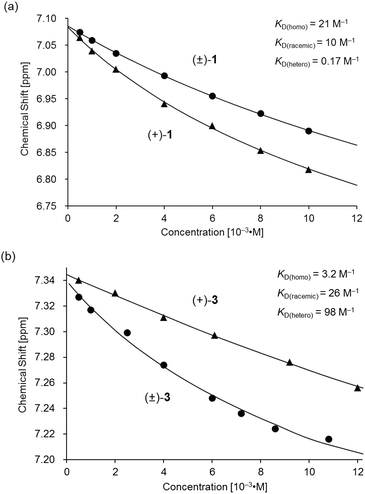 |
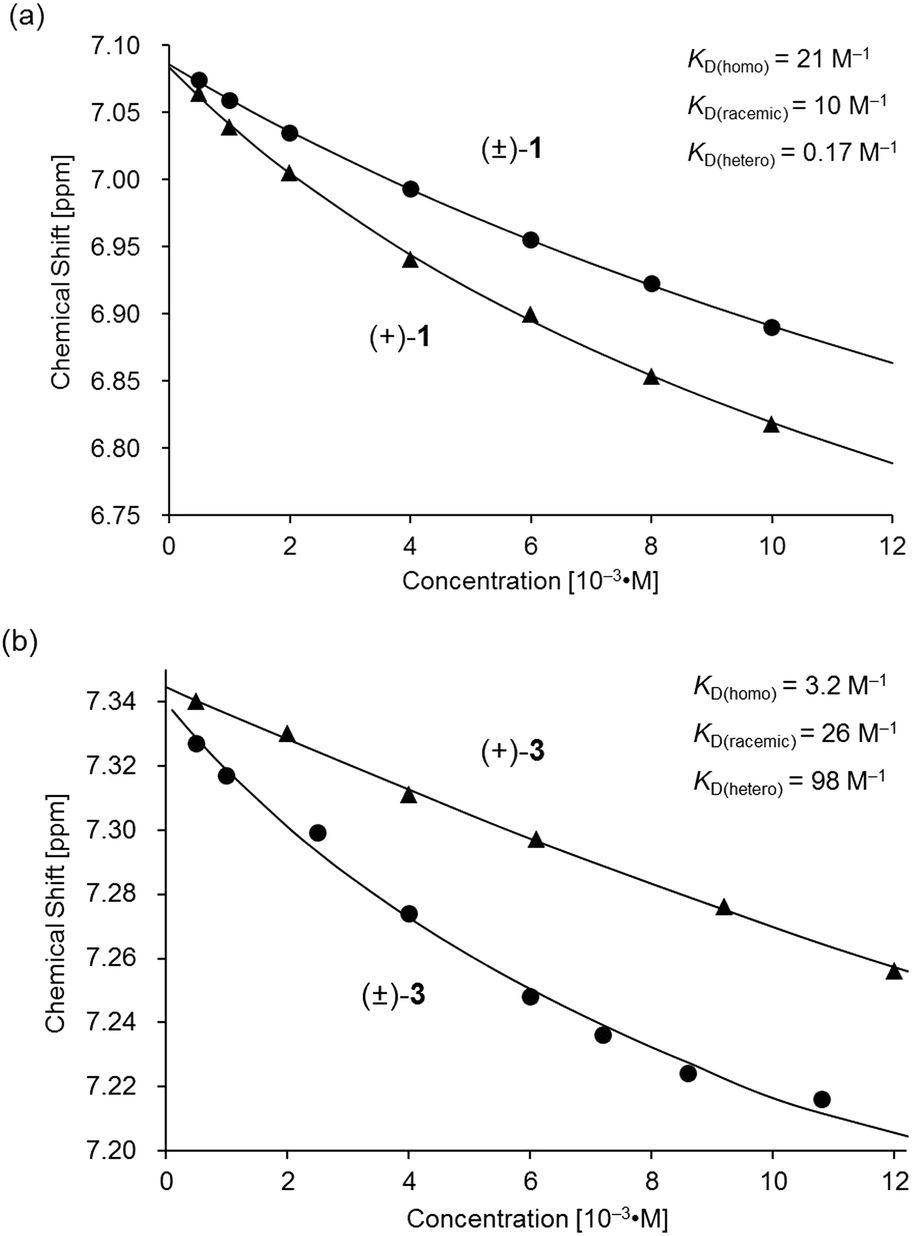
| | Fig. 7 Variations in the 1H NMR chemical shift of the H11 signal for (a) (±)- and (+)-1 and (b) (±)- and (+)-3 as functions of concentration in CDCl3 at 293 K. The plot of (±)- and (+)-3 in (b) is presented for comparison.11 | |
Based on a monomer-dimer equilibrium model,13 the equilibrium constants for the dimerization of (±)- and (+)-1 in CDCl3 [KD(racemic), KD(homo)] at 293 K were determined to be 10 and 21 M−1, respectively, by means of curve-fitting analysis14 of the concentration dependence of the H11/H22/H33/H44 chemical shifts (Fig. 7a). The heterochiral association constant KD(hetero) at 293 K was estimated to be 0.17 M−1, based on the relationship between KD(racemic) and the homo- and heterochiral association constants; KD(racemic) = 1/2 KD(homo) + 1/4 KD(hetero).6 Given that a similar analysis for 3 reportedly afforded KD(homo) and KD(hetero) values of 3.2 and 98, respectively (Fig. 7b),11 the KD(homo)/KD(hetero) ratio is estimated to be 1.2 × 102 for 1 and 0.033 for 3.
Mechanistic rationale for switching of the linker-dependence of the association chirality
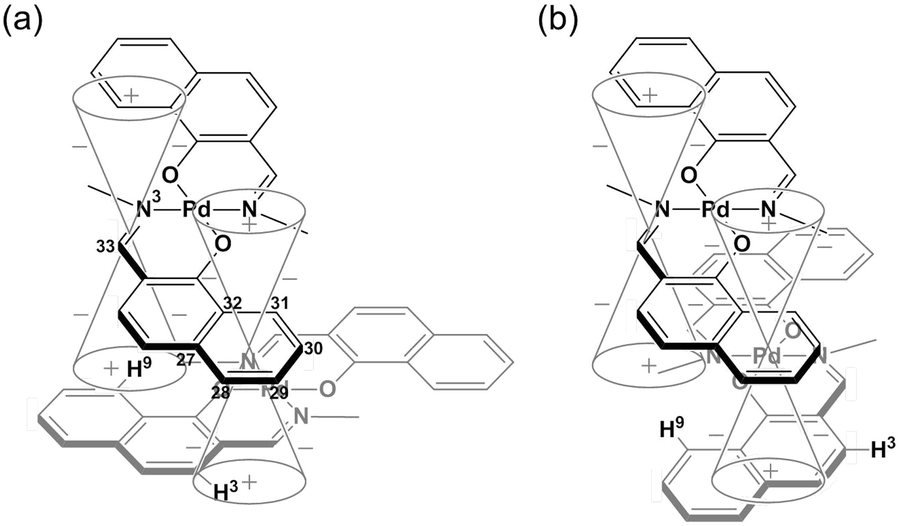
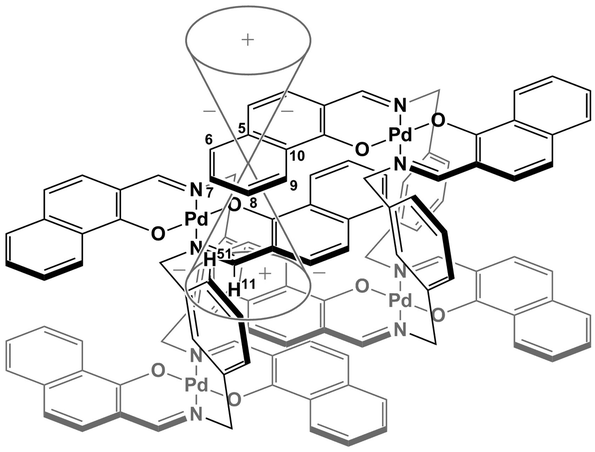
The specific homochiral association behaviors of 1 can be rationalized by assuming the equilibrated formation of interpenetrating homochiral dimers as a result of double outward intermolecular π-stacking interactions between aromatic rings of the coordination blades (Fig. 8a). In this process, the inner side of the upper blade of the rightmost molecule firmly adheres to the outer side of the upper blade of the leftmost molecule. In addition, the outer side of the lower blade of the right molecule is symmetrically in contact with the inner side of the lower blade of the left molecule. The extreme upfield shift of the imine moiety signal (H11/H22/H33/H44) and the aromatic signal (H49/H51/H57/H59) upon increasing the concentration (Fig. 6) can be explained by this interpenetrative associative dimer model (Fig. 9). According to this model, proton H11/H22/H33/H44 in the upper blade of the left molecule experiences strong shielding due to the ring current from the outer benzene ring [C(5)–C(6)–C(7)–C(8)–C(9)–C(10)] in the upper blade of the right molecule, while proton H49/H51/H57/H59 in the front linker of the left molecule is also shielded by the same outer benzene ring in the upper blade of the right molecule.
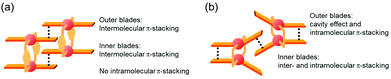 |
| | Fig. 8 Schematic representation of (a) the double outward stacking model for the dimer association of the rigid complex 1 and (b) the single inward stacking model for that of the flexible complex 3. | |
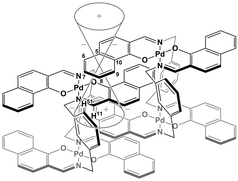 |
| | Fig. 9 Schematic representation of the interpenetrative association and the resulting shielding effect for (R)-1. | |
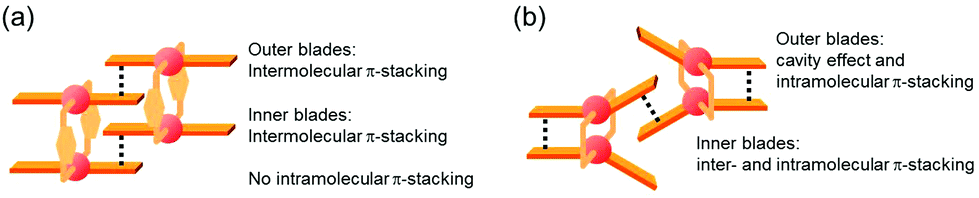
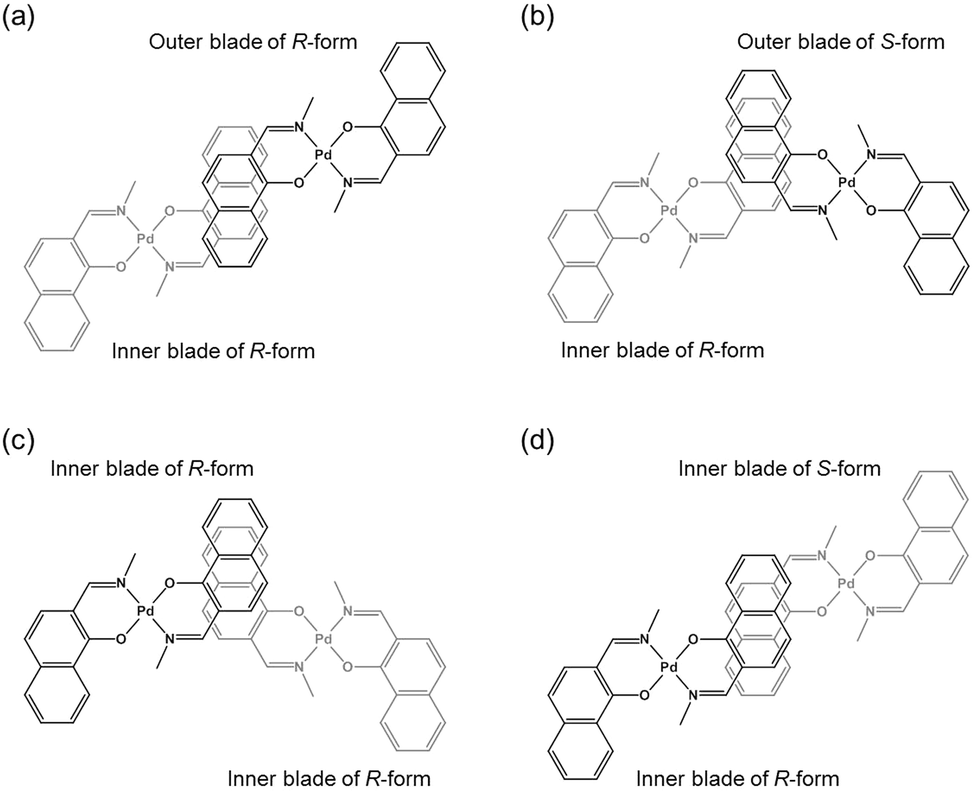
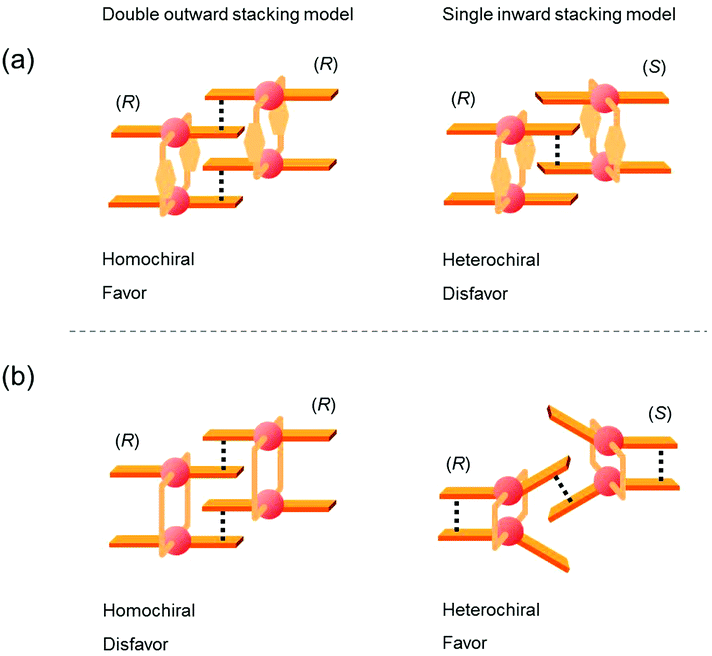
The double outward stacking mode gives a molecular-level explanation for the present homochiral association properties of 1. Fig. 10 shows overhead views of the outward and inward stacking motifs in the homochiral and heterochiral interpenetrative associations of 1. In the homochiral association, the outer penetrating blade of (R)-1 on the right molecule and the inner blade of (R)-1 on the left side are identical, which minimizes the steric and electrostatic repulsions between the two penetrating blades (Fig. 10a). This is in contrast to the heterochiral association (Fig. 10b), whereby stacking between outer and inner blades generates much more steric and electrostatic repulsion. This double outward stacking situation in the dimeric association of rigid complex 1 is quite different from the single inner stacking mode postulated for that of the flexible analogue 3.11 In this latter stacking mode, the inner blades of both the (R)- and (S)-forms in heterochiral association (Fig. 10d) reduce the steric and electrostatic repulsions much more than those of the (R)-forms in homochiral association (Fig. 10c).
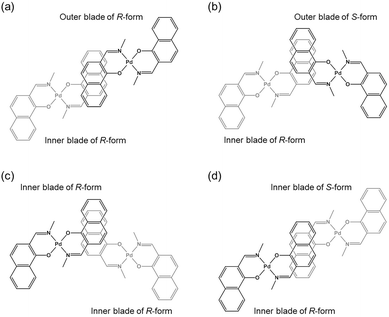 |
| | Fig. 10 Overhead views of (a, b) outward and (c, d) inward stacking motifs for the (a, c) homochiral and (b, d) heterochiral interpenetrative dimerizations of clothespin-shaped binuclear trans-bis(2-imino-1-naphthyloxy)Pd complexes, based on the (a, b) double outward and (c, d) single inward interpenetrative stacking dimerization models shown in Fig. 8. | |
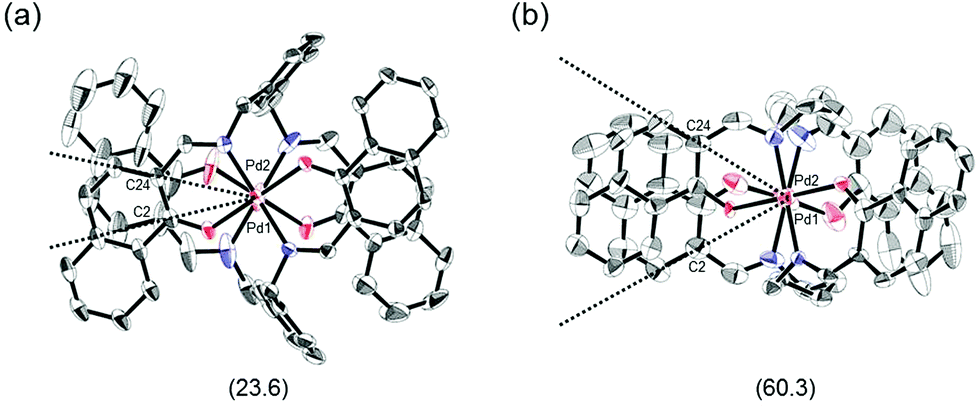
The proposed double outward stacking mode for the dimeric association of 1 can be justified on the basis of the conformational constraint arising from the rigid m-xylylene linkers. Fig. 11 shows Pd(1)–Pd(2) projections for ORTEP representations of m-xylylene-linked 1 and pentamethylene-linked 3. The dihedral C(2)–Pd(1)–Pd(2)–C(24) angles for 1 and 3, which express the tortuosity of the co-facial trans-bis(iminoaryloxy)Pd moieties, are 23.6° and 60.3°, respectively. The less twisted molecular structure of 1, with the planar coordination blades, strongly suggests that the highly rigid m-xylylene linkers in this complex induce conformational fixation, leading to a lack of intramolecular stacking stabilization between co-facial coordination blades (Fig. 2a, b and 11a). This is in contrast to 3, which can adopt a highly twisted conformation based on intramolecular stacking interactions between bent co-facial coordination blades on the basis of the highly flexible polymethylene linkers (Fig. 11b). Thus, we can say with a high degree of certainty that the planar coordination blades of 1, stabilized by d–π conjugation, result from compensation for the loss of intramolecular π-stacking stabilization upon the linker-induced conformational fixation.
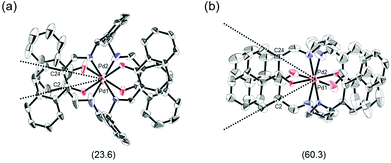 |
| | Fig. 11 Pd(1)–Pd(2) axis projections for (a) (R)-1, and (b) (R)-3 in the crystalline state. The dihedral C(2)–Pd(1)–Pd(2)–C(24) angles are given under each structure. The structure of (R)-3 is presented only for comparison.11 | |
Fig. 12 shows schematic representations of the origin of the contrasting association chirality behaviors of 1 and 3. On the basis of the discussion of the conformational rigidity of these clothespin-shaped complexes, it can be concluded that, during the interpenetrative dimeric association of 1, the double outward stacking mode (Fig. 12a, left) is preferred over the single inward stacking mode (Fig. 12a, right). This is because the H-shaped conformational unit 1, bearing highly aligned and rigid co-facial coordination blades (Fig. 11a), can obtain much greater stabilization from the dual π-stacking interactions as opposed to the single-point contact. This is in contrast to the association dimers of the flexible pentamethylene-linked 3 analogue incorporating a highly twisted A-shaped conformational unit. In this case, a single inward stacking mode is favored owing to the balance between inter- and intramolecular π-stacking stabilization (Fig. 12b, right), while the double outward stacking mode is disfavored because this mode sacrifices intramolecular π-stacking interactions in order to obtain firmly bound intermolecular double stacking interactions (Fig. 12b, left). Given the forgoing argument that the double outward stacking mode favors homochiral association and the single inward stacking mode promotes heterochiral association (Fig. 10), it is reasonably certain that the contrasting rigidity of the linkers is directly responsible for the different stacking modes, which in turn leads to the drastic change in association chirality in the solution state.
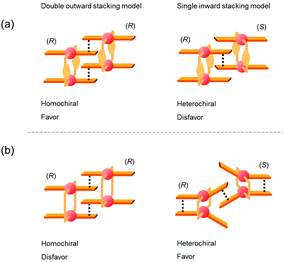 |
| | Fig. 12 Plausible models for the interpenetrative association dimers of (a) m-xylylene-linked 1 and (b) pentamethylene-linked 3. | |
Conclusions
We successfully synthesized the anti-forms of clothespin-shaped binuclear trans-bis(β-iminoaryloxy)palladium(II) complexes 1 and 2, doubly linked with rigid m-xylylene spacers. The structures and association properties of these rigid complexes were elucidated and compared with those of flexible pentamethylene-linked analogues. 1H NMR analyses revealed that 1 undergoes interpenetrative stacking association in CDCl3, whereby homochiral association occurs preferentially over heterochiral association with a KD(homo)/KD(hetero) ratio of 1.2 × 102. This is in contrast to the heterochiral association properties reported for the flexible pentamethylene-linked analogue 3, which has a KD(homo)/KD(hetero) value of 0.033. On the basis of XRD data and molecular models, the exceptional linker-dependence of the association chirality observed in this work was explained by assuming two contrasting interpenetrative association models; double outward stacking for the conformationally rigid complex 1 and single inner stacking for the flexible complex 3. This comparison study demonstrated that the rigidity or flexibility of the linkers in the present unique class of clothespin-shaped palladium complexes has a significant effect on the molecular mobility of the co-facial coordination blades. This in turn leads to the unprecedented phenomenon of remote controllability of the association chirality in the solution state. Research is currently underway to provide new molecular functions based on the controllable association properties of these clothespin-shaped analogues.
Experimental section
General
Palladium complexes (±)-1 and (±)-2 (anti-forms) were prepared by treating [Pd(OAc)2] with N,N′-bis(1-hydroxy-2-naphthylmethylene)-m-xylylenediamine and N,N′-bis(2-hydroxy-1-naphthylmethylene)-m-xylylenediamine, respectively, in the presence of N,N,N′,N′-tetramethylethylenediamine in boiling toluene. Melting points were determined using specimens on glass plates in conjunction with a Yanagimoto melting point apparatus. FT-IR spectra were obtained with a Bruker Equinox 55 spectrometer. NMR spectra were acquired using a Varian Unity-Inova 500 spectrometer (1H, 500 MHz; 13C, 125 MHz). Mass spectra were obtained with a JEOL JMS-700 spectrometer. Optical resolution of (±)-1 and (±)-2 was performed using a Japan Analytical Industry LC-9201 recycling preparative HPLC system with a Daicel Chiralpak IA column and CHCl3 as the mobile phase. Optical rotations were measured on a Jasco DIP-370 digital polarimeter and CD spectra were recorded with a Jasco J-805 spectropolarimeter.
Complex 1.
(Orange solid, 30%): mp >300 °C; IR (KBr): 3049, 2914, 1628, 1598, 1540, 1497, 1453, 1420, 1384, 1335, 1228, 1147, 889, 787, 740, 682 cm−1; 1H NMR (CDCl3, 500 MHz): δ 4.03 (d, J = 14.6 Hz, 4H, H45b, H52b, H53b, H60b), 5.78 (d, J = 14.6 Hz, 4H, H45a, H52a, H53a, H60a), 5.79 (d, J = 8.5 Hz, 4H, H3, H14, H25, H36), 6.33 (d, J = 8.5 Hz, 4H, H4, H15, H26, H37), 6.91 (ddd, J = 8.3, 6.8, 1.1 Hz, 4H, H8, H19, H30, H41), 6.99 (s, 4H, H11, H22, H33, H44), 7.18 (dd, J = 7.8, 1.2 Hz, 4H, H49, H51, H57, H59), 7.36 (ddd, J = 8.0, 6.8, 1.2 Hz, 4H, H7, H18, H29, H40), 7.39 (dd, J = 8.3, 1.2 Hz, 4H, H9, H20, H31, H42), 7.39 (dd, J = 7.8, 7.8 Hz, 2H, H50, H58), 7.43 (dd, J = 8.0, 1.1 Hz, 4H, H6, H17, H28, H39), 8.65 (s, 2H, H47, H55) ppm; 13C NMR (CDCl3, 125 MHz) δ 59.0, 112.3, 114.9, 123.6, 125.1, 125.4, 126.8, 127.0, 127.2, 127.88, 127.94, 128.8, 137.4, 141.1, 162.7, 163.5 ppm; HRMS (FAB): m/z calcd for C60H44N4O4106Pd2: 1096.1432 [M]+; found: 1096.1423. (+)-1: [α]25D = +872 ± 3 (c 0.110, CHCl3). (−)-1: [α]25D = −872 ± 3 (c 0.110, CHCl3).
Complex 2.
(Yellow solid, 18%): mp >300 °C; IR (KBr): 3052, 2924, 1618, 1539, 1509, 1434, 1416, 1363, 1342, 1307, 1255, 1186, 1143, 1098, 959, 821, 741 cm−1; 1H NMR (CDCl3, 500 MHz): δ 4.19 (d, J = 13.2 Hz, 4H, H45b, H52b, H53b, H60b), 5.83 (d, J = 13.2 Hz, 4H, H45a, H52a, H53a, H60a), 6.56 (d, J = 9.2 Hz, 4H, H10, H21, H32, H43), 6.85 (d, J = 9.2 Hz, 4H, H9, H20, H31, H42), 7.11 (ddd, J = 7.9, 7.0, 1.0 Hz, 4H, H6, H17, H28, H39), 7.27 (dd, J = 7.8, 1.5 Hz, 4H, H49, H51, H57, H59), 7.28 (dd, J = 7.9, 1.5 Hz, 4H, H7, H18, H29, H40), 7.32 (dd, J = 7.8, 7.8 Hz, 2H, H50, H58), 7.36 (ddd, J = 8.5, 7.0, 1.5 Hz, 4H, H5, H16, H27, H38), 7.70 (dd, J = 8.5, 1.0 Hz, 4H, H4, H15, H26, H37), 7.92 (s, 2H, H47, H55), 8.52 (s, 4H, H11, H22, H33, H44) ppm; 13C NMR (CDCl3, 125 MHz) δ 60.3, 109.5, 118.7, 121.5, 122.8, 126.45, 126.53, 127.0, 127.7, 128.3, 129.1, 134.2, 134.3, 140.2, 157.2, 165.0 ppm; HRMS (FAB): m/z calcd for C60H44N4O4106Pd2: 1096.1432 [M]+; found: 1096.1434. (+)-2: [α]25D = +686 ± 1 (c 0.110, CHCl3). (−)-2: [α]25D = −686 ± 1 (c 0.110, CHCl3).
XRD structure determination
Crystals suitable for XRD studies were analyzed using a Rigaku R-AXIS RAPID imaging plate diffractometer with graphite-monochromated MoKα radiation (λ = 0.71075 Å). The structures of (±)-1 and (±)-2 were solved using direct methods and refined by applying the full-matrix least-squares method. In subsequent refinement, the function ∑ω(Fo2 − Fc2)2 was minimized, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. The positions of non-hydrogen atoms were determined from difference Fourier electron density maps and refined anisotropically. All calculations were performed using the Crystal Structure crystallographic software package and illustrations were generated using ORTEP.15 Details of the structural determinations are provided in Fig. 2 and Table 1. CCDC 1477549 (for (±)-1) and 1477548 (for (±)-2) contain the supplementary crystallographic data for this paper.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Notes and references
-
(a) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS PubMed;
(b) L. M. Greig and D. Philp, Chem. Soc. Rev., 2001, 30, 287–302 RSC.
-
(a) Y. Ishida and T. Aida, J. Am. Chem. Soc., 2002, 124, 14017–14019 CrossRef CAS PubMed;
(b) O. Maury, J. Lacour and H. Le Bozec, Eur. J. Inorg. Chem., 2001, 201–204 CrossRef CAS;
(c) J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed;
(d) J. Guilleme, M. J. Mayoral, J. Calbo, J. Aragó, P. M. Viruela, E. Ortí, T. Torres and D. González-Rodríguez, Angew. Chem., Int. Ed., 2015, 54, 2543–2547 CrossRef CAS PubMed.
-
(a) K. Hanabusa, M. Yamada, M. Kimura and H. Shirai, Angew. Chem., Int. Ed. Engl., 1996, 35, 1949–1951 CrossRef CAS;
(b) M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2009, 38, 967–975 RSC;
(c) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed ; and references therein.
-
(a) D. M. Walba, F. Stevens, N. A. Clark and D. C. Parks, Acc. Chem. Res., 1996, 29, 591–597 CrossRef CAS;
(b) F. Vera, J. Barberá, P. Romero, J. L. Serrano, M. B. Ros and T. Sierra, Angew. Chem., Int. Ed., 2010, 49, 4910–4914 CrossRef CAS PubMed;
(c) F. Vera, J. L. Serrano and T. Sierra, Chem. Soc. Rev., 2009, 38, 781–796 RSC ; and references therein.
-
(a) C. Roche, H.-J. Sun, M. E. Prendergast, P. Leowanawat, B. E. Partridge, P. A. Heiney, F. Araoka, R. Graf, H. W. Spiess, X. Zeng, G. Ungar and V. Percec, J. Am. Chem. Soc., 2014, 136, 7169–7185 CrossRef CAS PubMed;
(b) Z.-G. Wang, B.-H. Zhou, Y.-F. Chen, G.-D. Yin, Y.-T. Li, A.-X. Wu and L. Isaacs, J. Org. Chem., 2006, 71, 4502–4508 CrossRef CAS PubMed;
(c) I. Weissbuch and M. Lahav, Chem. Rev., 2011, 111, 3236–3267 CrossRef CAS PubMed ; and references therein.
- D. Gut, A. Rudi, J. Kopilov, I. Goldberg and M. Kol, J. Am. Chem. Soc., 2002, 124, 5449–5456 CrossRef CAS PubMed.
- A. Nojiri, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2012, 51, 2137–2141 CrossRef CAS PubMed.
- M. Mizumura, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 5378–5381 CrossRef CAS PubMed.
- Solid-state phosphorescence:
(a) N. Komiya, M. Okada, K. Fukumoto, D. Jomori and T. Naota, J. Am. Chem. Soc., 2011, 133, 6493–6496 CrossRef CAS PubMed;
(b) N. Komiya, M. Okada, K. Fukumoto, K. Kaneta, A. Yoshida and T. Naota, Chem. – Eur. J., 2013, 19, 4798–4811 CrossRef CAS PubMed;
(c) N. Komiya, N. Itami and T. Naota, Chem. – Eur. J., 2013, 19, 9497–9505 CrossRef CAS PubMed;
(d) N. Komiya, M. Okada, K. Fukumoto, S. Iwata and T. Naota, Dalton Trans., 2014, 43, 10074–10085 RSC;
(e) K. Fukumoto, N. H.-T. Le, N. Komiya and T. Naota, Inorg. Chem. Commun., 2014, 50, 88–91 CrossRef CAS;
(f) N. Komiya, M. Okada, M. Hoshino, N. H.-T. Le and T. Naota, Eur. J. Inorg. Chem., 2014, 6085–6096 CrossRef CAS;
(g) N. Komiya, M. Okada, R. Inoue, S. Kawamorita and T. Naota, Polyhedron, 2015, 98, 75–83 CrossRef CAS.
- Molecular mobility: R. Inoue, S. Kawamorita and T. Naota, Chem. – Eur. J., 2016, 22, 5712–5726 CrossRef CAS PubMed.
- Association: M. Naito, H. Souda, H. Koori, N. Komiya and T. Naota, Chem. – Eur. J., 2014, 20, 6991–7000 CrossRef CAS PubMed.
- Ultrasound-induced aggregation:
(a) T. Naota and H. Koori, J. Am. Chem. Soc., 2005, 127, 9324–9325 CrossRef CAS PubMed;
(b) N. Komiya, T. Muraoka, M. Iida, M. Miyanaga, K. Takahashi and T. Naota, J. Am. Chem. Soc., 2011, 133, 16054–16061 CrossRef CAS PubMed;
(c) M. Naito, R. Inoue, M. Iida, Y. Kuwajima, S. Kawamorita, N. Komiya and T. Naota, Chem. – Eur. J., 2015, 21, 12927–12939 CrossRef CAS PubMed.
- We checked this assumption by Saunders-Hyne analysis. The monomer–dimer model gives the best fit over the full concentration range. M. Saunders and J. B. Hyne, J. Chem. Phys., 1958, 29, 1319–1323 CrossRef CAS.
- I. Horman and B. Dreux, Helv. Chim. Acta, 1984, 67, 754–764 CrossRef CAS.
-
M. N. Burnett and C. K. Johnson, Oak Ridge National Laboratory Report ORNL-6895, 1996.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1477548 and 1477549. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00315j |
| ‡ Present address: Chemistry Laboratory, The Jikei University School of Medicine, Kokuryo, Chofu, Tokyo 182-8570, Japan. |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.