
Cp*Co(III)-catalyzed, N–N bond-based redox-neutral synthesis of isoquinolines†
Jie
Wang
,
Shanke
Zha
,
Kehao
Chen
and
Jin
Zhu
*
Department of Polymer Science and Engineering, School of Chemistry and Chemical Engineering, State Key Laboratory of Coordination Chemistry, Nanjing National Laboratory of Microstructures, Nanjing University, Nanjing 210093, China. E-mail: jinz@nju.edu.cn
First published on 8th August 2016
Abstract
We report herein a redox-neutral Cp*Co(III)-catalyzed cyclization of acetophenone N-Boc hydrazones with alkynes for streamlined synthesis of isoquinolines, accomplished via formation of a C–C and a C–N bond along with N–N bond cleavage. Moreover, this rapid approach can be completed in 30 min with yields up to 85%.
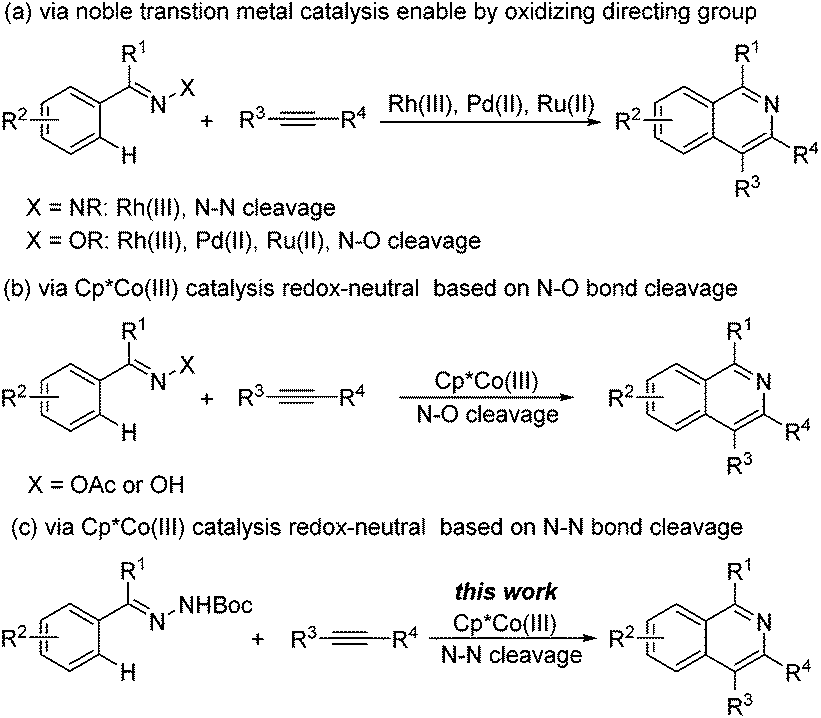
The wealth of isoquinoline skeletons in biologically and pharmacologically1 active molecules has provided a driving force for chemists to develop increasingly efficient methods towards their preparation. Traditional routes, which are demonstrated well by the Bischler–Napieralski reaction,2 the Pomeranz–Fritsch reaction3 and the Pictet–Gams reaction,4 typically need highly active substrates and harsh reaction conditions. Thus, the development of efficient transformations that exhibit mild conditions and good functional group tolerance is highly desirable. Metal-catalyzed cyclization reactions have been developed to permit mild and efficient syntheses of heterocycles,5 including isoquinolines. Initially, most approaches require the use of stoichiometric or superstoichiometric amounts of external oxidants, especially metal salts, to sustain the catalytic turnover, which indisputably results in lower atom efficiency by producing undesired waste byproducts and off-cycle side reactions.6 Since chemists were aware of its shortcomings, a new strategy employing internal oxidizing directing groups to regenerate the catalyst was developed. Shortly afterward, this “external-oxidant-free” strategy has created a renaissance for the streamlined synthesis of isoquinolines via Pd,7 Ru8 and Rh-catalyzed9 C–H bond activation. In these internal oxidant protocols, the N–O7,8,10 and N–N bonds11 have been recruited as a critical handle for both C–N cyclization and catalyst turnover (Scheme 1a). While these methods are effective, their synthetic application may be compromised by the relatively high cost and low abundance of these noble metals.
 | ||
| Scheme 1 Metal-catalyzed redox-neutral synthesis of isoquinolines. | ||
In light of the economic practicality of chemistry, the focus has shifted in recent years to the use of inexpensive and accessible base metal catalysts in lieu of the precious but often used noble transition metals for C–H activation processes. Undoubtedly, great progress has been accomplished with versatile cobalt catalysts which were reported by Kanai/Matsunaga,12 Sundararaju,13 Ellman,14 Glorius,15 Ackermann,16 Chang,17 and others.18 For isoquinolines synthesis, however, Cp*Co(III) catalysis has only showcased N–O bond cleavage19 for oxidative-directing groups to date (Scheme 1b). Although Cp*Co(III)-catalyzed N–N bond cleavage type reactions are reported in indole synthesis,20 isoquinoline synthesis has not been studied. To broaden the area of application for Cp*Co(III)-catalyzed isoquinoline synthesis, we focus on the exploration of new oxidizing motifs as potential directing groups. Here, we make the first report of a redox-neutral Cp*Co(III)-catalyzed synthetic approach for the direct synthesis of isoquinolines based on an N–N bond cleavage (Scheme 1c).
At the outset of our studies, we tested various reaction conditions for isoquinoline synthesis with acetophenone N-Boc hydrazone 1a and diphenylacetylene 2a (Table 1 and ESI Tables 1 and 2†). Preliminary experiments highlighted [Cp*Co(CO)I2]/AgOTf to be the catalyst system of choice and indicated that HFIP was the most suitable among a variety of solvents (Table SI-1†). For an additive, while acetates such as KOAc gave an unsatisfactory result (entry 1), acids proved to be superior, with HOAc being optimal (entries 2 and 3). The synthesis of product 3a occurred in the absence of silver(I) salts (entry 4), but was more effective when employing both Cp*Co(III) species and AgSbF6 (entries 5–9). Further systematic optimization with respect to the amount of HOAc, within the 10 mol%–200 mol% range (entries 9–14), enabled the pinpointing of its optimum quantity (20 mol%) under which condition this transformation was achieved in high yield and 3a was obtained in an 85% yield. When monitoring the reaction at different periods of time, we found that the reaction could reach 85% yield in 30 min (entries 11 and 15–18). The reaction temperature at 100 °C was just optimal as lower temperatures furnished reduced yields of product and higher temperatures did not further improve the yield either (ESI, Table 2†). With a lower catalyst loading, the reaction efficiency deteriorated and gave 3a in an unsatisfactory yield (entries 19 and 20).
|
|
||||
|---|---|---|---|---|
| Entry | Additive 1 (mol%) | Additive 2 (mol%) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (47 mg, 0.2 mmol), 2a (53 mg, 0.3 mmol), [Cp*Co(CO)I2] (9.6 mg, 10 mol%), HFIP (2 mL). b Isolated yields. c [Cp*Co(CO)I2] (1 mg, 1 mol%), AgSbF6 (1.4 mg, 2.0 mol%). d [Cp*Co(CO)I2] (4.8 mg, 5.0 mol%), AgSbF6 (6.9 mg, 10 mol%). | ||||
| 1 | AgOTf (20) | KOAc (100) | 12 | 44 |
| 2 | AgOTf (20) | PivOH (100) | 12 | 52 |
| 3 | AgOTf (20) | HOAc (100) | 12 | 66 |
| 4 | — | HOAc (100) | 12 | 42 |
| 5 | AgBF4 (20) | HOAc (100) | 12 | 67 |
| 6 | AgCO2CF3 (20) | HOAc (100) | 12 | 59 |
| 7 | AgOTs (20) | HOAc (100) | 12 | 62 |
| 8 | AgPF6 (20) | HOAc (100) | 12 | 66 |
| 9 | AgSbF6 (20) | HOAc (100) | 12 | 75 |
| 10 | AgSbF6 (20) | HOAc (10) | 12 | 81 |
| 11 | AgSbF6 (20) | HOAc (20) | 12 | 85 |
| 12 | AgSbF6 (20) | HOAc (50) | 12 | 68 |
| 13 | AgSbF6 (20) | HOAc (150) | 12 | 70 |
| 14 | AgSbF6 (20) | HOAc (200) | 12 | 54 |
| 15 | AgSbF6 (20) | HOAc (20) | 0.1 | 40 |
| 16 | AgSbF6 (20) | HOAc (20) | 0.5 | 85 |
| 17 | AgSbF6 (20) | HOAc (20) | 1 | 82 |
| 18 | AgSbF6 (20) | HOAc (20) | 5 | 83 |
| 19c | AgSbF6 (2) | HOAc (20) | 0.5 | 29 |
| 20d | AgSbF6 (10) | HOAc (20) | 0.5 | 67 |
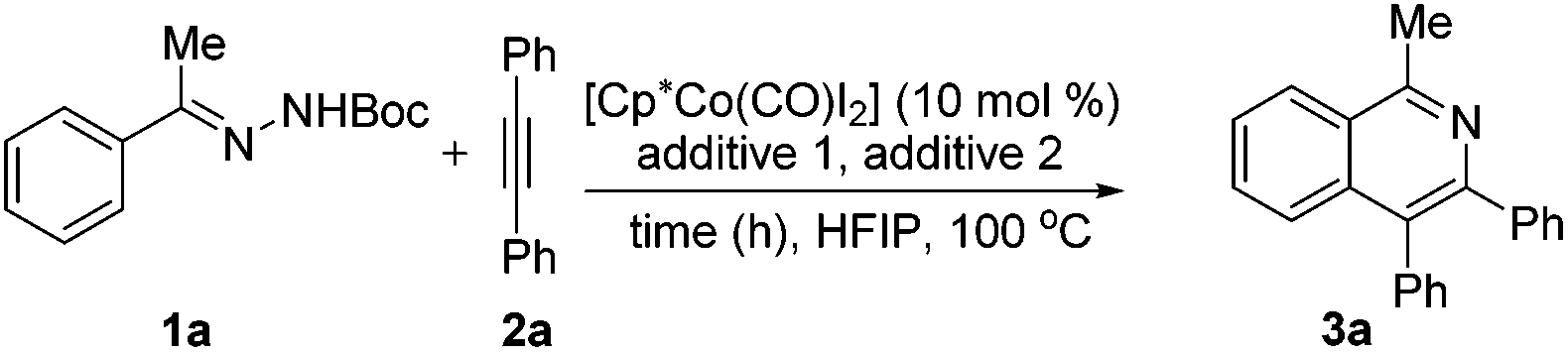
With the optimized conditions in hand, a broad range of acetophenone N-Boc hydrazones 1a–1u were screened to couple with diphenylacetylene 2a, as shown in Scheme 2. First, diverse monosubstituted acetophenone N-Boc hydrazones (1b–1j) which had been prepared beforehand, bearing various electron-rich and electron-deficient groups (F, Cl, Br, I, Me, OMe, CF3, CO2Me, and Ph) at the para-positions, all underwent smooth coupling with diphenylacetylene 2a, and the cyclization products (3b–3j) were isolated in yields of 71–89%. Notably, a strong electron-withdrawing group (NO2) at the para-position of acetophenone N-Boc hydrazone (1k) was tolerable and gave the corresponding isoquinoline product (3k) at a moderate yield (45%). The reaction of meta-substituted acetophenone N-Boc hydrazones 1l and 1m preferentially occurred at the less hindered position to afford the corresponding products 3l (72%) and 3m (69%) in good yields with single and opposite regioisomer selectivity. Also, the substrate 1n bearing a methoxy group at the ortho-position showed weaker reactivity in this reaction to give 3n in an acceptable yield (46%). In addition, benzophenone and 2,3,4-dihydronephthalen N-Boc hydrazone (1o and 1p) were smoothly converted to the corresponding products 3o and 3p in yields of 85% and 86%, respectively. Furthermore, our method is suitable not only for diverse monosubstituted acetophenone N-Boc hydrazones, but also for disubstituted (1q–1t) and even trisubstituted (1u) acetophenone N-Boc hydrazones, and their desired products (3q–3u) can be isolated in good yields and with exclusive regioselectivity.
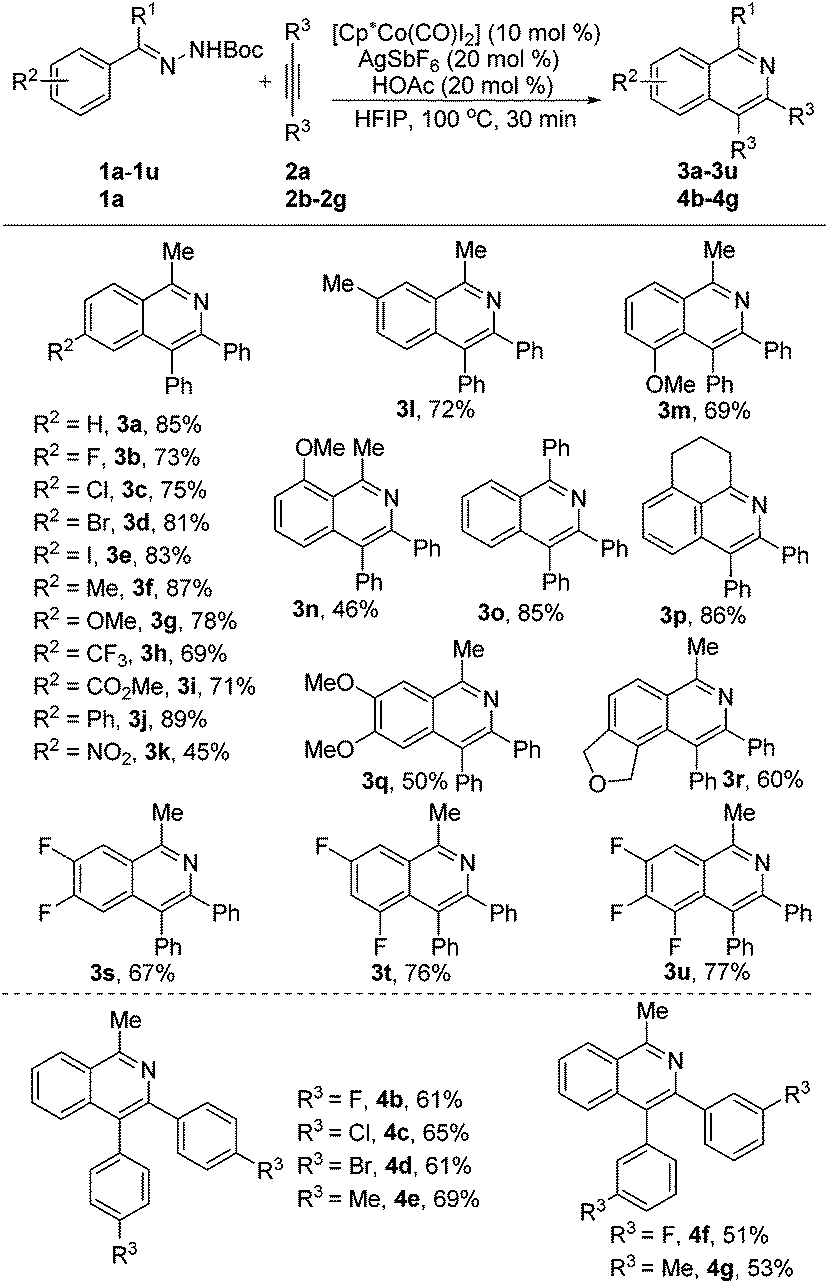 | ||
| Scheme 2 Scope of acetophenone N-Boc hydrazones and alkyne substrates. Reaction conditions: 1a–1u (0.2 mmol), 2a–2g (0.3 mmol), [Cp*Co(CO)I2] (9.6 mg, 10 mol%), AgSbF6 (13.7 mg, 20 mol%), HOAc (2.4 mg, 20 mol%), HFIP (2 mL). Isolated yields. | ||
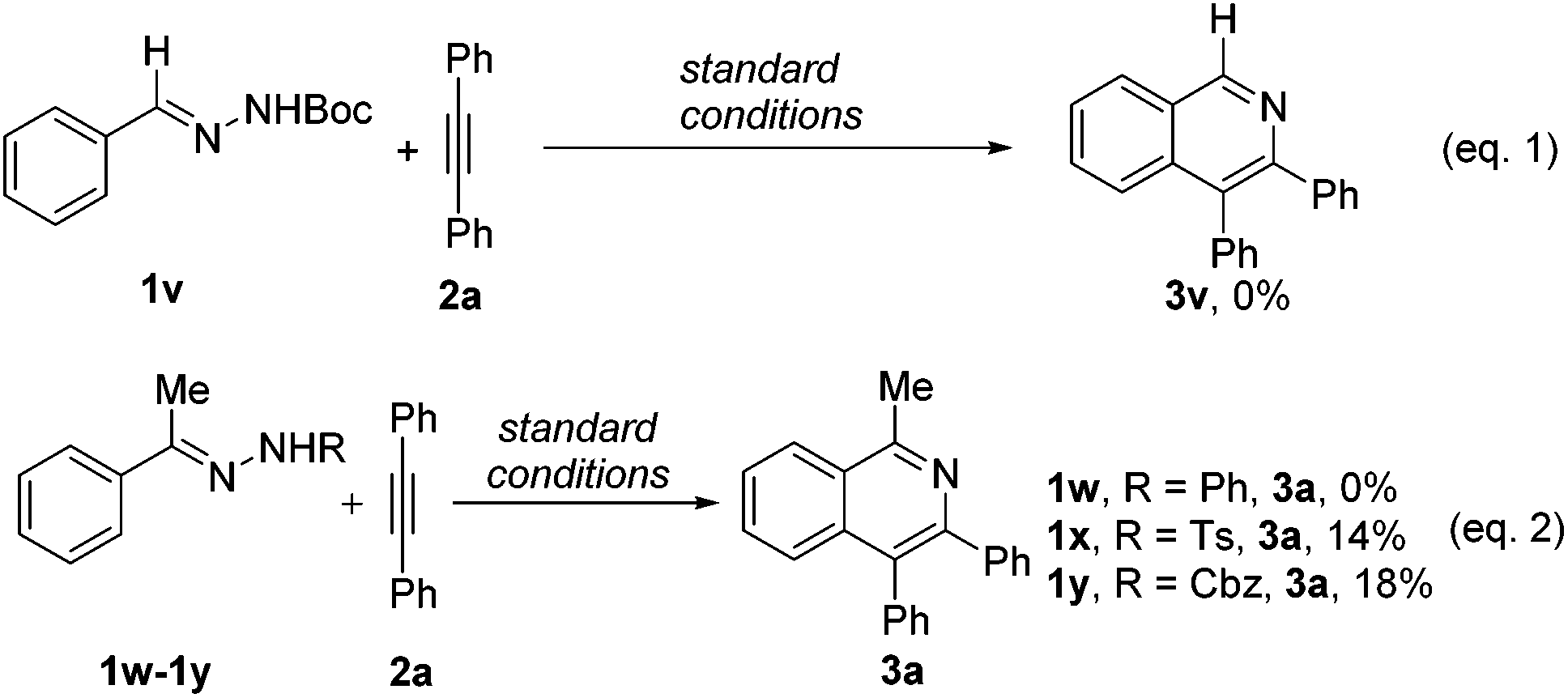
Subsequently, we briefly investigated the scope of the alkyne in the coupling with acetophenone N-Boc hydrazone 1a. Symmetrically substituted diarylacetylenes bearing halogen or methyl groups at the para- and meta-positions gave the products 4b–4g in 51%–69% yields (Scheme 2). The use of terminal alkynes, unsymmetrical alkyl/aryl internal alkynes, and dialkyl alkynes gave no corresponding products. And it is surprising to mention that benzaldehyde N-Boc hydrazone 1v with 2a under the same reaction conditions did not afford the expected isoquinoline product 3v (Scheme 3, eqn (1)). Then, we investigated the catalytic efficacy with different N-substitution patterns of acetophenone hydrazone (Scheme 3, eqn (2)). We began our study by using the bulkier N-Ph (1w). However, when we employed a Cp*Co(III)-catalyst with this substrate, no formation of the desired product 3a was observed apart from all the starting materials. But upon switching from the 1w to the N-Ts (1x) and N-Cbz (1y), the desired product 3a could be obtained in poor yields.
 | ||
| Scheme 3 Investigation of the catalytic efficacy with different N-substitution patterns. | ||
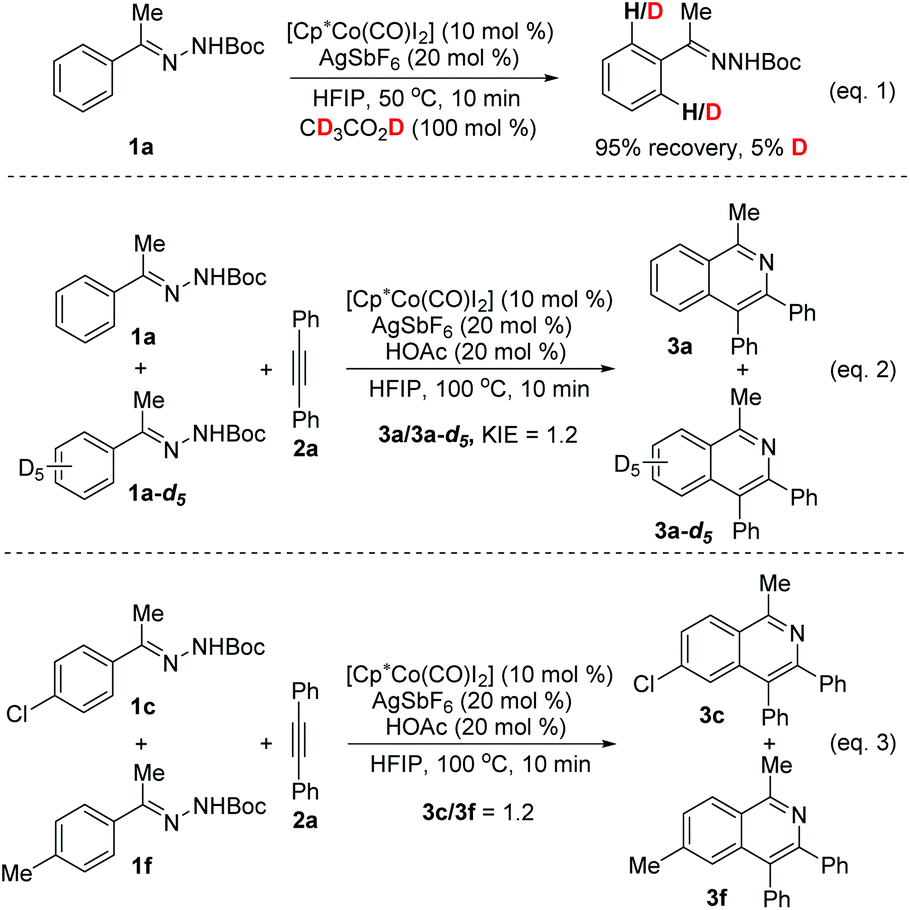
Several experiments were briefly performed to understand the reaction mechanism (Scheme 4). To probe the C–H activation process, H/D exchange between acetophenone N-Boc hydrazone 1a and CD3COOD (100 mol%) was performed (eqn (1)). After stirring for 10 min at 50 °C, 95% of 1a was recovered and only 5% deuterium incorporation was observed, which suggests that under the reaction conditions the C–H bond activation step is irreversible. A small KIE value of 1.2 was obtained (eqn (2)), indicating that C–H activation might not be involved in the rate-determining step. Intermolecular competition between 1c and 1f for the coupling with 2a under the optimized conditions could provide information on the electronic effects of the para-substituent, and 1H NMR analysis of the mixed product revealed that 3c and 3f were obtained in a 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (eqn (3)), indicating that the C–H activation probably occurs by a concerted metalation–deprotonation (CMD) mechanism.15d,19a
:1 ratio (eqn (3)), indicating that the C–H activation probably occurs by a concerted metalation–deprotonation (CMD) mechanism.15d,19a
 | ||
| Scheme 4 Mechanistic experiments. | ||
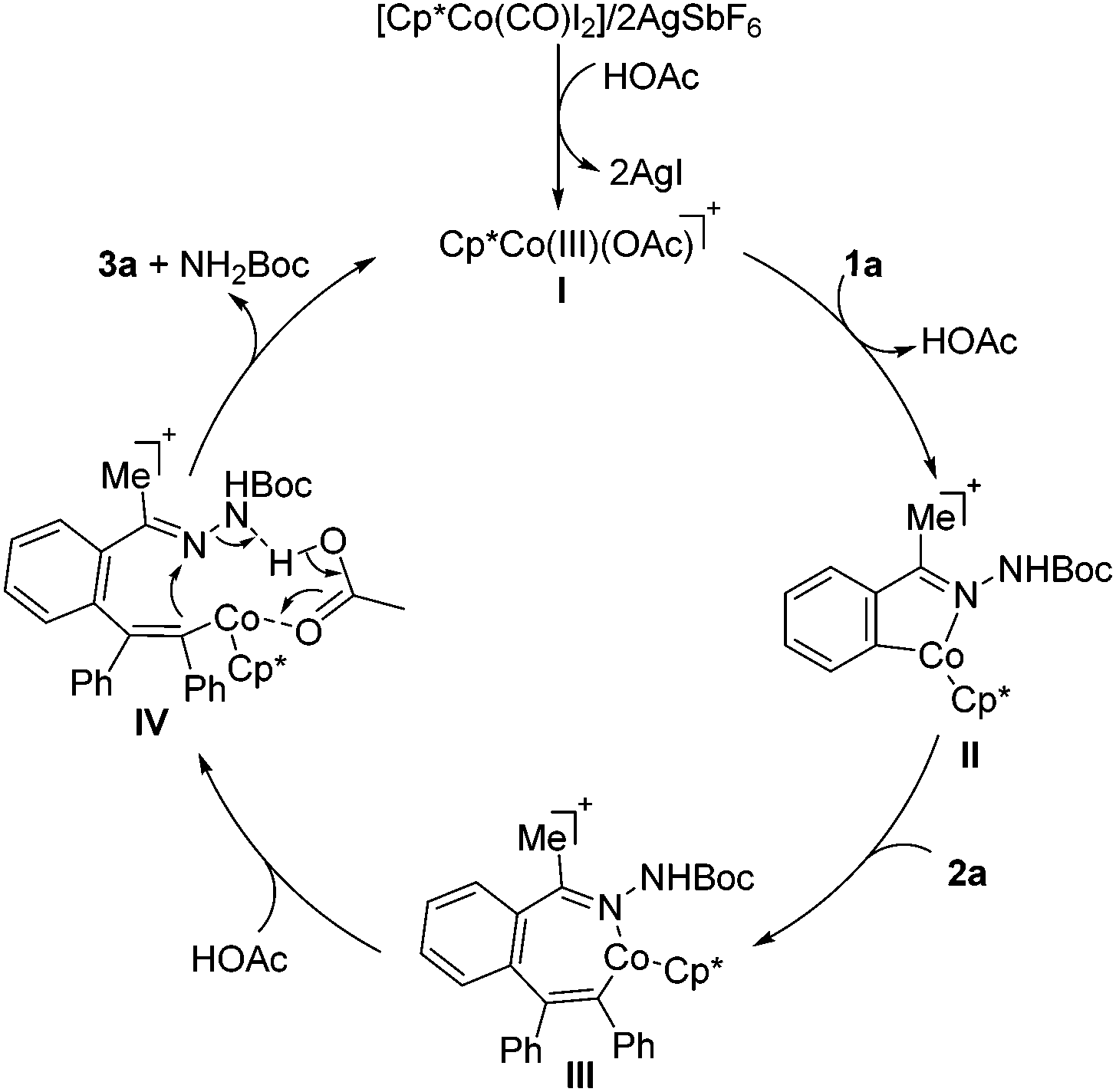
On the basis of our preliminary mechanistic experiments and literature precedents,9–20 the possible reaction pathway for the formation of isoquinolines is depicted in Scheme 5. After formation of the catalytically active cobalt species I, the C–H metalation takes place to form the cobaltacycle II. Then the seven-membered intermediate III can be generated through an alkyne insertion to further undergo an acetic acid-assisted proton migration,21 leading to the intermediate IV, followed by intramolecular substitution to form the C–N bond and break the N–N bond with the aid of acid. Finally, the product 3a is formed along with the regeneration of the active catalyst I for a new catalytic cycle.
 | ||
| Scheme 5 Proposed reaction pathway. | ||
In conclusion, we have developed a redox-neutral Cp*Co(III)-catalyzed cyclization of acetophenone N-Boc hydrazones with alkynes for streamlined synthesis of isoquinolines, accomplished via a C–C and C–N bond formation along with N–N bond cleavage. Moreover, this rapid approach can be completed in 30 min with yields up to 85%.
We gratefully acknowledge support from the National Natural Science Foundation of China (21425415, 21274058) and the National Basic Research Program of China (2015CB856303).
Notes and references
- (a) A. Morrell, S. Antony, G. Kohlhagen, Y. Pommier and M. Cushman, J. Med. Chem., 2006, 49, 7740 CrossRef CAS PubMed; (b) P. Fish, C. Barber, D. Brown, R. Butt, M. Collis, R. Dickinson, B. Henry, V. Horne, J. Huggins, E. King, M. O'Gara, D. McCleverty, F. McIntosh, C. Phillips and R. Webster, J. Med. Chem., 2007, 50, 2341 CrossRef CAS PubMed; (c) L. Chen, M. Conda-Sheridan, P. Reddy, A. Morrell, E. Park, T. Kondratyuk, J. Pezzuto, R. Breemen and M. Cushman, J. Med. Chem., 2012, 55, 5965 CrossRef CAS PubMed.
- W. Whaley and T. Govindachari, in Organic Reactions, ed. R. Adams, Wiley, New York, 1951, vol. 6, p. 151 Search PubMed.
- W. Gensler, in Organic Reactions, ed. R. Adams, Wiley, New York, 1951, vol. 6, pp. 191–206 Search PubMed.
- W. Whaley and T. Govindachari, in Organic Reactions, ed. R. Adams, Wiley, New York, 1951, vol. 6, pp. 74–150 Search PubMed.
- (a) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS PubMed; (b) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703 CrossRef CAS PubMed; (c) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (d) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (e) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596 CrossRef CAS PubMed; (f) X. Guo, D. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed; (g) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (h) W. Wu, Y. Liu and S. Bi, Org. Biomol. Chem., 2015, 13, 8251 RSC; (i) W. Chen and Z. Lin, Organometallics, 2015, 34, 309 CrossRef CAS.
- (a) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050 CrossRef CAS PubMed; (b) K. Morimoto, K. Hirano, T. Satoh and M. Miura, Chem. Lett., 2011, 40, 600 CrossRef CAS; (c) P. Villuendas and E. Urriolabeitia, J. Org. Chem., 2013, 78, 5254 CrossRef CAS PubMed; (d) D. Kim, J. Park and C. Jun, Adv. Synth. Catal., 2013, 355, 2667 CrossRef CAS; (e) S. Gupta, J. Han, Y. Kim, S. Lee, Y. Rhee and J. Park, J. Org. Chem., 2014, 79, 9094 CrossRef CAS PubMed; (f) K. Liu, S. Chen, X. Li and P. Liu, J. Org. Chem., 2016, 81, 265 CrossRef CAS PubMed; (g) W. Zhang, C. Wang, H. Lin, L. Dong and Y. Xu, Chem. – Eur. J., 2016, 22, 907 CrossRef CAS PubMed.
- (a) Z. Zhang and L. Yang, J. Org. Chem., 2014, 79, 7041 CrossRef CAS PubMed; (b) Z. Zhu, X. Tang, X. Li, W. Wu, G. Deng and H. Jiang, J. Org. Chem., 2016, 81, 1401 CrossRef CAS PubMed.
- (a) L. Ackermann and S. Fenner, Org. Lett., 2011, 13, 6548 CrossRef CAS PubMed; (b) C. Kornhaaß, J. Li and L. Ackermann, J. Org. Chem., 2012, 77, 9190 CrossRef PubMed; (c) R. Chinnagolla, S. Pimparkar and M. Jeganmohan, Org. Lett., 2012, 14, 3032 CrossRef CAS PubMed.
- (a) R. He, Z. Huang, Q. Zheng and C. Wang, Tetrahedron Lett., 2014, 55, 5705 CrossRef CAS; (b) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS PubMed; (c) P. Arockiam, C. Bruneau and P. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed.
- (a) K. Parthasarathy and C. Cheng, J. Org. Chem., 2009, 74, 9359 CrossRef CAS PubMed; (b) P. Too, Y. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed; (c) D. Zhao, F. Lied and F. Glorius, Chem. Sci., 2014, 5, 2869 RSC; (d) X. Li, M. Sun, Q. Jin, K. Liu and P. Liu, J. Org. Chem., 2016, 81, 3901 CrossRef CAS PubMed; (e) R. He, Z. Huang, Q. Zheng and C. Wang, Tetrahedron Lett., 2014, 55, 5705 CrossRef CAS.
- (a) Y. Wang, K. Toh, J. Lee and S. Chiba, Angew. Chem., Int. Ed., 2011, 50, 5927 CrossRef CAS PubMed; (b) S. Chuang, P. Gandeepan and C. Cheng, Org. Lett., 2013, 15, 5750 CrossRef CAS PubMed; (c) X. Huang, X. Yang, R. Song and J. Li, J. Org. Chem., 2014, 79, 1025 CrossRef CAS PubMed; (d) S. Zhang, D. Huang, G. Xu, S. Cao, R. Wang, S. Peng and J. Sun, Org. Biomol. Chem., 2015, 13, 7920 RSC.
- (a) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207 CrossRef CAS PubMed; (b) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Chem. – Eur. J., 2013, 19, 9142 CrossRef CAS PubMed; (c) B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Adv. Synth. Catal., 2014, 356, 1491 CrossRef CAS; (d) H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424 CrossRef CAS PubMed; (e) B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Chem. Commun., 2015, 51, 4659 RSC; (f) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944 CrossRef CAS PubMed.
- (a) D. Kalsi and B. Sundararaju, Org. Lett., 2015, 17, 6118 CrossRef CAS PubMed; (b) M. Sen, B. Emayavaramban, N. Barsu, J. Premkumar and B. Sundararaju, ACS Catal., 2016, 6, 2792 CrossRef CAS; (c) N. Barsu, M. Rahman, M. Sen and B. Sundararaju, Chem. – Eur. J., 2016, 22, 9135 CrossRef CAS PubMed.
- (a) J. Hummel and J. Ellman, J. Am. Chem. Soc., 2015, 137, 490 CrossRef CAS PubMed; (b) J. Hummel and J. Ellman, Org. Lett., 2015, 17, 2400 CrossRef CAS PubMed.
- (a) D. Yu, T. Gensch, F. de Azambuja, S. Vásquez-Céspedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722 CrossRef CAS PubMed; (b) D. Zhao, J. Kim, L. Stegemann, C. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508 CrossRef CAS PubMed; (c) J. Kim, S. Greßies and F. Glorius, Angew. Chem., Int. Ed., 2016, 128, 5667 CrossRef; (d) D. Zhao, S. Vásquez-Céspedes and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 1657 CrossRef CAS PubMed.
- (a) N. Sauermann, M. González and L. Ackermann, Org. Lett., 2015, 17, 5316 CrossRef CAS PubMed; (b) W. Ma and L. Ackermann, ACS Catal., 2015, 5, 2822 CrossRef CAS; (c) R. Mei, J. Loup and L. Ackermann, ACS Catal., 2016, 6, 793 CrossRef CAS; (d) H. Wang, M. Moselage, M. J. González and L. Ackermann, ACS Catal., 2016, 6, 2705 CrossRef CAS; (e) J. Li, M. Tang, L. Zang, X. Zhang, Z. Zhang and L. Ackermann, Org. Lett., 2016, 18, 2742 CrossRef CAS PubMed.
- (a) A. Pawar and S. Chang, Org. Lett., 2015, 17, 660 CrossRef CAS PubMed; (b) P. Patel and S. Chang, ACS Catal., 2015, 5, 853 CrossRef CAS; (c) J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103 CrossRef CAS PubMed.
- (a) X. Liu, S. Zhang, J. Wu, Q. Li and H. Wang, Tetrahedron Lett., 2015, 56, 4093 CrossRef CAS; (b) X. Liu, S. Zhang, C. Jiang, J. Wu, Q. Li and H. Wang, Org. Lett., 2015, 17, 5404 CrossRef CAS PubMed; (c) Z. Zhang, B. Liu, C. Wang and B. Shi, Org. Lett., 2015, 17, 4094 CrossRef CAS PubMed; (d) Z. Zhang, B. Liu, J. Xu, S. Yan and B. Shi, Org. Lett., 2016, 18, 1776 CrossRef CAS PubMed; (e) N. Yoshikai, ChemCatChem, 2015, 7, 732 CrossRef CAS; (f) L. Kong, S. Yu, X. Zhou and X. Li, Org. Lett., 2016, 18, 588 CrossRef CAS PubMed; (g) F. Wang, H. Wang, Q. Wang, S. Yu and X. Li, Org. Lett., 2016, 18, 1306 CrossRef CAS PubMed.
- (a) B. Sun, T. Yoshino, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2015, 54, 12968 CrossRef CAS PubMed; (b) H. Wang, J. Koeller, W. Liu and L. Ackermann, Chem. – Eur. J., 2015, 21, 15525 CrossRef CAS PubMed; (c) M. Sen, D. Kalsi and B. Sundararaju, Chem. – Eur. J., 2015, 21, 15529 CrossRef CAS PubMed; (d) K. Muralirajan, R. Kuppusamy, S. Prakash and C. Cheng, Adv. Synth. Catal., 2016, 358, 774 CrossRef CAS; (e) S. Zhang, X. Liu, S. Chen, D. Tan, C. Jiang, J. Wu, Q. Li and H. Wang, Adv. Synth. Catal., 2016, 358, 1705 CrossRef CAS; (f) G. Sivakumar, A. Vijeta and M. Jeganmohan, Chem. – Eur. J., 2016, 22, 5899 CrossRef CAS PubMed.
- (a) A. Lerchen, S. Vásquez-Céspedes and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 3208 CrossRef CAS PubMed; (b) Y. Liang and N. Jiao, Angew. Chem., Int. Ed., 2016, 55, 4035 CrossRef CAS PubMed.
- (a) G. Liu, Y. Shen, Z. Zhou and X. Lu, Angew. Chem., Int. Ed., 2013, 52, 6033 CrossRef CAS PubMed; (b) Y. Shen, G. Liu, Z. Zhou and X. Lu, Org. Lett., 2013, 15, 3366 CrossRef CAS PubMed; (c) H. Zhang, K. Wang, B. Wang, H. Yi, F. Hu, C. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 13234 CrossRef CAS PubMed; (d) J. Li and Z. Qiu, J. Org. Chem., 2015, 80, 10686 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00367b |
| This journal is © the Partner Organisations 2016 |