
On the scope of oxidation of tertiary amines: Meisenheimer rearrangements versus Cope elimination in 2-(cyanoethyl)-2-azanorbornanes†
Carlos A. D.
Sousa
 *a,
Ivo E.
Sampaio-Dias
b,
Xerardo
García-Mera
c,
Carlos F. R. A. C.
Lima
de and
José E.
Rodríguez-Borges
*b
*a,
Ivo E.
Sampaio-Dias
b,
Xerardo
García-Mera
c,
Carlos F. R. A. C.
Lima
de and
José E.
Rodríguez-Borges
*b
aREQUIMTE/LAQV, Departamento de Química e Bioquímica da Universidade do Porto, Rua do Campo Alegre, 4169-007 Porto, Portugal. E-mail: carlos.sousa@fc.up.pt; jrborges@fc.up.pt
bREQUIMTE/UCIBIO, Departamento de Química e Bioquímica da Universidade do Porto, Rua do Campo Alegre, 4169-007 Porto, Portugal
cDepartamento de Química Orgánica, Facultade de Farmacia, Universidade de Santiago de Compostela, E-15782 Santiago de Compostela, Spain
dCIQ-UP, Departamento de Química e Bioquímica, Faculdade de Ciências da Universidade do Porto, Portugal
eDepartamento de Química & QOPNA, Universidade de Aveiro, Campus de Santiago, Aveiro, Portugal
First published on 10th September 2016
Abstract
In this work, rearrangement reactions subsequent to the oxidation of tertiary amines were studied in 2-(cyanoethyl)-2-azanorbornane/ene systems. [1,2]- and [2,3]-Meisenheimer rearrangements, as well as the Cope elimination reaction, were observed with virtually complete selectivity. It was found that 2-(cyanoethyl)-2-azanorbornanes afford N-hydroxylamines through Cope elimination reactions and 2-(cyanoethyl)-2-azanorbornenes are prone to Meisenheimer rearrangements. In addition, the endo/exo configuration of 2-azanorbornenes plays a key role in the Meisenheimer rearrangement outcome. All the synthesized compounds were fully characterized by NMR spectroscopy and HRMS.
1. Introduction
Generally, organic reactions result in products that are predicted by commonly accepted mechanisms. However, in many occasions reactions do not afford exclusively the expected products and lead to other ones ensuing from mechanistically different pathways. These unexpected reactions are commonly referred to as rearrangement reactions, which are a broad class of organic reactions where the skeleton of the molecule is rearranged to give a structural isomer of the original molecule.1 Currently, there are many known mechanisms of rearrangement reactions,2,3 but there are also examples where products are formed through rearrangements difficult to explain.3–8 Nitrogen compounds, such as amines, nitriles or oximes, are relatively prone to rearrangement reactions. Examples of such rearrangements induced by oxidation of tertiary amines are the Meisenheimer rearrangement3,9 and the Cope elimination.2,3The Meisenheimer rearrangement is the thermal rearrangement of certain tertiary amine N-oxides to the corresponding O-substituted-N,N-disubstituted hydroxylamines.3 This reaction may proceed via a [1,2]- or [2,3]-rearrangement: the [1,2]-Meisenheimer rearrangement probably proceeds via a homolytic dissociation–recombination mechanism, whereas the [2,3]-Meisenheimer rearrangement is a concerted sigmatropic process that goes through a five-membered transition state, and is common when one of the substituents is allylic. Thus, the [2,3]-shift is usually much faster than the [1,2]-shift.3 There are several synthetic procedures making use of Meisenheimer rearrangements for the preparation of valuable bioactive molecules such as (+)-tanikolide,10 (R)-sulcatol,11 the 12-(S)-carba-eudistomin skeleton,12 the O-allyl-hydroxylamine derivative of the norfloxacin (NFLX) prodrug13,14 and magallanesine.15
In its turn, Cope elimination is a rearrangement in which trialkylamine-N-oxides having hydrogens in the β-position produce an olefin and the corresponding N,N-dialkylhydroxylamine. The reaction proceeds through a stereoselective syn elimination and the mechanism also involves a five-membered cyclic transition state. The Cope elimination found some useful synthetic applications, such as in the preparation of conformationally biased mimics of mannopyranosylamines,16 secondary hydroxylamines,17 enantiospecific synthesis of the taxoid intermediate (1S)-10-methylenecamphor18 or in virosaine A and bubbialidine.19 It has also been used in the synthesis of 3,4-disubsituted isoxazoles,20 and in the synthesis of functionalized isoxazolidines,21 spirocyclic lactams and lactones.22
When compared to the well-established reaction mechanisms, in which a synthetic route may be outlined based on the predictable products of each reaction step, rearrangements such as Meisenheimer and Cope elimination are less used in synthetic procedures, probably due to the difficulty to predict the reaction outcome. For example, Meisenheimer rearrangements and Cope elimination may be competitive since both reactions result from oxidation of tertiary amines; also, oxidations of amines may compete with other undesired oxidations such as of double bonds. Furthermore, the structure and the degree of functionalization of a molecule may decisively influence the outcome of such reactions. Hence, deeper knowledge on these rearrangements is essential towards their regular employment as reliable synthetic procedures. There are examples of classical synthetic procedures for obtaining valuable molecules that have been significantly simplified by taking advantage of the Meisenheimer rearrangement.10,14,15
Despite the importance of such rearrangements in synthetic organic chemistry, there are only a few independent studies in which specific molecules were used,12,23–27 thus leading to observations and conclusions that cannot be generalized. To the best of our knowledge, an integrative study that combines the possibility of more than one kind of rearrangement in the same molecule by oxidation of tertiary amines – such as [1,2]- or [2,3]-Meisenheimer rearrangements or Cope elimination – was never reported. In this study we aim to narrow this gap by adding some insights about the oxidation of tertiary amines in which these different rearrangements may potentially occur.
2-azanorbornane (or its unsaturated analogue, 2-azanorbornene) is a versatile chiral aza-Diels–Alder adduct with its application in stereoselective synthesis and several of its derivatives demonstrate biological activity.28 Our research group has been working on these bicyclic scaffolds, particularly as precursors for molecules of biologic interest.29–35 Due to the rigid nature of its bicyclic skeleton, we selected the system methyl 2-(cyanoethyl)-2-azabicyclo[2.2.1]heptane-3-carboxylate (or its unsaturated hept-5-ene analogue) as a model molecule to explore the tendency for the different types of rearrangements stated before after oxidation.
2. Results and discussion
2.1. Synthesis of the 2-(cyanoethyl)-2-azanorbornane/ene systems
We started by preparing the cycloadducts (±)-methyl 2-(cyanoethyl)-2-azabicyclo[2.2.1]hept-5-ene-3-exo-carboxylate (1) and (±)-methyl 2-(cyanoethyl)-2-azabicyclo[2.2.1]hept-5-ene-3-endo-carboxylate (2), through the aza-Diels–Alder reaction between cyclopentadiene (CPD) and the in situ generated imine from the condensation of methyl glyoxylate and 2-cyanoethylamine, using a standard described method that usually affords the exo isomer as the major one (Scheme 1).35–38 However, only compound 1 was obtained in extremely low yields and thus this route was rejected as an option for the preparation of 1/2. Considering this, compounds 1/2 were prepared via the aza-Diels–Alder reaction between CPD and the imine prepared in situ from methyl glyoxylate and ammonium chloride to afford a mixture of (±)-methyl 2-azabicyclo[2.2.1]hept-5-ene-3-exo-carboxylate (3) and (±)-methyl 2-azabicyclo[2.2.1]hept-5-ene-3-endo-carboxylate (4) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio.32,39 These cycloadducts were then treated with acrylonitrile to afford the corresponding tertiary amines via Michael addition (Scheme 1). In this way, the endo bicyclo 2 was obtained in an overall fair yield (≈45%) and its exo isomer 1 was achieved as a minor compound (≈13%).
:4 ratio.32,39 These cycloadducts were then treated with acrylonitrile to afford the corresponding tertiary amines via Michael addition (Scheme 1). In this way, the endo bicyclo 2 was obtained in an overall fair yield (≈45%) and its exo isomer 1 was achieved as a minor compound (≈13%).
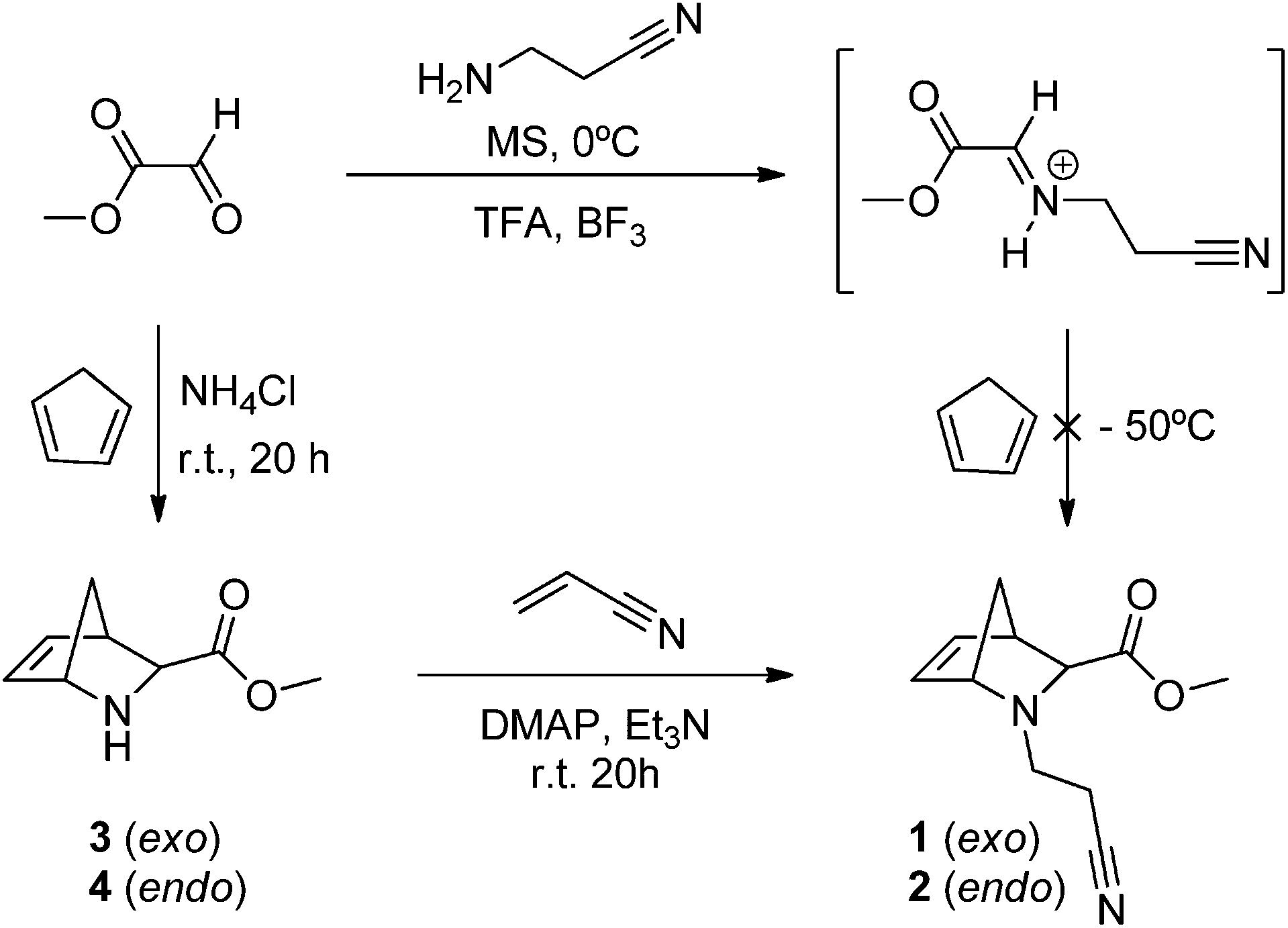 | ||
| Scheme 1 Synthetic route to the preparation of N-(cyanoethyl)-2-azanorbornenes 1/2. | ||
In order to study the influence of the double bond on the oxidative rearrangements of 1/2, we also prepared the corresponding saturated compounds 5/6. Since the exo isomer 1 is obtained as a minor adduct from the procedure represented in Scheme 1, an alternative for the synthesis of its saturated analogue is necessary. Hence, the aza-Diels–Alder reaction between CPD and the imine prepared in situ from methyl glyoxylate and benzylamine was performed according to the literature,36,38 leading to the corresponding exo (7)/endo (8) adducts in good yield with a 4:1 diastereomeric ratio. Subsequent catalytic hydrogenation of 7 using 10% Pd/C and H2 gave the corresponding exo-azabicyclo[2.2.1]heptane 9 which was then reacted with acrylonitrile to generate the desired (±)-methyl 2-(cyanoethyl)-2-azabicyclo[2.2.1]heptane-3-exo-carboxylate (5) in good yield, as represented in Scheme 2.
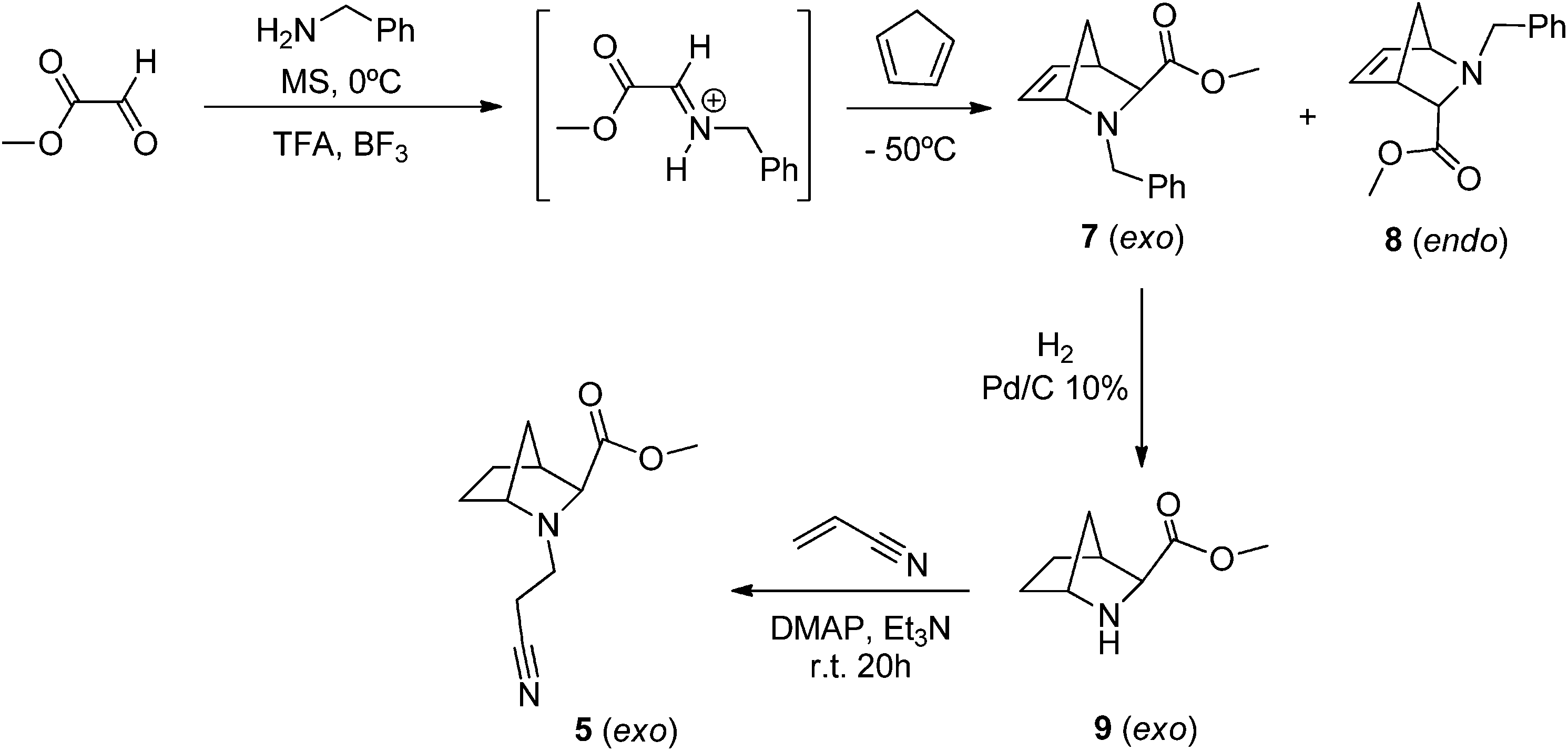 | ||
| Scheme 2 Synthetic route to the preparation of exo N-(cyanoethyl)-2-azanorbornane 5. | ||
On the other hand, the endo isomer of 5 (6) was effectively obtained from 2 through catalytic hydrogenation using 10% Pd/C and triethylsilane (TES) in MeOH as an in situ source of H2, at room temperature (Scheme 3).
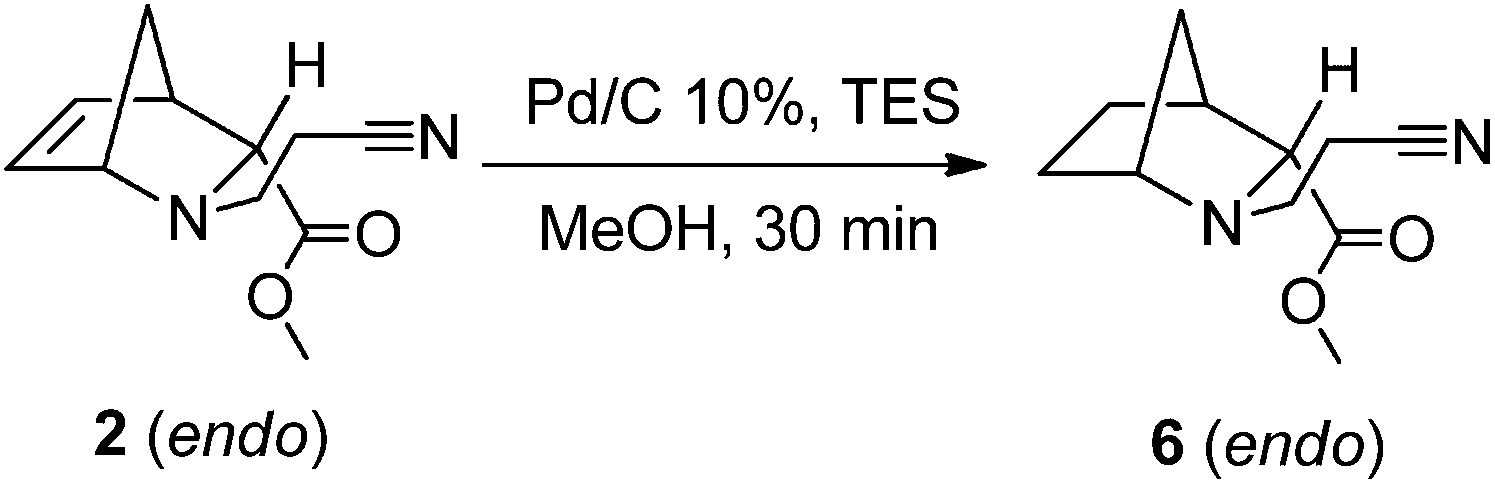 | ||
| Scheme 3 Catalytic hydrogenation of 2 to obtain the corresponding endo N-(cyanoethyl)-2-azanorbornane 6, at controlled room temperature (21 °C). | ||
2.2. Study of the rearrangements induced by oxidation of compounds 1/2 and 5/6
Compound 1 was oxidized with equimolar amounts of m-CPBA in dry dichloromethane, under an inert atmosphere at 0 °C. Despite the presence of a double bond, its oxidation to form the respective epoxide was not expected, since the oxidation of tertiary amines is known to be much faster. In fact, oxidation of the double bond of similar aza-bicyclic compounds was only reported in Boc-protected amines or amides.40–43It was likely that the oxidation of 1 with m-CPBA would lead to the respective N-oxide 10, which could undergo [1,2]- or [2,3]-Meisenheimer rearrangements, or even Cope elimination. However, a spontaneous [1,2]-Meisenheimer rearrangement was observed from 10, leading exclusively to oxazabicyclo[3.2.1]octene 11 with no evidence of epoxide or Cope elimination (Scheme 4). On the other hand, in the course of NMR analyses for full characterization, a complete transformation of 11 into its isomeric oxazabicyclo[3.3.0]octene 12 was observed in CDCl3 after 6 hours at room temperature (93% conversion was observed in 2 h, see the ESI, Fig. S41†). A similar transformation was not observed in other common solvents such as CHCl3, CH2Cl2, MeOH, iPrOH, acetone, benzene, CH3CN or CD3CN. In fact, compound 11 was found to be quite stable when stored under an inert atmosphere at room temperature. Refluxing 11 in acetonitrile or chloroform for 48 h led to the formation of 12 with 76–83% yield. The proposed mechanism for the formation of 11 is illustrated in Scheme 4. m-CPBA oxidizes the nitrogen atom of 1; as 1 has an exo configuration, it is likely that the 2-cyanoethyl group prefers the endo configuration due to steric hindrance. Thus, N-oxide 10 will predominantly adopt the configuration in which the oxygen atom lies to the same side of the methylene bridge. In this way, the oxygen atom is not able to attack the double bond and only the homolytic bond cleavage of C1–N may occur, leading to a [1,2]-Meisenheimer rearrangement to form oxazabicyclo[3.2.1]octene 11.
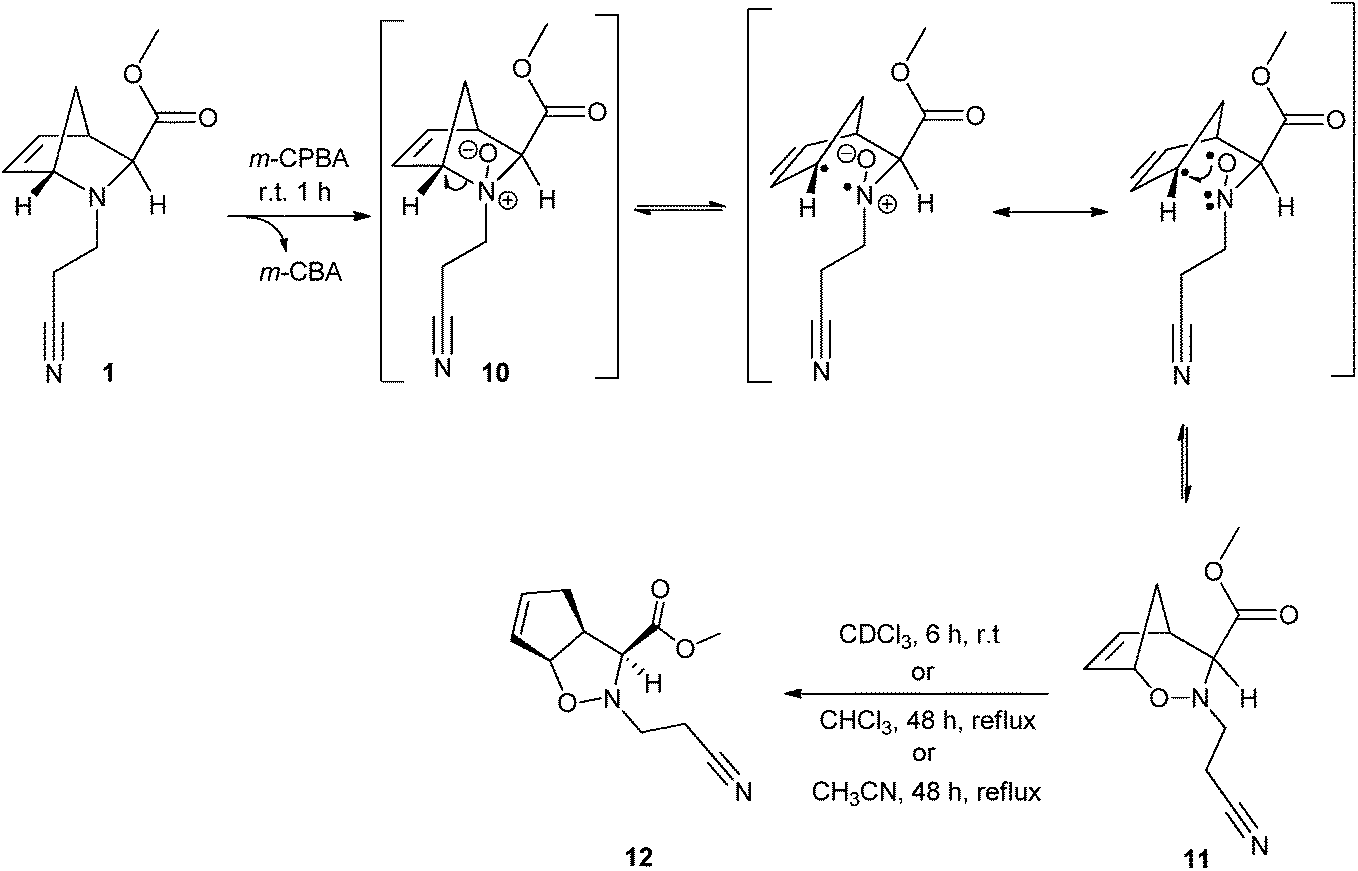 | ||
| Scheme 4 Reaction mechanism for the formation of the oxazabicyclo[3.2.1]octene 11 from oxidation of 1. Compound 11 rearranged into oxazabicyclo[3.3.0]octene 12 in i – CDCl3, 6 h, room temperature, 100%; ii – CHCl3, 48 h, reflux, 76%; iii – CH3CN, 48 h, reflux, 83%. | ||
Bailey and co-workers reported a similar observation with an analogous exo aza-bicyclo compound, and they hypothesized the formation of the oxazabicyclo[3.3.0]octene directly from the Meisenheimer intermediate.44 Conversely, this study demonstrates that oxazabicyclo[3.3.0]octene 12 results entirely from compound 11, probably via homolytic cleavage of its C1–O bond to afford the thermodynamically more stable adduct. This rearrangement is particularly favored in the presence of CDCl3.
The thermodynamic feasibility of the reactions presented in Scheme 4 was further evaluated by computational chemistry calculations at the B3LYP/6-311++G(d,p) level of theory. The results evidence that the reactions 10 → 11 and 11 → 12 are significantly exothermic, with calculated enthalpies of the reaction, ΔrH, at T = 298.15 K, of −65 and −33 kJ mol−1, respectively. By comparison, in the case of the endo adducts (Scheme 5) the conversions of 14 → 11endo and 11endo → 13 have calculated ΔrH, at T = 298.15 K, of −81 and −46 kJ mol−1, respectively, in which 11endo represents the endo analogue of 11; note that the intermediate 11endo was not observed experimentally and is referred here just for comparison. These results support the thermodynamic spontaneity of these reactions and that 12 and 13 are the more stable adducts. The detailed computational results are presented in the ESI.†
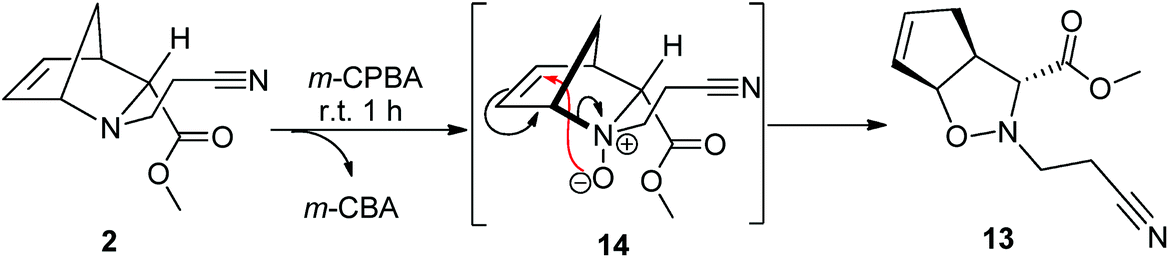 | ||
| Scheme 5 [2,3]-Meisenheimer rearrangement of the in situ prepared N-oxide 14 into oxazabicyclo[3.3.0]octene 13. | ||
A similar study under the same conditions was performed using the endo bicyclo 2, which generated the oxazabicyclo[3.3.0]octene 13 as a unique product. No traces of co-products such as the epoxide, the Cope elimination product or 2-oxa-3-zabicyclo[3.2.1]oct-6-ene were detected. The mechanism that describes this reaction is depicted in Scheme 5. In this case, as the N-oxide 14 oxygen atom prefers the endo position, it is able to attack at bicyclo C5 and thus undergoes a concerted sigmatropic process. Considering the exclusive formation of oxazabicyclo[3.3.0]octene 13, one may conclude that [2,3]-Meisenheimer rearrangement is both kinetically and thermodynamically favored over [1,2]-Meisenheimer rearrangement.
To obtain more insights into the mechanism of the reactions depicted in Schemes 4 and 5 a conformational analysis at the B3LYP/6-311++G(d,p) level of theory was carried out for compounds 1, 2, 10 and 14, focusing on the relative position of the cyanoethyl group bonded to the nitrogen. In all these molecules the cyanoethyl group can be oriented like an anti-configuration relative to the methylene bridge (as shown for 1 in Scheme 4) or like a syn-configuration (as shown for 2 in Scheme 5). The stereochemical configuration of 1 and 2 influences the reaction outcome by favoring the attack from the side opposite to the cyanoethyl group. The computational results indicate that anti-1 (cyanoethyl down) is about 20 kJ mol−1 more stable than its syn conformer (cyanoethyl up), while syn is the most stable configuration of 2 by approximately 5 kJ mol−1. Hence, regardless of the anti-conformers (cyanoethyl down) being more stable than the syn for both the intermediates 10 and 14 (as indicated by computational calculations presented in the ESI†), oxidation will occur preferentially on the more stable isomers and yield the intermediates with the corresponding configuration: anti-10 and syn-14, in agreement with the experimental observations.
The application of the same oxidation protocol to 2-azanorbornanes 5/6 led to the formation of the corresponding N-hydroxylamines 15/16. The reactions proceeded smoothly and very cleanly (TLC) in 2 hours at room temperature. Again, N-oxides 17/18 were not isolated (although observed by TLC) which suggests a spontaneous Cope elimination in good yield, with no traces of any other compounds ensuing from Meisenheimer rearrangements being identified (Scheme 6).
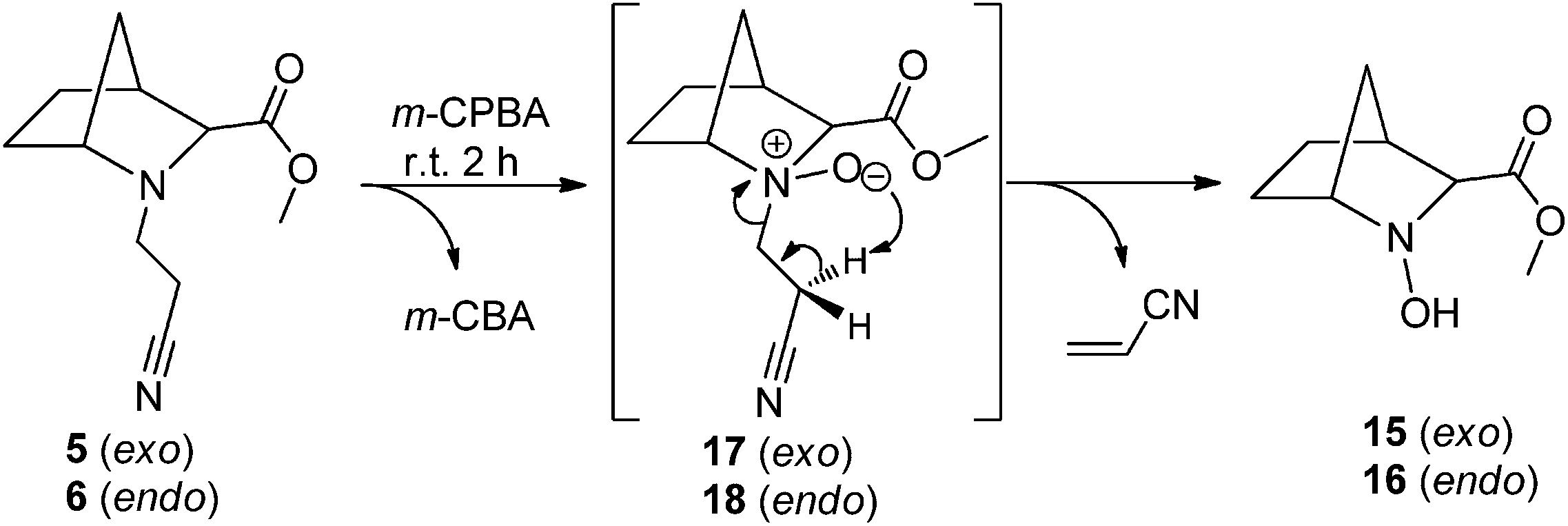 | ||
| Scheme 6 Preparation of N-hydroxylamines 15/16via Cope elimination. | ||
From these results, it is clear that stereochemical effects associated with the relative position of the 2-azanorbornane's methylene bridge and the N-oxide oxygen atom do not interfere with the outcome of the Cope elimination reaction.
In order to assess the influence of the highly stereochemical constraints of the 2-azanorbornane ring in the reaction outcome, the same study using the related L-proline methyl ester (19) was performed. The treatment of 19 with acrylonitrile afforded the desired methyl N-(2-cyanoethyl)-L-prolinate (20) and the subsequent oxidation protocol with m-CPBA generated the corresponding N-hydroxylamine 21 as the single product with very good yield,17 with no traces of Meisenheimer rearrangement being detected (Scheme 7).
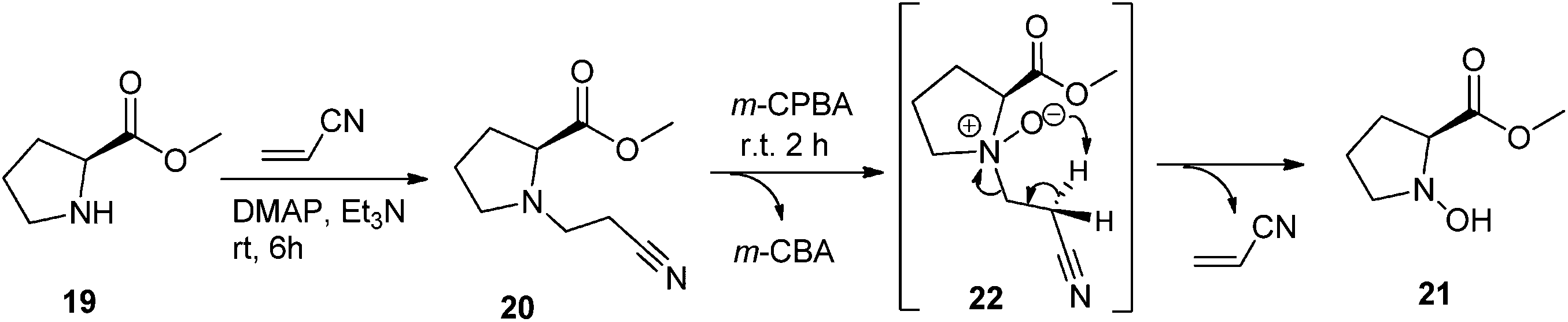 | ||
| Scheme 7 Synthesis of methyl N-hydroxyl-L-prolinate 21. | ||
In this case, N-oxide 22 may be isolable;17,45 nevertheless, our aim was not N-oxide isolation and thus its formation was only followed by TLC. Again, N-hydroxylamine 21 formed smoothly without the need of heat, in contrast with previous statements.45
Considering the results taken from the oxidations of azacycloalkanes 5, 6 and 19, it seems clear that the Cope elimination is preferred over the [1,2]-Meisenheimer rearrangement; however, opposite results were obtained with 2-azanorbornenes 1/2. The presence of a double bond at the C5–C6 position further constricts the bicyclic ring, particularly at C1 and C4. More importantly, the double bond helps to stabilize the radical by conjugative resonance stabilization, which is more effective than hyperconjugation. To have a more quantitative idea about the influence of the double bond on the stabilization energy of the radical, the homolytic bond dissociation energy was calculated using B3LYP/6-311++G(d,p) for the molecules shown in Scheme 8. The results suggest that the double bond should stabilize the radical by about 54 kJ mol−1.
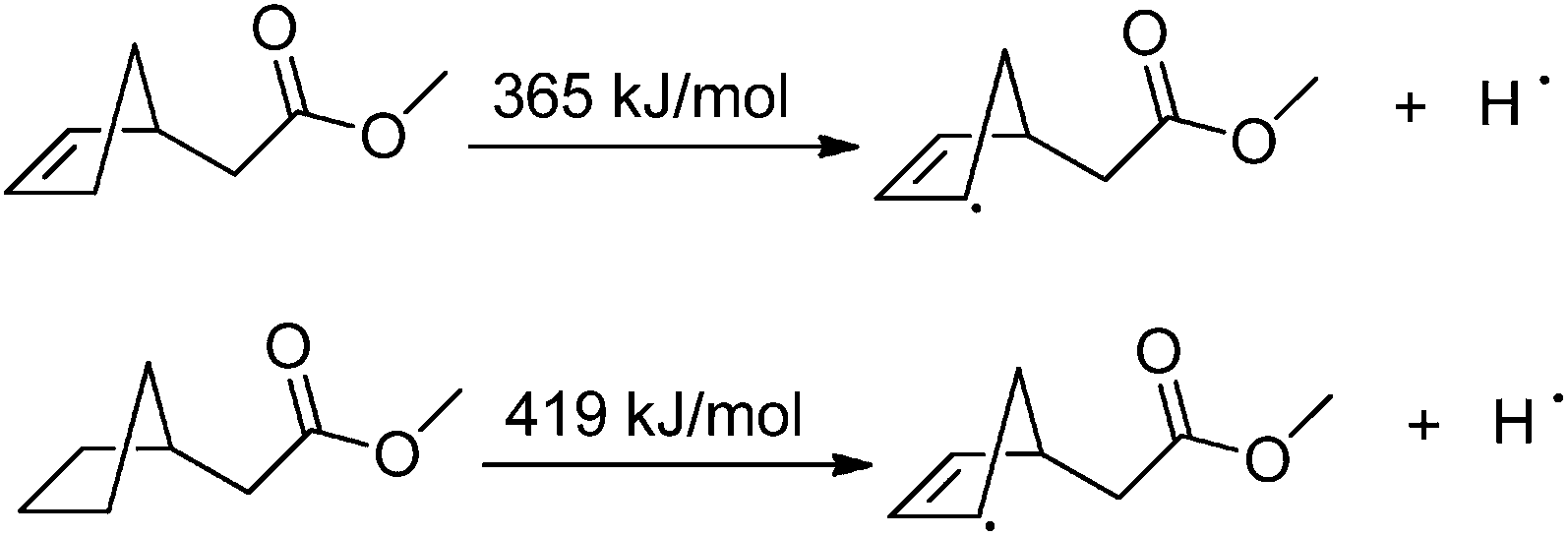 | ||
| Scheme 8 Calculated homolytic bond dissociation energies, at the B3LYP/6-311++G(d,p) level of theory, for the molecular species used to evaluate the effect of the double bond in compounds 1/2 and 5/6. | ||
For these reasons, the C1–N bond is more prone to homolytic cleavage in 2-azanorbornenes 1/2. This kind of homolysis was already observed for similar bicyclic compounds during the process of catalytic hydrogenation, in which a minor byproduct originated from the ring opening at C1–N was identified.46 On the other hand, catalytic hydrogenolysis of N-benzyl or N-phenylethyl-2-azanorbornanes under similar conditions does not lead to ring opening.47,48 In this work, we also tested the catalytic hydrogenation of a mixture of 1/2 using 10% Pd/C and TES in MeOH, in which a higher load of 10% Pd/C was used. As the in situ production of hydrogen is highly exothermal, the temperature of the reaction mixture reached more than 50 °C, which probably explains the formation of compound 23 as the major one and the saturated 2-azanorbornanes as minors (Scheme 9). This observation further supports that the presence of a double bond favors the homolytic cleavage of C1–N in 2-azanorbornenes, thus explaining the formation of 11 through [1,2]-Meisenheimer rearrangement from 10 rather than Cope elimination, as well as the formation of 12 from 11 (Scheme 10).
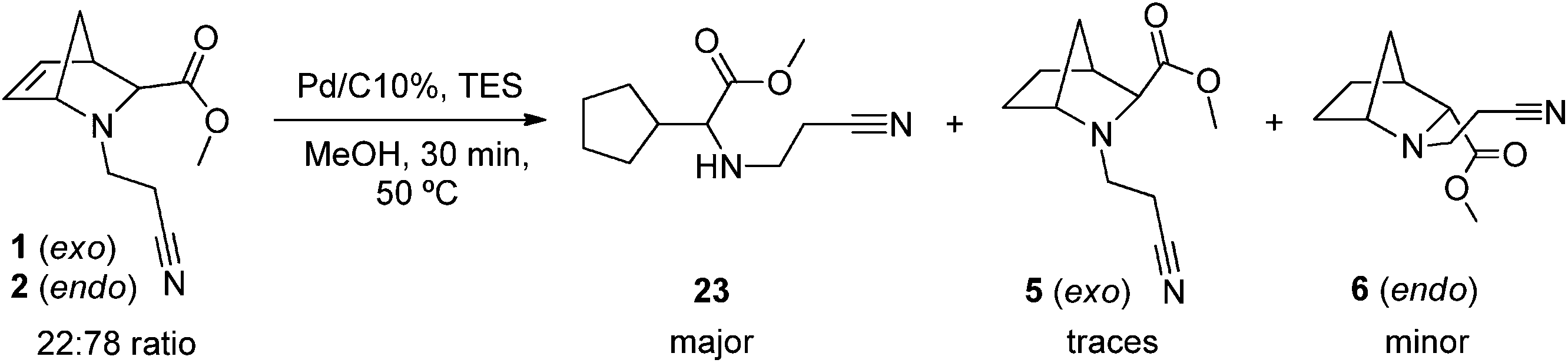 | ||
| Scheme 9 Catalytic hydrogenation of a mixture of 1/2 to obtain the corresponding exo/endo mixture of N-(cyanoethyl)-2-azanorbornanes 5/6 as minor products, and compound 23 as the major one (85%) at non-controlled temperature (>50 °C). | ||
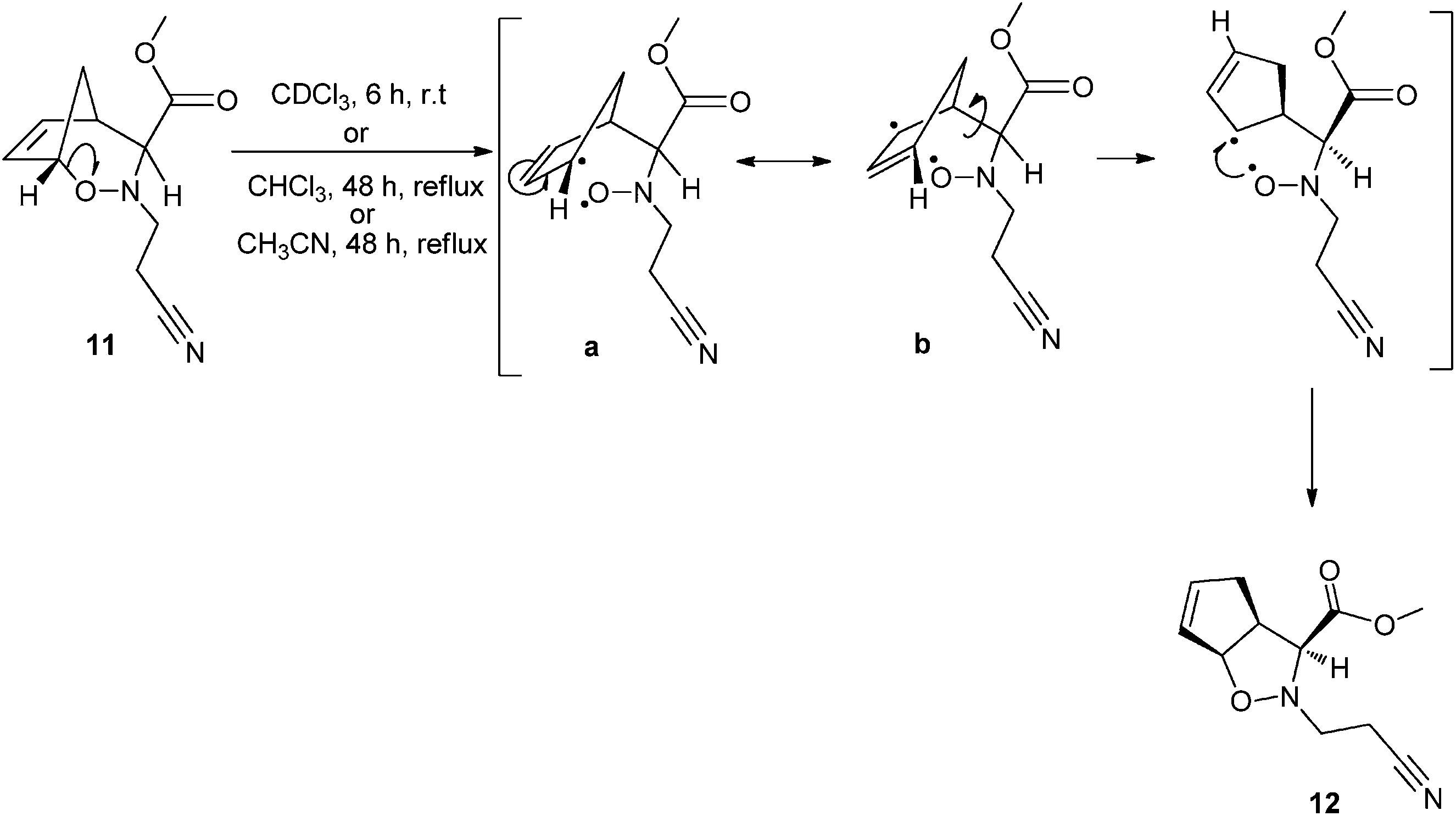 | ||
| Scheme 10 Proposed mechanism for the formation of oxazabicyclo[3.3.0]octene 12 from oxazabicyclo[3.2.1]octene 11: the homolytic dissociation of the C1–O bond yields a stabilized state transition due to allylic resonance stabilization, in which the resonance structure b is thermodynamically preferred over a. | ||
The mechanism presented in Scheme 10 was further supported by evaluating the energy profile for hindered internal rotation in compound 11 as depicted in Fig. 1. The computational results indicate that conversion of 11 into 12 has an energy barrier of about 60 kJ mol−1. This explains why compound 1 yields 11 as a stable intermediate, which is subsequently converted into 12 after heating (Scheme 4). However, for compound 2, when the respective intermediate 14 is formed its C(H2)–C(H)–C(H)(COOCH3)–N dihedral angle is already above the potential energy maximum (>90°). Hence the molecule can easily relax and converge to the equilibrium structure of compound 13 (a diastereomer of 12) without passing by an intermediate analogous to 11. It can be concluded that the difference in the reaction mechanisms presented in Schemes 4 and 5 ultimately results from the conformational preferences of compounds 1 and 2 with respect to the relative position of the cyanoethyl group, as discussed before. When this group is anti to the methylene bridge the oxygen attacks from the top and the stable intermediate 11 is formed because the high rotational barrier prevents its immediate conversion to 12. Conversely, when it is syn the oxygen binds from the bottom and the molecule does not need to surpass a significant energetic barrier in order to yield the thermodynamic most stable product 13. These results further support that conversion of 11 into 12 results from the cleavage of the significantly labile C–O bond and subsequent molecular rearrangement.
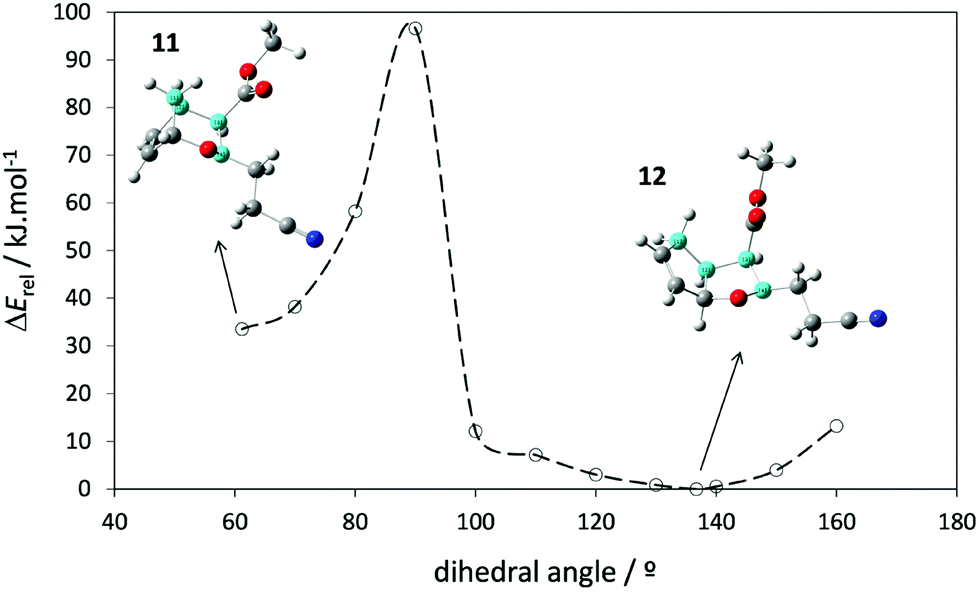 | ||
| Fig. 1 Potential energy profile for hindered internal rotation around the C(H2)–C(H)–C(H)(COOCH3)–N dihedral angle (highlighted in blue) in 11, calculated at the B3LYP/6-311++G(d,p) level of theory (the C–O bond breaks during internal rotation). | ||
From a synthetic point of view, these results also demonstrate that it is not possible to obtain the unsaturated N-hydroxylamine analogues of 15/16via the oxidation–Cope elimination methodology from 1/2, since the Meisenheimer rearrangements are favored. Hence, the synthesis of such N-hydroxy-2-azanorbornenes has to proceed via an alternative pathway already described by our research group.31,33
3. Conclusion
2-(Cyanoethyl)-2-azanorbornane/ene systems were oxidized with m-CPBA to form the corresponding N-oxide, which immediately underwent Meisenheimer rearrangements or the Cope elimination reaction.The oxidation of 3-exo-2-(cyanoethyl)-2-azanorbornene (1) led to the corresponding N-oxide (10) in which the oxygen atom lies to the same side of the methylene bridge. Thus, this configuration does not allow a direct attack of the oxygen atom to the C5 of the bicyclic ring in order to undergo a [2,3]-sigmatropic Meisenheimer rearrangement. In this way, only the oxazabicyclo[3.2.1]octene 11 is formed through homolytic dissociation of the C1–N bond and subsequent [1,2]-Meisenheimer rearrangement. However, oxazabicyclo[3.2.1]octene 11 is also prone to homolytic dissociation of its C1–O bond, which was induced by either thermal conditions or the presence of deuterated chloroform (the reasons for this observation are still unclear). Due to radical conjugative resonance stabilization, oxazabicyclo[3.3.0]octene 12 is preferably formed after recombination.
On the other hand, the oxidation of 3-endo-2-(cyanoethyl)-2-azanorbornene (2) led to the corresponding N-oxide (14) which spontaneously rearranged into oxazabicyclo[3.3.0]octene 13. The opposite positioning of the oxygen atom regarding the bicyclic methylene bridge allows a direct [2,3]-Meisenheimer rearrangement to afford 13 as the single product.
Regarding the 2-(cyanoethyl)-2-azanorbornanes 5/6, it was demonstrated that their N-oxides do not undergo Meisenheimer rearrangements in any extent, but they rather undergo Cope elimination to afford N-hydroxylamines 15/16 quite effectively.
In conclusion, stereochemical factors, relative stabilization of radicals or the presence of conjugative resonance stabilization may dictate the outcome of rearrangements promoted by N-oxidation of tertiary amines. In this work, Meisenheimer rearrangements and Cope elimination products were never simultaneously detected under the same oxidation protocol, denoting a complete selectivity for the particular systems studied. In saturated 2-azanorbornane N-oxides, Cope elimination prevails over [1,2]-Meisenheimer rearrangement. However, if a conjugative allylic bond is present, the preference order changes to [2,3]-Meisenheimer rearrangement > [1,2]-Meisenheimer rearrangement > Cope elimination.
4. Experimental
4.1. General notes
All chemicals were of reagent grade and were obtained from Sigma-Aldrich or Fluka and used without further purification. CPD was obtained by bidistillation of dicyclopentadiene and used immediately. Flash chromatography was performed on silica gel (Merck 60, 230–240 mesh), and analytical TLC was carried out on pre-coated silica gel plates (Merck 60 F254, 0.25 mm) using UV light and/or an ethanolic solution of phosphomolybdic acid (followed by gentle heating) for visualization. The melting point was determined on an electrothermal melting point apparatus and is uncorrected. 1H-, 13C{1H} and DEPT-NMR spectra were recorded on a Bruker Avance III 400 at CEMUP (Centro de Materiais da Universidade do Porto). Peak assignments were made by using DEPT, HSQC and by comparison with analogous molecules previously reported by our group.31,33,35,49 The NMR spectra were calibrated using TMS as an internal standard. Mass spectra were recorded on a LTQ Orbitrap XL mass spectrometer (Thermo Fischer Scientific, Bremen, Germany) controlled by using LTQ Tune Plus 2.5.5 and Xcalibur 2.1.0 (CEMUP).4.2. Experimental procedures
(±)-Methyl 2-azabicyclo[2.2.1]hept-5-ene-3-exo-carboxylate (3) and methyl (±)-methyl 2-azabicyclo[2.2.1]hept-5-ene-3-endo-carboxylate (4) were synthesized following the procedures early described.33,39![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) aHbCH2CN), 2.64–2.44 (m, 3H, CHabC2CN), 2.29 (br s, 1H, H-3), 1.86 (m, 1H, H-7syn), 1.52–1.34 (m, 1H, H-7anti); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 173.7 (C,
aHbCH2CN), 2.64–2.44 (m, 3H, CHabC2CN), 2.29 (br s, 1H, H-3), 1.86 (m, 1H, H-7syn), 1.52–1.34 (m, 1H, H-7anti); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 173.7 (C, ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O2CH3), 136.9 (CH, C-5), 133.1 (CH, C-6), 118.6 (C, CN), 65.5 (CH), 65.4 (CH), 52.1 (CH3, CO2H3), 50.6 (CH2), 48.1 (CH), 46.8 (CH2), 18.2 (CH2, H2CN); HRMS-ESI: calculated for [C11H15N2O2]+ (M + H+) 207.11280, found 207.11284.
aHbCH2CN), 2.92 (ddd, J = 12.2, 8.0, 6.5 Hz, 1H, CHabCH2CN), 2.61 (qdd, J = 16.8, 8.2, 6.4 Hz, 2H, CH2C2CN), 1.81–1.74 (m, 1H, H-7syn), 1.66–1.59 (m, 1H, H-7anti); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 173.1 (C, O2CH3), 138.3 (CH, C-5), 135.6 (CH, C-6), 118.8 (C, CN), 68.5 (CH), 66.3 (CH), 53.2 (CH2), 52.1 (CH3, CO2H3), 47.8 (CH), 45.8 (CH2), 18.8 (CH2, H2CN); HRMS-ESI: calculated for [C11H15N2O2]+ (M + H+) 207.11280, found 207.11280.
O2CH3), 136.9 (CH, C-5), 133.1 (CH, C-6), 118.6 (C, CN), 65.5 (CH), 65.4 (CH), 52.1 (CH3, CO2H3), 50.6 (CH2), 48.1 (CH), 46.8 (CH2), 18.2 (CH2, H2CN); HRMS-ESI: calculated for [C11H15N2O2]+ (M + H+) 207.11280, found 207.11284.
aHbCH2CN), 2.92 (ddd, J = 12.2, 8.0, 6.5 Hz, 1H, CHabCH2CN), 2.61 (qdd, J = 16.8, 8.2, 6.4 Hz, 2H, CH2C2CN), 1.81–1.74 (m, 1H, H-7syn), 1.66–1.59 (m, 1H, H-7anti); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 173.1 (C, O2CH3), 138.3 (CH, C-5), 135.6 (CH, C-6), 118.8 (C, CN), 68.5 (CH), 66.3 (CH), 53.2 (CH2), 52.1 (CH3, CO2H3), 47.8 (CH), 45.8 (CH2), 18.8 (CH2, H2CN); HRMS-ESI: calculated for [C11H15N2O2]+ (M + H+) 207.11280, found 207.11280.
(±)-Methyl 2-benzyl-2-azabicyclo[2.2.1]hept-5-ene-3-exo-carboxylate (7) and (±)-methyl 2-benzyl-2-azabicyclo[2.2.1]hept-5-ene-3-endo-carboxylate (8) were synthesized following the procedures described earlier.26,28,38
:1 (12 ml), a catalytic amount of 10% Pd–C was added and the mixture was left under a H2 atmosphere (4 bar) and mechanical agitation. After 72 h the catalyst was filtered off through a celite pad and silica gel and washed with AcOEt and then MeOH to recover the amine 9. The methanolic filtrate was then concentrated by rotary evaporation and used in the next reaction without further purifications.
1H NMR (400 MHz, CDCl3): δ = 3.71 (s, 3H, CO2CH3), 3.48 (br s, 1H, H-3), 2.90 (dddd, J = 13.9, 12.5, 7.7, 6.9 Hz, 2H, C2CH2CN), 2.68 (br s, 1H), 2.63 (d, J = 4.0 Hz, 1H), 2.61–2.45 (m, 2H, CH2C2CN), 1.87–1.63 (m, 3H), 1.47–1.26 (m, 3H); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 173.4 (C, O2CH3), 118.7 (C, CN), 70.7 (CH), 61.5 (CH), 52.2 (CH3, CO2H3), 48.1 (CH2, H2CH2CN), 42.2 (CH), 36.8 (CH2, C-7), 28.8 (CH2), 23.0 (CH2), 18.4 (CH2, CH2H2CN); HRMS-ESI: calculated for [C11H17N2O2]+ (M + H+) 209.12845, found 209.12842.
:6), affording 6 as a yellow oil (0.96 g, 95%).
1H NMR (400 MHz, CDCl3): δ = 3.73 (s, 3H, CO2CH3), 3.30 (dd, J = 3.8, 1.7 Hz, 1H), 3.24 (br s, 1H), 3.01 (ddd, J = 12.3, 8.2, 6.4 Hz, 1H, CHCH2CN), 2.77 (br s, 1H), 2.68 (ddd, J = 12.3, 7.9, 6.5 Hz, 1H, CHCH2CN), 2.49 (qdd, J = 16.7, 8.0, 6.4 Hz, 2H, CH2C2CN), 1.73–1.53 (m, 3H), 1.50–1.36 (m, 2H), 1.35–1.24 (m, 1H); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 173.5 (C, O2CH3), 118.8 (C, CN), 72.0 (CH), 64.2 (CH), 53.3 (CH2, H2CH2CN), 51.9 (CH3, CO2H3), 41.9 (CH), 35.9 (CH2, C-7), 310. (CH2), 23.4 (CH2), 18.4 (CH2, CH2H2CN); HRMS-ESI: calculated for [C11H17N2O2]+ (M + H+) 209.12845, found 209.12832.
:1), affording 11 as a yellow oil (0.1660 g, 83%).
1H NMR (400 MHz, CD3CN): δ = 6.49–6.40 (m, 1H), 6.10–6.03 (m, 1H), 4.61 (br s, 1H, H-1), 3.68 (s, 3H, CO2CH3), 3.61 (dt, J = 3.5, 1.2 Hz, 1H, H-4), 3.53 (ddd, J = 13.5, 5.8, 4.6 Hz, CaHbCH2CN), 3.08–3.01 (m, J = 4.9, 3.6, 1.3, 0.6 Hz, 1H, H-5), 2.83 (dddd, J = 13.4, 9.1, 5.9, 0.7 Hz, 1H, CHabCH2CN), 2.49 (dddd, J = 17.0, 10.5, 7.5, 5.2, 2H, CH2C2CN), 1.87–1.82 (m, 1H, H-8syn), 1.65 (dddd, J = 10.8, 4.3, 2.9, 1.2 Hz, 1H, H-8anti); 13C{1H} NMR and DEPT (101 MHz, CD3CN): δ = 172.6 (C, O2CH3), 139.0 (CH), 131.2 (CH), 120.3 (C, CN), 81.6 (CH, C-1), 65.7 (CH, C-4), 51.9 (CH3, CO2H3), 51.4 (CH2, H2CH2CN), 42.4 (CH, C-5), 39.6 (CH2, C-8), 17.2 (CH2, CH2H2CN); HRMS-ESI: calculated for [C11H15N2O3]+ (M + H+) 223.10772, found 223.10761.
1H NMR (400 MHz, CDCl3): δ = 5.96–5.90 (m, 1H, H-8), 5.63 (dq, J = 5.7, 2.2 Hz, 1H, H-7), 5.14–5.08 (m, 1H, H-1), 3.70 (s, 3H, CO2CH3), 3.47–3.39 (m, 2H, H-4 + CaHbCH2CN), 3.34 (dddd, J = 12.1, 8.4, 6.5, 0.5 Hz, 1H, CHabCH2CN), 2.77 (ddd, J = 12.2, 8.6, 5.5 Hz, 1H, CH2CaHbCN), 2.71–2.56 (m, 2H, CH2CHabCN + H-5), 2.49–2.38 (m, 1H, H-6syn), 2.22–2.12 (m, 1H, H-anti); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 169.3 (C, CO2CH3), 136.2 (C-8), 129.2 (C-7), 118.4 (C, CN), 86.8 (CH, C-1), 72.3 (CH, C-4), 52.4 (CH2, H2CH2CN), 52.2 (CH3, CO2CH3), 46.5 (CH, C-5), 35.3 (CH2, C-6), 16.4 (CH2, H2CN); HRMS-ESI: calculated for [C11H15N2O3]+ (M + H+) 223.10772, found 223.10765.
1H NMR (400 MHz, CDCl3): δ = 5.89 (dtd, J = 2.8, 2.2, 0.6 Hz, 1H, H-8), 5.73 (ddd, J = 5.7, 4.3, 2.2 Hz, 1H, H-7), 5.10 (dt, J = 7.7, 1.8 Hz, 1H, H-1), 3.73 (s, 3H, CO2CH3), 3.35 (dddd, J = 9.8, 8.6, 7.5, 2.7 Hz, 1H, CaHbCH2CN), 3.22 (dt, J = 12.5, 7.5 Hz, 1H, CHabCH2CN), 3.14 (d, J = 7.1 Hz, 1H, H-4), 3.02–2.88 (m, 1H, CH2CaHbCN), 2.74–2.54 (m, 3H, CH2CHabCN + H-5 + H-6syn), 2.47–2.37 (m, 1H, H-6anti); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 170.0 (C, CO2CH3), 135.4 (C-8), 129.4 (C-7), 118.3 (C, CN), 88.0 (CH, C-1), 74.9 (CH, C-4), 52.6 (CH3, CO2CH3), 52.5 (CH2, H2CH2CN), 48.8 (CH, C-5), 37.2 (CH2, C-6), 16.8 (CH2, H2CN); HRMS-ESI: calculated for [C11H15N2O3]+ (M + H+) 223.10772, found 223.10771.
1H NMR (400 MHz, CDCl3): δ = 7.38 (br s, 1H, N–O), 3.71 (s, 3H, CO2CH3), 3.62 (br s, 1H, H-3), 3.15 (br s, 1H, H-1), 2.52 (br s, 1H, H-4), 2.38 (br s, 1H, H-6), 1.77 (d, J = 9.9 Hz, 1H, H-7), 1.73–1.63 (m, 1H, H-5), 1.62–1.53 (m, 1H, H-5), 1.46–1.35 (m, 1H, H-6), 1.34–1.29 (m, 1H, H-7); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 173.1 (C, CO2CH3), 73.1 (CH, C-1), 65.0 (CH, C-3), 52.0 (CH3, CO2H3), 41.4 (CH, C-4), 34.7 (CH2, C-7), 29.2 (CH2, C-5), 20.3 (CH2, C-6); HRMS-ESI: calculated for [C8H14NO3]+ (M + H+) 172.09682, found 172.09699.
1H NMR (400 MHz, CDCl3): δ = 8.19 (br s, 1H, N–O), 3.81 (dd, J = 4.3, 1.6 Hz, 1H, H-3), 3.71 (s, 3H, CO2CH3), 3.58 (d, J = 4.1 Hz, 1H, H-1), 2.73 (br s, 1H, H-4), 2.14 (ddt, J = 10.0, 3.9, 2.0 Hz, 1H, H-6), 1.60 (ddd, J = 16.1, 8.0, 3.9 Hz, 1H, H-7), 1.49–1.30 (m, 3H, H-5 + H-6 + H-7), 1.13–1.05 (m, 1H, H-5); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 172.5 (C, CO2CH3), 76.9 (CH, C-3), 68.5 (CH, C-1), 51.9 (CH3, CO2H3), 40.3 (CH, C-4), 35.5 (CH2, C-6), 25.8 (CH2, C-7), 23.0 (CH2, C-5); HRMS-ESI: calculated for [C8H14NO3]+ (M + H+) 172.09682, found 172.09702; mp = 65–68 °C;
1H NMR (400 MHz, CDCl3): δ = 3.73 (s, 3H, CO2CH3), 3.34 (dd, J = 8.8, 5.2 Hz, 1H, H-2), 3.18 (td, J = 7.8, 3.7 Hz, 1H, H-5), 3.07 (dt, J = 12.5, 7.2 Hz, 1H, CaHbCH2CN), 2.83 (dt, J = 12.5, 7.1 Hz, 1H, CHabCH2CN), 2.61–2.49 (m, 3H, H-5 + CH2C2CN), 2.20–2.06 (m, 1H), 2.03–1.80 (m, 3H); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 174.0 (C, CO2CH3), 118.6 (C, CN), 65.2 (CH, C-2), 53.0 (CH2, H2CH2CN), 51.8 (CH3, CO2CH3), 49.7 (CH2), 29.1 (CH2), 23.3 (CH2), 17.5 (CH2, CH2H2CN); HRMS-ESI: calculated for [C9H15N2O2]+ (M + H+) 183.11280, found 183.11288.
1H NMR (400 MHz, CDCl3): δ = 7.51 (br s, 1H, N–OH), 3.73 (s, 3H, CO2CH3), 3.66 (t, J = 8.0 Hz, 1H, H-2), 3.39–3.27 (m, 1H, H-5), 2.94 (dd, J = 16.9, 8.3 Hz, 1H, H-5), 2.29–2.13 (m, 1H), 1.98–1.77 (m, 3H); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 173.13 (C, CO2CH3), 69.65 (CH, C-2), 57.42 (CH2, C-5), 52.01 (CH3, CO2CH3), 25.99 (CH2), 20.74 (CH2); HRMS-ESI: calculated for [C6H12NO3]+ (M + H+) 146.08117, found 146.08115.
:1) (1.00 g, 4.85 mmol) in MeOH (15 ml) and 10% Pd–C (≈50 mg), TES (7.8 ml, 49 mmol) was added at once, without external temperature controlling. It was verified that the formation of H2 was significantly more vigorous and the system reached temperatures of around 50 °C. After the typical work-up protocol, the residue was subjected to column chromatography (eluent: Hex → Et2O), which afforded 23 as a yellow oil (0.8669 g, 85%).
1H NMR (400 MHz, CDCl3): δ = 3.71 (s, 3H, CO2CH3), 3.06 (d, J = 7.9 Hz, 1H, H-2), 2.96 (dt, J = 12.1, 6.8 Hz, 1H, CaHbCH2CN), 2.68 (dt, J = 12.1, 6.6 Hz, 1H, CHabCH2CN), 2.51–2.40 (m, 2H, CH2C2CN), 2.13–1.94 (m, 2H), 1.75 (dddd, J = 8.9, 7.7, 4.1, 1.9 Hz, 1H), 1.67–1.44 (m, 5H), 1.41–1.29 (m, 2H); 13C{1H} NMR and DEPT (101 MHz, CDCl3): δ = 175.34 (C, CO2CH3), 118.54 (C, CN), 65.41 (CH, C-2), 51.79 (CH3, CO2CH3), 43.89 (CH2, H2CH2CN), 43.09 (CH, C-3), 29.43 (CH2), 29.17 (CH2), 25.34 (CH2), 25.20 (CH2), 19.07 (CH2, CH2H2CN); HRMS-ESI: calculated for [C11H19N2O2]+ (M + H+) 211.14410, found 211.14421.
4.3. Computational details
All quantum chemical calculations were performed using the Gaussian 09 software package.50 Optimized geometries and respective electronic energies, Eel, were computed at the B3LYP/6-311++G(d,p) level of theory. For some cases frequency calculations at the same level of theory were carried out. These confirmed that the structures correspond to true minima (no imaginary frequencies were found), and allowed calculation of the enthalpies at T = 298 K, H298 K, by considering the contributions of Eel, zero-point energy (ZPE) and thermal enthalpy to T = 298.15 K; no scaling factors were used (this is a reasonable approximation for relative comparisons of molecular energetics). The torsional potential profile for the internal rotation defined by the C(H2)–C(H)–C(H)(COOCH3)–N dihedral in compound 11 was evaluated using B3LYP/6-311++G(d,p) by varying the dihedral angle from 60° to 160° in 10° increments and keeping it frozen while optimizing the remaining degrees of freedom. All calculations were performed without symmetry restrictions.Acknowledgements
The work was funded by FCT/MEC and FEDER under Program PT2020 (projects UID/QUI/50006/2013-POCI/01/0145/FEDER/007265 and UID/MULTI/04378/2013-POCI/01/0145/FEDER/007728). C. A. D. Sousa, I. E. Sampaio-Dias and C. F. R. A. C. Lima thank FCT for their grants SFRH/BPD/80100/2011, SFRH/BD/93632/2013 and SFRH/BPD/77972/2011, respectively.References
- M. B. Smith and J. March, March's Advanced Organic Chemistry, Wiley-Interscience, 6th edn, 2007 Search PubMed.
- J. J. Li, Name Reactions - A Collection of Detailed Mechanisms and Synthetic Applications, Springer, Heidelberg, 4 edn, 2009 Search PubMed.
- L. Kürti and B. Czakó, Strategic Applications of Named Reactions in Organic Synthesis, Elsevier Academic Press, USA, 2005 Search PubMed.
- C. A. D. Sousa, M. L. C. Vale, J. E. Rodríguez-Borges and X. García-Mera, J. Mol. Struct., 2012, 1007, 31–35 CrossRef CAS.
- I. Cikotiene, M. Jonusis and V. Jakubkiene, Beilstein J. Org. Chem., 2013, 9, 1819–1825 CrossRef PubMed.
- R. W. Sabnis, D. W. Rangnekar and N. D. Sonawane, J. Heterocycl. Chem., 1999, 36, 333–345 CrossRef CAS.
- G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045–1075 RSC.
- A. Jacobs, Understanding Organic Reaction Mechanisms, Cambridge University Press, 1997 Search PubMed.
- J. Meisenheimer, Chem. Ber., 1919, 52, 1667–1677 CrossRef.
- Y. Xie, M. Sun, H. Zhou, Q. Cao, K. Gao, C. Niu and H. Yang, J. Org. Chem., 2013, 78, 10251–10263 CrossRef CAS PubMed.
- S. G. Davies and G. D. Smyth, Tetrahedron: Asymmetry, 1996, 7, 1005–1006 CrossRef CAS.
- T. Kurihara, Y. Sakamoto, J. Tsukamoto, H. Ohishi, S. Harusawa and R. Yoneda, J. Chem. Soc., Perkin Trans. 1, 1993, 81–87 RSC.
- H. Kondo, F. Sakamoto, T. Uno, Y. Kawahata and G. Tsukamoto, J. Med. Chem., 1989, 32, 671–674 CrossRef CAS PubMed.
- A. Theodorou, D. Limnios and C. G. Kokotos, Chem. – Eur. J., 2015, 21, 5238–5241 CrossRef CAS PubMed.
- R. Yoneda, Y. Sakamoto, Y. Oketo, S. Harusawa and T. Kurihara, Tetrahedron, 1996, 52, 14563–14576 CrossRef CAS.
- L. Remen and A. Vasella, Helv. Chim. Acta, 2002, 85, 1118–1127 CrossRef CAS.
- I. A. O'Neil, E. Cleator and D. J. Tapolczay, Tetrahedron Lett., 2001, 42, 8247–8249 CrossRef.
- A. García Martínez, E. Teso Vilar, A. García Fraile, S. De la Moya Cerero and B. Lora Maroto, Tetrahedron: Asymmetry, 2002, 13, 17–19 CrossRef.
- H. Miyatake-Ondozabal, L. M. Bannwart and K. Gademann, Chem. Commun., 2013, 49, 1921–1923 RSC.
- Q. Jia, P. M. S. Benjamin, J. Huang, Z. Du, X. Zheng, K. Zhang, A. H. Conney and J. Wang, Synlett, 2013, 79–84 CAS.
- I. A. O'Neil, V. E. Ramos, G. L. Ellis, E. Cleator, A. P. Chorlton, D. J. Tapolczay and S. B. Kalindjian, Tetrahedron Lett., 2004, 45, 3659–3661 CrossRef.
- G. L. Ellis, I. A. O'Neil, V. E. Ramos, S. B. Kalindjian, A. P. Chorlton and D. J. Tapolczay, Tetrahedron Lett., 2007, 48, 1687–1690 CrossRef CAS.
- J. Blanchet, M. Bonin, L. Micouin and H. Husson, Tetrahedron Lett., 2000, 41, 8279–8283 CrossRef CAS.
- L. E. H. Buston, I. Coldham and K. R. Mulholland, Tetrahedron: Asymmetry, 1998, 9, 1995–2009 CrossRef.
- A. Guarna, E. G. Occhiato, M. Pizzetti, D. Scarpi, S. Sisi and M. Sterkenburg, Tetrahedron: Asymmetry, 2000, 11, 4227–4238 CrossRef CAS.
- K. C. Majumdar, B. Roy, P. K. Basu and P. Biswas, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2006, 45, 719–725 Search PubMed.
- Z. Mucsi, A. Szabó, I. Hermecz, Á. Kucsman and I. G. Csizmadia, J. Am. Chem. Soc., 2005, 127, 7615–7631 CrossRef CAS PubMed.
- E. Wojaczyńska, J. Wojaczyński, K. Kleniewska, M. Dorsz and T. K. Olszewski, Org. Biomol. Chem., 2015, 13, 6116–6148 Search PubMed.
- J. E. Rodríguez-Borges, M. L. C. Vale, F. R. Aguiar, M. J. Alves and X. García-Mera, Synthesis, 2008, 971–977 CrossRef.
- J. E. Rodríguez-Borges, S. Gonçalves, M. L. C. Vale, X. García-Mera, A. Coelho and E. Sotelo, J. Comb. Chem., 2008, 10, 372–375 CrossRef PubMed.
- C. A. D. Sousa, M. L. C. Vale, J. E. Rodríguez-Borges, X. García-Mera and J. Rodríguez-Otero, Tetrahedron Lett., 2008, 49, 5777–5781 CrossRef CAS.
- C. A. D. Sousa, M. L. C. Vale, J. E. Rodríguez-Borges and X. García-Mera, New J. Chem., 2010, 34, 2546–2551 RSC.
- C. A. D. Sousa, M. L. C. Vale, X. García-Mera and J. E. Rodríguez-Borges, Tetrahedron, 2012, 68, 1682–1687 CrossRef CAS.
- C. A. D. Sousa, F. Rizzo-Aguiar, M. L. C. Vale, X. García-Mera, O. Caamaño and J. E. Rodríguez-Borges, Tetrahedron Lett., 2012, 53, 1029–1032 CrossRef CAS.
- C. A. D. Sousa, M. A. Maestro, X. Garcia-Mera and J. E. Rodriguez-Borges, RSC Adv., 2014, 4, 57768–57772 RSC.
- L. Stella and H. Abraham, Tetrahedron Lett., 1990, 31, 2603–2606 CrossRef CAS.
- X. García-Mera, J. E. Rodríguez-Borges, M. L. C. Vale and M. J. Alves, Tetrahedron, 2011, 67, 7162–7172 CrossRef.
- F. Fernández, X. García-Mera, J. E. Rodríguez-Borges and M. L. C. Vale, Tetrahedron Lett., 2003, 44, 431–433 CrossRef.
- M. Hursthouse, K. M. A. Malik, D. E. Hibbs, S. M. Roberts, A. J. H. Seago, V. Sik and R. Storer, J. Chem. Soc., Perkin Trans. 1, 1995, 2419–2425 RSC.
- B. M. Domínguez and P. M. Cullis, Tetrahedron Lett., 1999, 40, 5783–5786 CrossRef.
- C. D. Cox, J. R. Malpass, J. Gordon and A. Rosen, J. Chem. Soc., Perkin Trans. 1, 2001, 2372–2379 RSC.
- D. M. Hodgson, C. R. Maxwell, R. Wisedale, I. R. Matthews, K. J. Carpenter, A. H. Dickenson and S. Wonnacott, J. Chem. Soc., Perkin Trans. 1, 2001, 3150–3158 RSC.
- M. Ishikura, M. Matsumoto and A. Murakam, Heterocycles, 2004, 64, 241–248 CrossRef CAS.
- P. D. Bailey, I. M. McDonald, G. M. Rosair and D. Taylor, Chem. Commun., 2000, 2451–2452 RSC.
- H. T. Nagasawa, J. G. Kohlhoff, P. S. Fraser and A. A. Mikhail, J. Med. Chem., 1972, 15, 483–486 CrossRef CAS PubMed.
- D. A. Alonso, S. K. Bertilsson, S. Y. Johnsson, S. J. M. Nordin, M. J. Södergren and P. G. Andersson, J. Org. Chem., 1999, 64, 2276–2280 CrossRef CAS.
- V. I. Tararov, R. Kadyrov, Z. Kadyrova, N. Dubrovina and A. Borner, Tetrahedron: Asymmetry, 2002, 13, 25–28 CrossRef CAS.
- S. Venkatraman, F. G. Njoroge, W. Wu, V. Girijavallabhan, A. J. Prongay, N. Butkiewicz and J. Pichardo, Bioorg. Med. Chem. Lett., 2006, 16, 1628–1632 CrossRef CAS PubMed.
- C. A. D. Sousa, J. E. Rodríguez-Borges and X. Garcia-Mera, Tetrahedron Lett., 2014, 55, 4628–4631 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00330c |
| This journal is © the Partner Organisations 2016 |