
Highly selective synthesis of 6-substituted benzothiophenes by Sc(OTf)3-catalyzed intermolecular cyclization and sulfur migration†
Xiaoming
Wang
,
Tobias
Gensch
and
Frank
Glorius
*
Westfälische Wilhelms- Universität Münster, Organisch-Chemisches Institut, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de; Web: http://www.uni-muenster.de/Chemie.oc/glorius/index.html Fax: (+49) 251-83-33202
First published on 12th September 2016
Abstract
A series of 6-substituted benzo[b]thiophenes was efficiently synthesized using a Sc(OTf)3-catalyzed intermolecular cyclization between para-substituted N-(arylthio)succinimides and alkynes taking advantage of a unique and selective 1,2-sulfur migration. Investigations with DFT shed light on the migration process occuring via an electrophilic ipso cyclization of a vinyl cation to a key spirocyclic thiete intermediate.
Introduction
Benzannulated heterocyclic compounds, such as benzofuran, indole or benzo[b]thiophene, have diverse applications in medicinal chemistry and materials science and have duly attracted a great deal of interest in industry and academia.1 Among many strategies for the synthesis of these heterocyclic compounds, the intermolecular coupling of aromatic substrates with alkynes via C–H functionalization2 presents a straightforward route from readily available starting materials that benefits from high step and atom economy. However, the site selectivity in the C–H functionalization step can be problematic when unsymmetrical meta-substituted substrates are used, resulting in a mixture of the corresponding products (Scheme 1a). Thus, the development of novel and efficient methods for the highly selective synthesis of 6-substituted products will be highly desirable.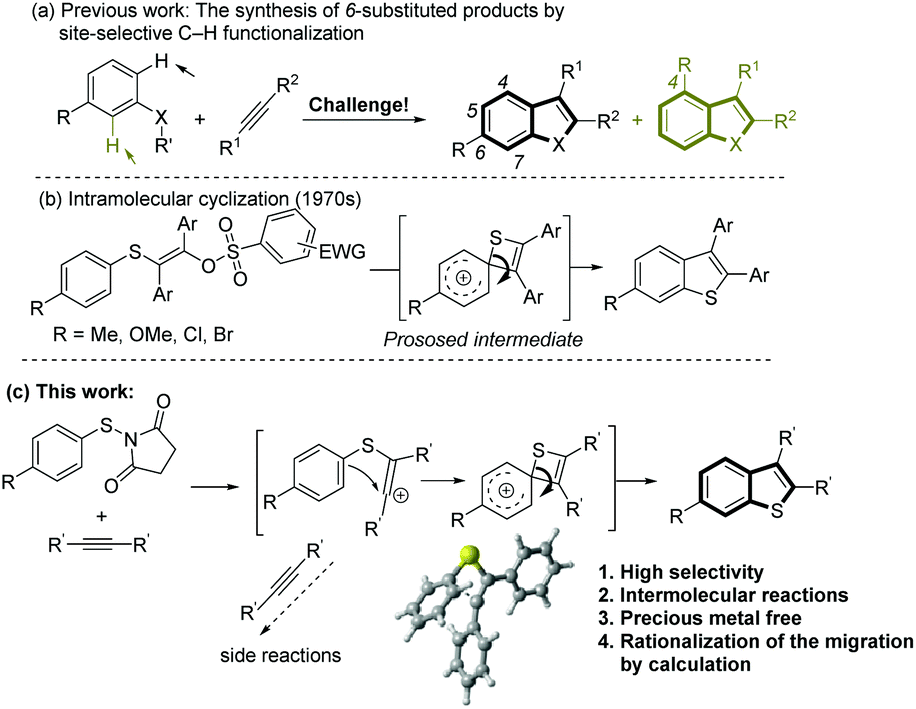 | ||
| Scheme 1 The synthesis of 6-substituted benzothiophenes by sulfur migration. | ||
Although polysubstituted benzo[b]thiophenes are of considerable importance in industry and academia,3 compared with the synthesis of other heteroarenes such as benzofuran and indole,2 intermolecular cyclization strategies for the formation of the benzothiophene skeleton by C–H functionalization are limited. Recently, oxidative radical cyclizations, involving benzenesulfanyl radical addition onto alkynes, have been successfully employed.4 However, the selective synthesis of 6- over 4-substituted products from meta-substituted starting materials is riddled by low selectivities. As early as the 1968, an unusual rearrangement to 6-substituted benzothiophenes was observed in the cyclization of specific arylthiovinyl sulfonic esters with a para substituent.5 Initiated by a key vinyl cation, a 1,2-sulfur migration from a thiete/Wheland intermediate was proposed to explain the rearrangement (Scheme 1b). However, this kind of intramolecular cyclization has not been applied in organic synthesis because of the difficulties in the preparation of the arylthiovinyl sulfonic esters, very low atom-economy and a lack of understanding about the formation of the thiete intermediate.
Recently, N-(arylthio)succinimides (as well as N-arylthio-phthalimides) have been utilized as electrophilic sulfenylating agents in the arylthiolation of (hetero)aromatic C–H bonds and functionalization of olefins and alkynes.6 Thus, we proposed the reaction of N-(arylthio)succinimide with alkynes would generate the key vinyl cation that could further react with the nucleophilic phenyl group. A challenge in this intramolecular cyclization process is the potential for side reactions by the vinyl cation.
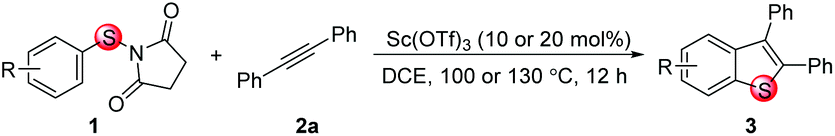
Here, we present a highly selective and practical synthesis of 6-substituted benzothiophenes by Sc(OTf)3-catalyzed intermolecular cyclization of para-substituted N-(arylthio)succinimides with diarylalkynes (Scheme 1c). Insight on the formation of the spirocyclic thiete intermediate is provided by DFT calculations. The cyclization to the thiete is facilitated by the short distance between the vinyl cation and the ipso carbon, so very little structural distortion is required in the transition state. When this Wheland complex is not stabilized by the substituents, the competing cyclization to the ortho carbon, resulting in the formation of a five membered thiocycle, can become more favored. In the course of the computational study, an alternative pathway was discovered that could result in an overall aryl migration. Consecutive experiments confirmed the feasibility of this pathway. During the preparation of this manuscript, a similar silver-mediated annulation of N-(arylthio)succinimides with alkynes was reported that was proposed to proceed via oxidative radical annulation.7
Results and discussion
Our investigation commenced by studying the reaction of para-fluorine substituted substrate 1a with 2a using Lewis acids or Brønsted acids as catalysts in DCE at 100 °C (Table 1). The reactions using CF3CO2H or MsOH as the catalyst didn't give any desired product. To our delight, with BF3·OEt2 as the catalyst (Table 1, entry 3), the reaction afforded 6-fluorine substituted benzothiophene 3a with high regioselectivity (>99/1), albeit in low yield (29%). The structural assignment was confirmed by X-ray crystal structure analysis. This sulfur migration involves C–S bond cleavage and the formation of a new C–S bond at the ortho-position.5,8,9|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Ratio of isomersb | Yield (%)c |
| a Reactions were carried out using catalyst (10 mol%), 1a (0.15 mmol), and 2a (0.1 mmol) in DCE (2.5 mL) for 12 h at 100 °C under an argon atmosphere. b The ratio of the isomers was determined by GC. c GC yield. d Isolated yield in 0.2 mmol scale reaction at 130 °C. | ||||
| 1 | CF3CO2H | DCE | — | 0 |
| 2 | MsOH | DCE | — | 0 |
| 3 | BF3·OEt2 | DCE | >99/1 | 29 |
| 4 | TMSOTf | DCE | >99/1 | 32 |
| 5 | Al(OTf)3 | DCE | >99/1 | 22 |
| 6 | Zn(OTf)2 | DCE | — | 0 |
| 7 | Yt(OTf)3 | DCE | — | 0 |
| 8 | In(OTf)3 | DCE | >99/1 | 77 |
| 9 | Sc(OTf)3 | DCE | >99/1 | 78 (82)d |
| 10 | Sc(OTf)3 | Toluene | >99/1 | 6 |
| 11 | Sc(OTf)3 | CH3CN | — | <5 |
| 12 | — | DCE | — | 0 |
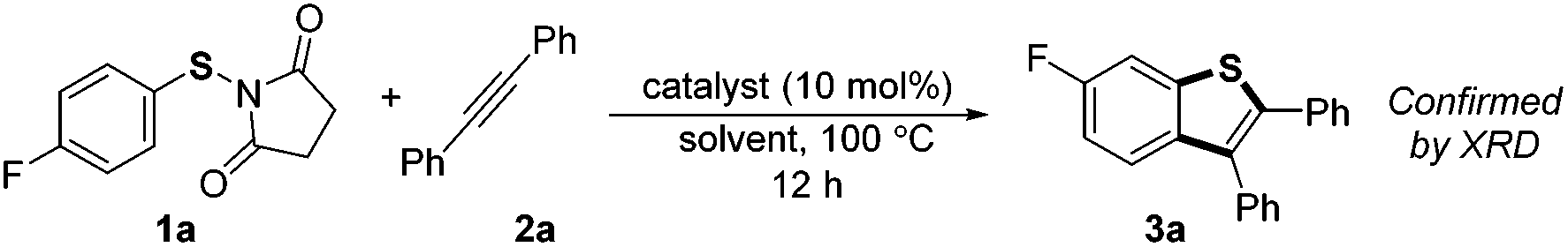
Further optimization of the conditions by testing several kinds of Lewis acids and solvents revealed that the reaction with Sc(OTf)3 as the catalyst and DCE as solvent gave the highest yield (Table 1, entry 9, 82%). A control reaction conducted under these conditions without Sc(OTf)3 did not give any desired product (Table 1, entry 12).
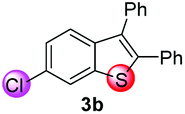
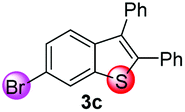
Under the optimized reaction conditions, the scope of N-(arylthio)succinimides 1 was investigated using the internal alkyne diphenylacetylene (2a) with Sc(OTf)3 as the catalyst. As shown in Table 2, the cyclization of substrates 1 bearing different substituents with 2a proceeded smoothly to afford the products 3. Both para-electron-donating and electron-withdrawing substituents on the phenyl ring of the substrates gave the products resulting from sulfur migration in high selectivity (>93/7). As such, this method represents a highly efficient and practical route to access 6-substituted benzo[b]thiophenes, which are typically difficult to obtain with high regioselectivity from meta-substituted starting materials (Table 2, entries 1–5). It is worth mentioning that this method was compatible with important functional groups on the phenyl ring of the N-(arylthio)succinimides such as halogens (F, Cl and Br) and methoxy groups. For example, π-extended multiply arylated benzo[b]thiophene derivatives have recently attracted considerable interest in the field of organic electronics.10 Thus, halogen substituted benzo[b]thiophenes such as 3b and 3c obtained by our method have potential value for the synthesis of such materials by subsequent cross-coupling. With unsubstituted substrate 1f, the reaction also afforded the desired product 3f in 68% yield. However, meta-substituted substrates (1g and 1h) afforded the corresponding products without sulfur migration as the major products, indicating a sensitivity of the reaction mechanism to the substitution pattern. This is in agreement with the results of the intramolecular cyclization of arylthiovinyl sulfonic esters.5
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Ratio of isomersb | Yieldc (%) |
| a Reactions were carried out using Sc(OTf)3 (10 or 20 mol%), 1 (0.3 or 0.4 mmol), and 2a (0.2 mmol) in DCE (5 mL) for 12 h at 100 °C or 130 °C under an argon atmosphere. b The ratio of the isomers was determined by GCMS or NMR. c Yield of isolated products for all the isomers is given. For details, see ESI. | ||||
| 1 |
|
|
>99/1 | 82 |
| 2 |
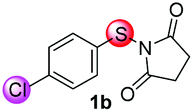
|
|
93/7 | 79 |
| 3 |
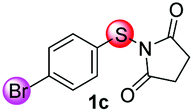
|
|
95/5 | 74 |
| 4 |
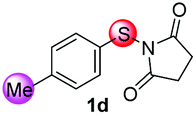
|
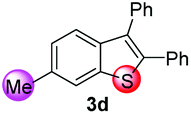
|
>99/1 | 67 |
| 5 |
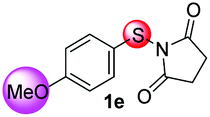
|
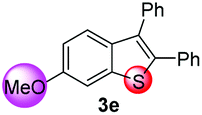
|
>99/1 | 56 |
| 6 |
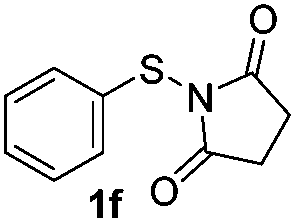
|
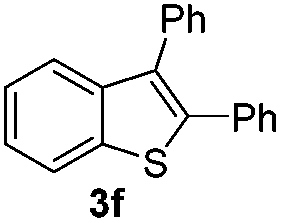
|
— | 68 |
| 7 |
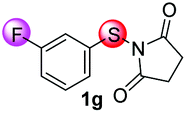
|
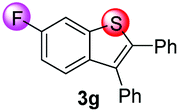
|
68/29/2/1 | 61 |
| 8 |
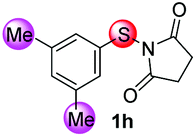
|
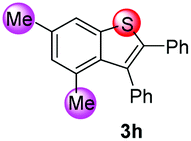
|
88/12 | 70 |
We next examined the scope of internal alkynes in the annulations with 1 (Scheme 2). Unsubstituted substrate 1f was chosen to test the reactivity first. A variety of symmetric diaryl substituted alkynes could be readily converted into the corresponding products 3i–m. The diaryl alkynes with F or Cl substituents at the para position gave the products 3i and 3j in moderate yields (53 and 56%). Substituents at the meta or ortho-positions of the phenyl group of the alkynes did not affect the process with the corresponding products 3k and 3l. In addition to symmetrical alkynes, the unsymmetrical alkyne ethyl 3-phenylpropiolate also gave the desired product 3o in 60% yield with complete regioselectivity. With para-fluorine substituted substrate 1a, the corresponding 6-fluorine substituted benzothiophenes 3n–3r resulting from 1,2-sulfur migration were obtained in high selectivities with moderate to good yields.
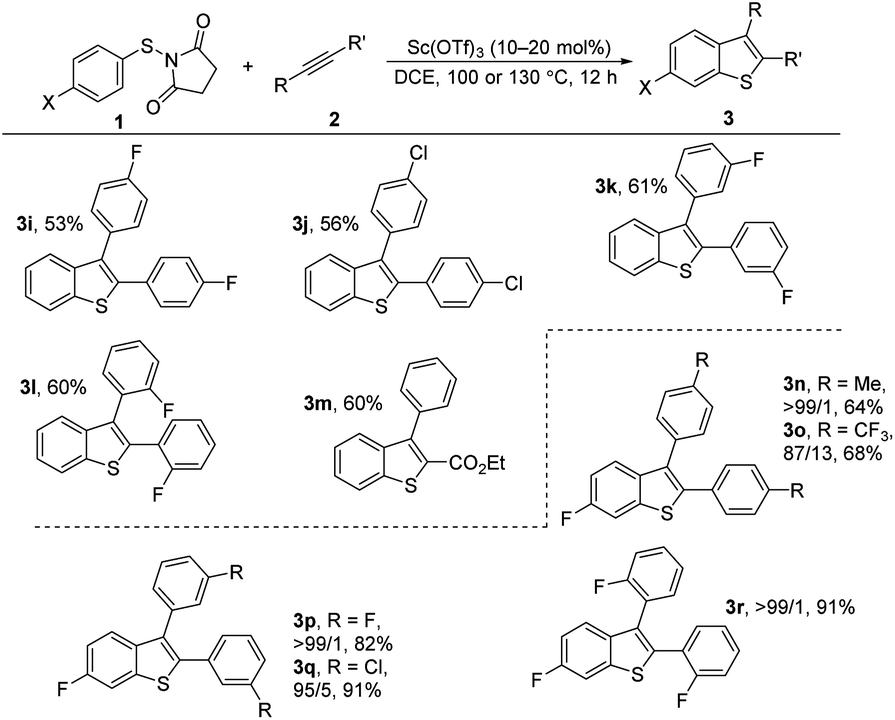 | ||
| Scheme 2 Sc(OTf)3-catalyzed cyclization of N-(arylthio)succinimides 1f or 1a with internal alkynes 2. Reactions were carried out using Sc(OTf)3 (10 or 20 mol%), 1 (0.3 or 0.4 mmol), and 2 (0.2 mmol) in DCE (5 mL) for 12 h at 100 °C or 130 °C under an argon atmosphere. Yield of isolated products is given. The ratio of the isomers was determined by NMR. For details, see ESI.† | ||
To better understand the mechanism of the process, a series of experiments based on unsubstituted substrate 1f were carried out (see the ESI†). Although N-(phenylthio)succinimide is a well-known electrophilic sulfenylating agent, the formation of a benzenesulfanyl radical is still possible9 because 1,2-diaryldisulfide can be detected by GCMS in some cases. Upon using 1,2-diphenyldisulfide as the substrate under the standard conditions with 2a, product 3f was formed in only 6% yield and most of the starting material was recovered. In addition, the reaction of 1f with 2a was tested in the presence of BHT (2,6-di-tert-butyl-4-methylphenol) as a radical scavenger and the reaction still afforded the product 3f, albeit in a lower yield of 30%. Moreover, in a competition reaction, 1f reacted exclusively with diphenylacetylene 2a, and not with dimethyl but-2-ynedioate, which is typically a better radical acceptor.4 All of these results suggest that a radical process is unlikely to be the major pathway involved in this reaction.7 The intermolecular kinetic isotope effect (KIE) was investigated using deuterated N-(phenylthio)succinimide, with an inverse secondary isotope effect (KIE 0.91)11 being observed. This result indicates that C–H bond cleavage is not itself the rate-determining step, but rather a change in the hybridization of a carbon from sp2 to sp3 is involved in a kinetically relevant step.11
For further insight, the mechanism was also studied computationally at the IEFPCM(DCE)/M06-2X/6-311G** level of DFT (Fig. 1).12 The thiirenium cation II can be formed from the reaction of 1f with 2a in the presence of a Lewis acid.13 An electrophilic attack on the arene becomes possible after isomerization to the vinyl cation III. In this structure, the electrophilic carbocation is positioned next to the arene with a relatively short distance (2.76 Å) to the ipso-carbon. Due to this proximity, the cyclization to a four-membered spirothiete intermediate (IVa) requires very little structural distortion and can occur readily. Conversely, the cyclization directly to a benzothiophenylium (V′) has a higher barrier by 4.6 kcal mol−1 and requires a greater structural reorganization. The thiete, which exists in two isomers (IVa and IVb) with a slightly different orientation of the cyclohexadienyl ring, has a low barrier of 2.6 kcal mol−1 for a 1,2-sulfur shift to the benzothiophenylium V. Deprotonation of V would give the product with the observed regiochemistry for para-substituted starting materials. The transition state structures of both electrophilic cyclizations from vinyl cation III (III–IVa and III–V′) are product-like. Thus, the barriers of these late transition states will be intimately affected by the ability of substituents at the phenyl ring to stabilize the forming Wheland intermediate. Consequently, para-directing substituents which are para to sulfur favor the ipso-cyclization to the thiete IVa and thus the pathway involving 1,2 migration. On the other hand, the direct formation of the five-membered intermediate V′ is favored by para-directing groups which are meta to the sulfur.
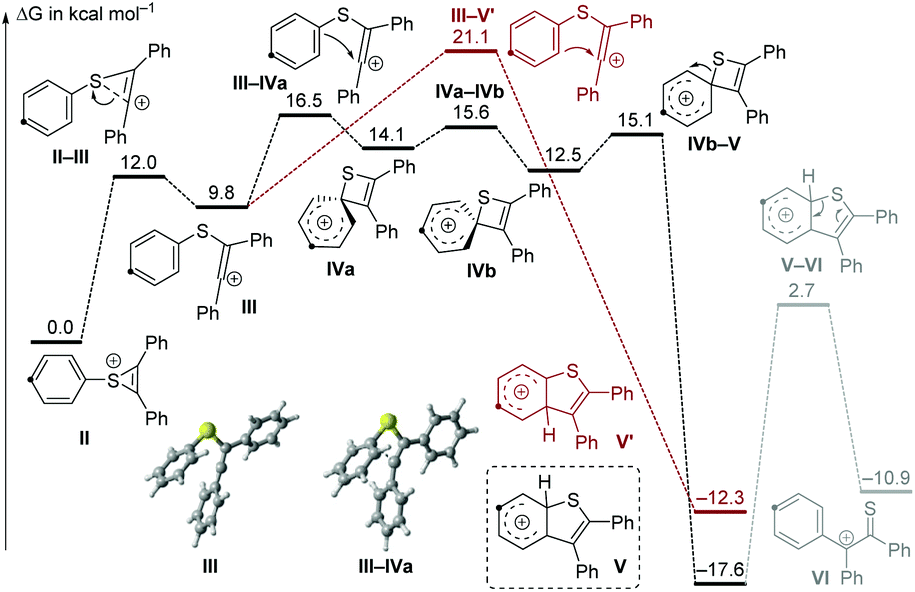 | ||
| Fig. 1 Free energy diagram (T = 100 °C) for the reactivity of thiirenium intermediate II. The same carbon has been highlighted in all structures for clarity. For computational details, see the ESI.† | ||
In the course of the computational study, a thioketone/benzylic cation structure was found (VI), as well as a transition state connecting this with the benzothiophenylium intermediate V. From VI, two alternative cyclization directions are possible, depending on which arene reacts with the sulfur. Wondering if this structure had any relevance to our process, we used perdeuterated alkyne 2a-d10 to check for any side products bearing only 9 deuterium atoms (Scheme 3). Using substrate 1b, no such product was detected on careful inspection of the GCMS trace. We reasoned, however, that steric bulk adjacent to the sulfur atom may lead to a change in the course of the reaction and favor a pathway involving intermediate VI. Indeed, the sterically hindered 2,4-dimethyl substituted substrate 1i resulted in three products, one of which only contained 9 deuterium atoms. This compound 3s′′-d9 was isolated in 9% yield and analysis of its structure confirmed that the xylyl ring is situated at the 3-position of the benzothiophene. This experimental evidence lends credibility to the existence of the intermediate V and the conclusions from the computations.
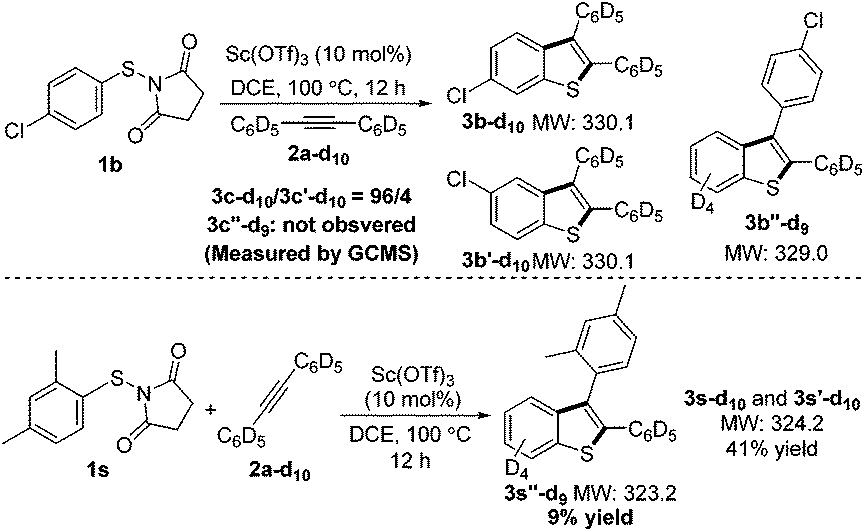 | ||
| Scheme 3 Aryl migration experiments. | ||
On the basis of these results and reports in the literature,5,6 a possible mechanism is suggested in Scheme 4. Firstly, the succinimide moiety of the sulfenylating agent 1 coordinates to the Lewis acid, generating an electrophilic intermediate I, which can undergo a nucleophilic attack by the alkyne to produce the thiirenium ion intermediate II,13 which is in equilibrium with the vinyl cation III. The cyclization to the four-membered thiete intermediate IV5,14 is favored over the formation of the five-membered cation V′ due to the proximity of the ipso-C to the cation in III. A 1,2-shift of the sulfur atom delivers cation V.8 Subsequent deprotonation affords the final product 3, resulting from formal aryl migration. It should be noted that the involvement of other processes, possibly involving radicals, cannot be totally excluded at this stage. The energetic difference between III–IVa and III–V′ is dependent on the stabilization of the forming Wheland intermediates, so that the reaction selectivity is influenced by the effect of the substituents at the phenylthio ring.
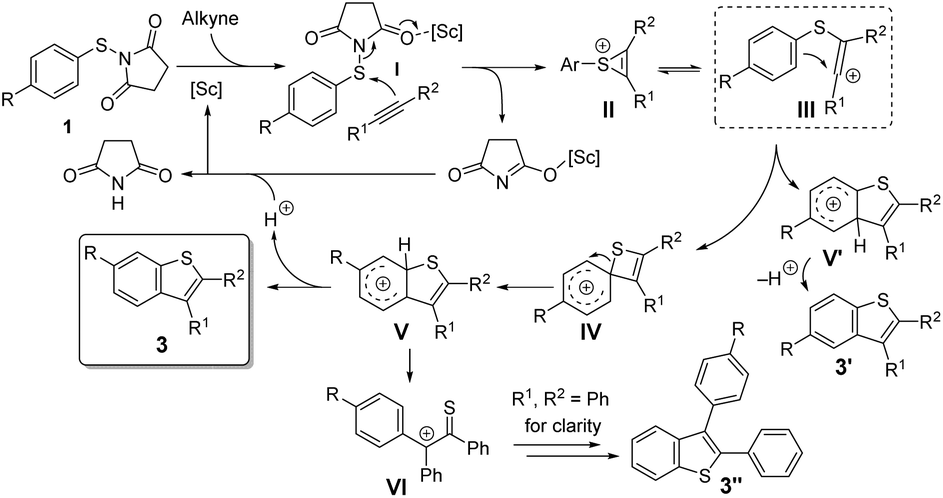 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed a Sc(OTf)3-catalyzed cyclization of N-(arylthio)succinimides with diarylalkynes leading to 2,3-diaryl benzo[b]thiophenes, which are key units in biologically active molecules and organic optoelectronic materials. Especially, the present methodology provides a highly efficient and practical route to access 6-substituted benzo[b]thiophenes from para-substituted substrates, involving an unusual 1,2-sulfur migration to afford isomerized products with high selectivity. Insight from DFT suggests that the 1,2-sulfur migration occurs from a four-membered thiete intermediate resulting from electrophilic cyclization of a vinyl cation with a short contact between the cation and the ipso carbon.Acknowledgements
This work was supported by the European Research Council under the European Community's Seventh Framework Program (FP7 2007-2013)/ERC Grant agreement no 25936 and the Alexander von Humboldt Foundation (X. Wang). We also thank Dr Christian Mück-Lichtenfeld, Dr Matthew N. Hopkinson and Dr Lisa Candish for helpful discussions.Notes and references
- (a) T. Eicher and S. Hauptmann, The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications, Wiley-VCH, Weinheim, 2nd edn, 2003 Search PubMed; (b) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed; (c) F. Mathey, Acc. Chem. Res., 2004, 37, 954 CrossRef CAS PubMed.
- (a) P. Thansandote and M. Lautens, Chem. – Eur. J., 2009, 15, 5874 CrossRef CAS PubMed; (b) T. Satoh and M. Miura, Synthesis, 2010, 3395 CrossRef CAS; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (d) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed; (e) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (f) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (g) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (h) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (i) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (j) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed.
- (a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed; (b) T. Y. Zhang, J. O'Toole and C. S. Proctor, Sulfur Rep., 1999, 22, 1 CrossRef CAS; (c) B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937 CrossRef CAS PubMed. Selected examples for the synthesis of benzothiophenes, see: (d) K. Inamoto, Y. Arai, K. Hiroya and T. Doi, Chem. Commun., 2008, 5529 RSC; (e) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188 CrossRef CAS PubMed; (f) T. Inami, Y. Baba, T. Kurahashi and S. Matsubara, Org. Lett., 2011, 13, 1912 CrossRef CAS PubMed.
- (a) K. Undheim and R. Lie, Acta Chem. Scand., 1973, 27, 595 CrossRef CAS; (b) L. Benati, P. C. Montevecchi and P. Spagnolo, J. Chem. Soc., Perkin Trans. 1, 1991, 2103 RSC; (c) K. Inamoto, Y. Arai, K. Hiroya and T. Doi, Chem. Commun., 2008, 5529 RSC; (d) K. Liu, F. Jia, H. Xi, Y. Li, X. Zheng, Q. Guo, B. Shen and Z. Li, Org. Lett., 2013, 15, 2026 CrossRef CAS PubMed; (e) D. Yang, K. Yan, W. Wei, L. Tian, Q. Li, J. You and H. Wang, RSC Adv., 2014, 4, 48547 RSC; (f) D. Wan, Y. Yang, X. Liu, M. Li, S. Zhao and J. You, Eur. J. Org. Chem., 2016, 55 CrossRef CAS; (g) K. C. Majumdar and P. Debnath, Tetrahedron, 2008, 64, 9799 CrossRef CAS.
- (a) G. Capozzi, G. Melloni, G. Modena and M. Piscitelli, Tetrahedron Lett., 1968, 37, 4039 CrossRef; (b) G. Capozzi, G. Melloni, G. Modena and U. Tonellato, J. Chem. Soc. D, 1969, 1520 RSC; (c) G. Capozzi, G. Melloni and G. Modena, J. Chem. Soc. C, 1970, 2621 RSC; (d) G. Melloni and G. Modena, J. Chem. Soc., Perkin Trans. 1, 1972, 218 RSC; (e) G. Capozzi, G. Melloni and G. Modena, J. Org. Chem., 1970, 35, 1217 CrossRef CAS; (f) M. S. Newman, Acc. Chem. Res., 1972, 5, 354 CrossRef CAS.
- Selected examples see: (a) H. Wang, D. Huang, D. Cheng, L. Li and Y. Shi, Org. Lett., 2011, 13, 1650 CrossRef CAS PubMed; (b) S. E. Denmark, D. J. P. Kornfilt and T. Vogler, J. Am. Chem. Soc., 2011, 133, 15308 CrossRef CAS PubMed; (c) P. Saravanan and P. Anbarasan, Org. Lett., 2014, 16, 848 CrossRef CAS PubMed; (d) S. E. Denmark and A. Jaunet, J. Org. Chem., 2014, 79, 140 CrossRef CAS PubMed; (e) S. E. Denmark and H. M. Chi, J. Am. Chem. Soc., 2014, 136, 8915 CrossRef CAS PubMed; (f) T. Hostier, V. Ferey, G. Ricci, D. G. Pardo and J. Cossy, Org. Lett., 2015, 17, 3898 CrossRef CAS PubMed; (g) S. E. Denmark, E. Hartmann, D. J. P. Kornfilt and H. Wang, Nat. Chem., 2014, 6, 1056 CrossRef CAS PubMed; (h) C. Saluzzo, Bull. Soc. Chim. Fr., 1994, 131, 831 CAS.
- E. Ramesh, M. Shankar, S. Dana and A. K. Sahoo, Org. Chem. Front., 2016, 3, 1126 RSC.
- (a) A. W. Sromek and V. Gevorgyan, Top. Curr. Chem., 2007, 274, 77 CrossRef CAS; (b) D. J. Fox, D. House and S. Warren, Angew. Chem., Int. Ed., 2002, 41, 2462 CrossRef CAS; (c) S. Kobayashi, T. Kitamura, H. Taniguchi and W. Schnabel, Chem. Lett., 1984, 2101 CrossRef CAS; (d) A. W. Sromek and V. Gevorgyan, Top. Curr. Chem., 2007, 274, 77 CrossRef CAS.
- (a) Y. Unoh, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 12975 CrossRef CAS PubMed; (b) Y.-R. Chen and W.-L. Duan, J. Am. Chem. Soc., 2013, 135, 16754 CrossRef CAS PubMed; (c) W. Ma and L. Ackermann, Synthesis, 2014, 2297 CAS; (d) A. Studer and M. Bossart, Tetrahedron, 2001, 57, 9649 CrossRef CAS.
- (a) G. Barbarella, L. Favaretto, A. Zanelli, G. Gigli, M. Mazzeo, M. Anni and A. Bongini, Adv. Funct. Mater., 2005, 15, 664 CrossRef CAS; (b) M.-S. Kim, B.-K. Choi, T.-W. Lee, D. Shin, S. K. Kang, J. M. Kim, S. Tamura and T. Noh, Appl. Phys. Lett., 2007, 91, 251111 CrossRef; (c) J. H. Jeon, N.-J. Lee, J.-H. Lee and M. C. Suh, Dyes Pigm., 2014, 111, 116 CrossRef CAS.
- (a) S. E. Scheppele, Chem. Rev., 1972, 72, 511 CrossRef; (b) M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857 CrossRef PubMed.
- (a) M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed; (b) Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; (c) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (d) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS; (e) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed . See the ESI† for full details on the calculations.
- (a) V. Luccbini, G. Modena and L. Pasquato, J. Am. Chem. Soc., 1993, 115, 4527 CrossRef; (b) W. Adam, S. G. Bosio, B. Fröhling, D. Leusser and D. Stalke, J. Am. Chem. Soc., 2002, 124, 8316 CrossRef CAS PubMed; (c) M. Fachini, V. Lucchini, G. Modena, M. Pasi and L. Pasquato, J. Am. Chem. Soc., 1999, 121, 3944 CrossRef CAS; (d) R. Destro, V. Lucchini, G. Modena and L. Pasquato, J. Org. Chem., 2000, 65, 3367 CrossRef CAS PubMed; (e) L. Bend and P. C. Montevecchi, Tetrahedron, 1993, 49, 5363 Search PubMed; (f) L. Benati and P. C. Montevecchi, J. Chem. Soc., Perkin Trans. 1, 1989, 1105 RSC.
- (a) D. C. Dittmer and F. A. Davis, J. Am. Chem. Soc., 1965, 87, 2064 CrossRef CAS; (b) D. C. Dittmer, K. Takahashi and F. A. Davis, Tetrahedron Lett., 1967, 8, 4061 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the NMR spectra for all products. CCDC 1498887. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00477f |
| This journal is © the Partner Organisations 2016 |