
Diastereoselective synthesis of 2,5-disubstituted-3-hydroxy-tetrahydrofurans through a counterion-directed Tsuji–Trost reaction†
Xavier
Caumes
a,
Louis
Jeanne-Julien
a,
Chakib
Khelifi
a,
Vincent
Gandon
*bc and
Emmanuel
Roulland
*a
aUMR 8638 COMETE, CNRS/Université Paris Descartes, Faculté de Pharmacie, 4, avenue de l'Observatoire, 75270, Paris Cedex 06, France. E-mail: emmanuel.roulland@parisdescartes.fr; Tel: +33153739751
bInstitut de Chimie Moléculaire et des Matériaux d'Orsay, CNRS UMR 8182, Univ. Paris-Sud, Université Paris-Saclay, bâtiment 420, 91405 Orsay cedex, France. E-mail: vincent.gandon@u-psud.fr; Fax: +33169154747; Tel: +33169153931
cInstitut de Chimie des Substances Naturelles, CNRS UPR 2301, Univ. Paris-Sud, Université Paris-Saclay, 1, av. de la Terrasse, 91198 Gif-sur-Yvette, France
First published on 2nd September 2016
Abstract
Tsuji–Trost Counterion Directed Catalysis (CDC) allows the diastereoselective synthesis of 5-alkyl-3-hydroxy-2-vinyl tetrahydrofurans in high yields using easily accessible 4,6-dihydroxyalk-2-en-1-yl esters as substrates. The mechanism of this reaction has been investigated by relying on DFT predictions and experimental confirmations based on deuterium labeling and modification of the pKa of counterions which demonstrates that the favored transition state is tightly organized through the formation of a network of H-bonds.
In reactions proceeding under Counterion-Directed Catalysis (CDC), ions present in the reaction medium play a determining role by establishing interactions with charged key reaction intermediates controlling for instance the stereochemical outcome of the transformation. Originally applying to organo-catalysis,1 this concept has been extended to transition-metal-catalysis.2 For instance, Toste3 used chiral phosphate gold complexes to achieve highly stereoselective cyclizations. CDC also applies to palladium catalysis and reactions in which a cationic π-allyl-PdII intervenes – namely Tsuji–Trost reactions – show themselves to be particularly good candidates even if to date only a limited number of examples have been reported.4 The first one was described by List who designed a direct enantioselective α-allylation of aldehydes using a chiral phosphate counterion. A mechanism was proposed in which the counterion establishes a H-bond with the nucleophile while an electrostatic interaction takes place with the charged π-allyl-PdII moiety.4a In 2014, the mechanism of this particular example was investigated by Sunoj et al.5 This DFT study reports a transition state model capable of rationalizing the chiral counterion-induced enantioselectivity. It was computationally found that the chiral phosphate acts as a counterion in the stereodetermining step and not as a conventional ligand, and the existence of an unusual H-bond involving one H of the π-allylpalladium moiety was suggested. In 2012, our group has studied the mechanism of the Tsuji–Trost reaction in the case of an O-nucleophile leading to a diastereoselective cyclization of allyl acetate 1 into 5-vinyl-3-hydroxy-2-alkyl tetrahydrofurans 2 (Scheme 1).6 Our DFT calculations predicted the crucial formation of a network of H-bonds involving the carboxylate counterion and the hydroxyl groups in the favored transition state [πR]‡. However, surprisingly DFT did not predict the formation of an electrostatic interaction with the charged π-allyl-PdII; it rather suggested the unusual formation of one additional H-bond involving the internal H of the π-allyl-PdII moiety which was unprecedented. This mechanism accounted well for the unexpectedly high diastereoselectivity of the transformation and the involvement of the counterion into various H-bonds was experimentally confirmed by a series of experimental observations. Thus, the observation that the dr diminishes with the pKa of the conjugated acid of the counterion matches the fact that weak carboxylic acids are more prone than strong ones to make H-bonds, which demonstrates the existence of the latter. Concerning the H-bond involving the internal H of the π-allyl-PdII moiety, by replacing the latter hydrogen atom by a deuterium atom we observed that cyclization occurred with dr erosion which proved the existence of such an H-bond. This reflected the fact that the strength of an O⋯D bond is slightly weaker than that of an O⋯H bond in this case. In addition, the protection of the directing OH as a methyl ether – or its replacement by a hydrogen atom7 – leads to a total loss of selectivity and strongly diminishes both the rate and yield of the reaction. Another important observation we made previously in our first attempts to synthesize tetrahydrofurans THF 2 from allylacetate 18 is that the use of Trost's chiral ligands9 led to a surprisingly strong mismatch effect precluding an efficient synthesis of the desired tetrahydrofuran. This constitutes an important scope limitation of this classical method; therefore the use of a diastereoselective approach becomes very pertinent when targeting such hydroxy-THF. THF10 as well as tetrahydropyrans (THP)11 are prevalent heterocyclic motifs often encountered in natural products, many of them being endowed with interesting biological properties. In this context, exploring the scope of this easily applicable Tsuji–Trost-CDC cyclization is therefore particularly relevant. This approach was first implemented with success through the achievement of our total synthesis of (+)-oocydin A (3)8 as well as for the synthesis of the C1–C8 fragment of obtusallene III (4)6 (Fig. 1). However, our previous study6 was restricted to the study of the synthesis of 2-alkyl-3-hydroxy-5-vinyl THF ([1.0]-THF) (Scheme 2). Therefore, the obvious scope extension consists of verifying whether the CDC cyclization mode remains effective while the OH directing group is shifted to another position. Our method would potentially open a stereoselective access to 5-alkyl-3-hydroxy-2-vinyltetrahydrofurans ([0.1]-THF), 6-alkyl-4-hydroxy-2-vinyltetrahydropyrans ([1.1]-THP) and 2-vinyl-3-hydroxy-6-alkyltetrahydropyrans ([0.2]-THP) (Scheme 2). As we can anticipate by taking the Baldwin rules into account, the [2.0] couple will not lead to a THP but rather to a THF bearing an OH on its side chain. In comparison with [1.0]-THF previously studied, synthesizing [0.1]-THF by CDC consists of shifting the directing OH group on the cyclization precursor of just one notch. In that case, we expected CDC to occur still since we anticipated that the required network of H-bonds would form leading to a well-organized transition state. Therefore, both syn- and anti-1,3-diol cyclization precursors 7a–d and 8a–d (Scheme 3) were straightforwardly synthesized through a divergent approach from known (syn/anti)-β-hydroxyepoxide 5.12 The latter were transformed into syn- and anti-1,3-diol 6-syn and 6-anti using the Falck–Mioskowski reaction,13 the two diols being separable by preparative HPLC. Cross-metathesis on 6-syn and 6-anti ensured the introduction of various leaving groups on the allyl moiety delivering then 7a–d and 8a–d, a series of various allylic esters. The cyclization was first explored on allyl acetates 7b and 8b under the conditions we used in our previous study.6 We were pleased to observe that cyclization occurred here also in high yield and with a good selectivity. However, surprisingly, and contrary to the case of [1.0]-THF, in [0.1]-THF the vinyl function is preferentially installed cis to the OH directing group.14 This occurs whatever the starting material is a syn- or an anti-diol (9-cis from 7b and 10-cis from 8b). This latter observation, plus the good measured dr, and the high reaction rate, constituted a first set of reliable clues indicating that this reaction occurs in a CDC mode.
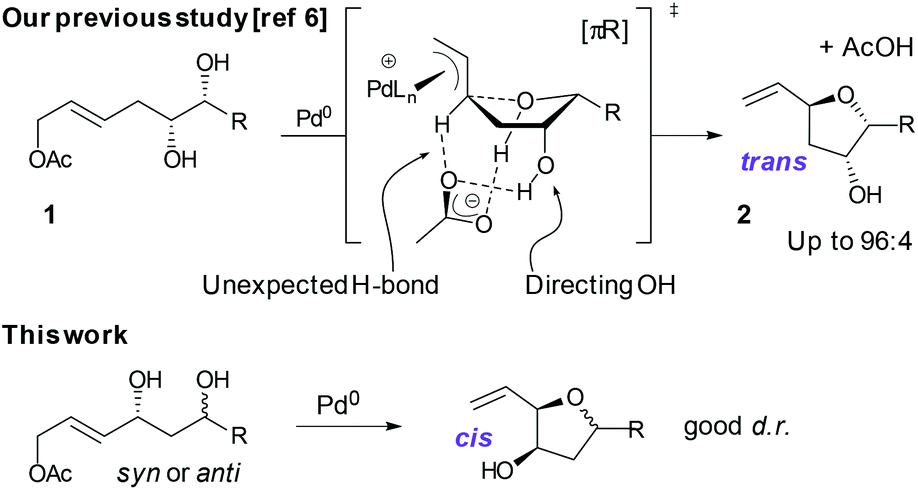 | ||
| Scheme 1 CDC concept applies to the Tsuji–Trost reaction for the synthesis of hydroxy-tetrahydrofurans. | ||
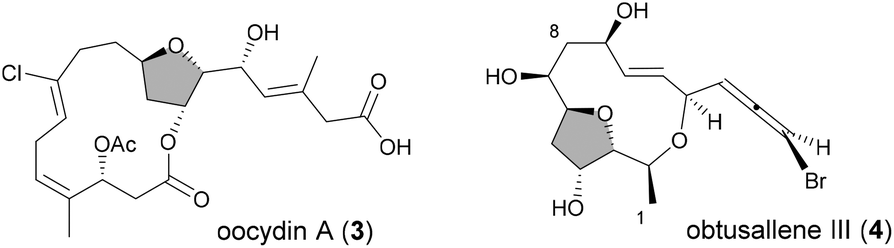 | ||
| Fig. 1 Synthesized by using the CDC approach. | ||
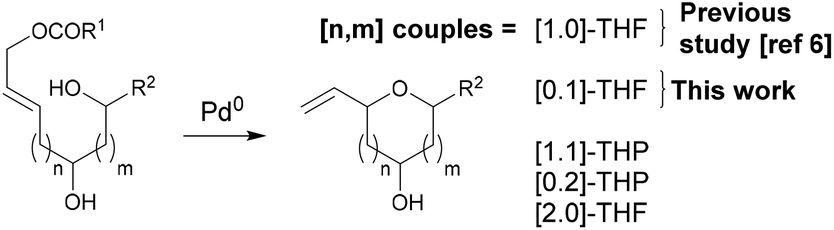 | ||
| Scheme 2 Potential application of CDC-Tsuji–Trost reaction to the syntheses of hydroxylated THF and THP. | ||
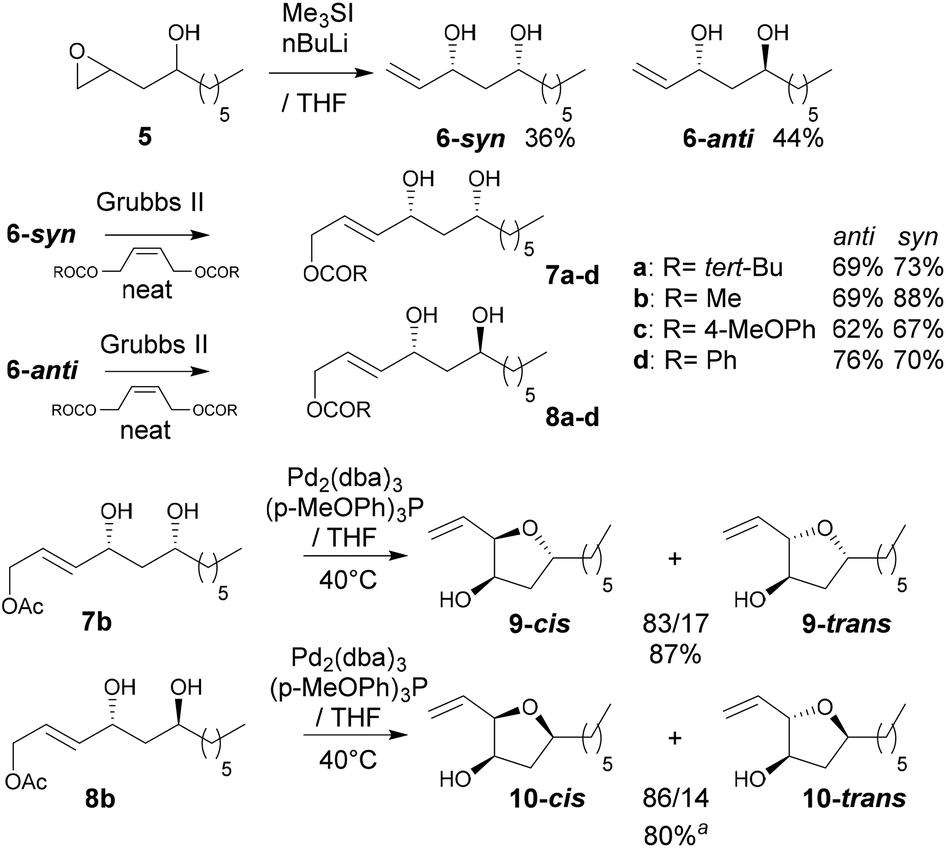 | ||
Scheme 3 Syntheses of syn- and anti-1,3-diols and cyclization onto [1.0]-THF. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The corresponding 4-methoxybenzoate ester was synthesized to allow HPLC separation of the diastereoisomers. The corresponding 4-methoxybenzoate ester was synthesized to allow HPLC separation of the diastereoisomers. | ||
A set of experiments was performed to see if the pKa of the counterion has an impact on the dr which is a reliable way to prove that a H-bond network is formed within the two possible transition states favoring one over the other. In Table 1 are summarized a series of cyclization reactions for which the carboxylic counterion acidity varies for both syn- and anti-starting diols. In this set of experiments, we observed a clear variation of the dr as a function of the pKa of the counterion. The case of the pivalate leaving group (entry 1) seems apart as it allowed only slow conversions likely because its hindrance strongly slows down the rate of π-allyl-PdII formation or destabilizes the network of H-bonds. Nonetheless and as expected, it appears that counterions conjugated to weaker acids lead to better dr than those conjugated with stronger ones. We may also notice that the relationship between pKa and dr is stronger in the syn-diol series (7a–d) than in the anti-diol series (8a–d). These observations are similar to the ones previously made in the case of [1.0]-THF,6 and strongly support the assumption that the counterion organizes the transition state through the formation of a network of H-bonds.
|
|
||||
|---|---|---|---|---|
| Entry | Leaving group X− | Starting diol | dr (cis/trans) conversion reaction time | HX pKa in H2O |
| a Incomplete conversion and formation of palladium black. | ||||
| 1 | tert-BuCO2− | syn (7a), anti (8a) | 9: 86/14, 37%, 5 h;a10: 81/19, 68%, 24 ha | 5.01 |
| 2 | MeCO2− | syn (7b), anti (8b) | 9: 83/17, 87%, 1 h; 10: 86/14, 84%, 2 h | 4.76 |
| 3 | p-MeOPhCO2− | syn (7c), anti (8c) | 9: 80/20, 100%,1 h; 10: 85/15, 90%, 2 h | 4.47 |
| 4 | PhCO2− | syn (7d), anti (8d) | 9: 75/25, 100%, 1 h; 10: 84/16, 100%, 1 h | 4.20 |
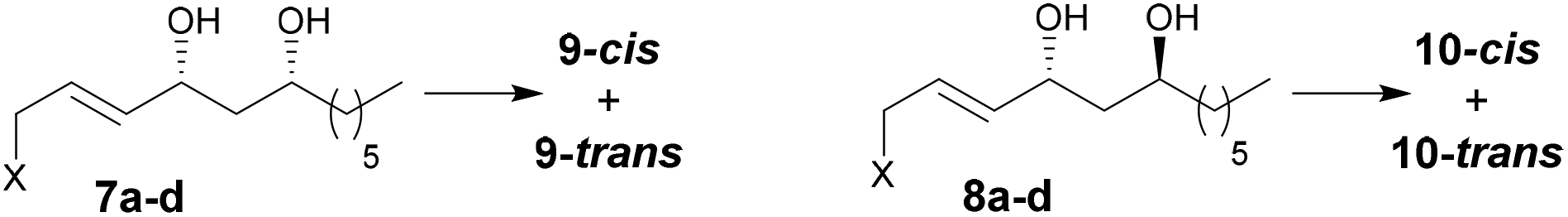
However, the reasons for which the directing OH favors the installation of the vinyl group cis in the case of [0.1]-THF remained unclear in particular since we previously observed the opposite in the case of [1.0]-THF, therefore DFT computations were carried out. To allow direct comparison with our previous study,6 we used the same level of theory. All computations were performed with the Gaussian '09 software package.15 Optimizations were carried out using the B3LYP functional. The atoms H, C, O, and P were described with the 6-31+G(d,p) basis set. The Pd atom was described by the quasi relativistic ECP28MWB basis set ((8s7p6d2f1g)/[6s5p3d2f1g]).16 Gibbs free energies were obtained from the frequency calculation at the optimization level (298 K). Pd(PH3)2 was used as active species instead of Pd[(p-MeOPh)3P]2 as we did in our previous study. The n-hexyl group was replaced by a methyl substituent. As in our previous study, it should be mentioned that we were unable to obtain a cyclization transition state in the absence of the acetate counterion, be it with the B3LYP or the M06 functional, the ECP28MWB or the LAND2DZ basis set. We started our investigations with the [πR]-1 and [πS]-1 conformers (Scheme 4). As in our previous study,6 we were unable to obtain a cyclization transition state in the absence of the acetate counterion.17 On the other hand, having the acetate H-bonded to the two alcohol functionalities allowed us to locate [πR]-1‡ and [πS]-1‡ on the potential energy surface. The two strong H-bonds between the acetate oxygens and the hydrogens of the alcohol functions are also present in the starting π-allyl complexes [πR]-1 and [πS]-1. In [πR]-1, a third H-bond is established between the rear oxygen atom of the acetate and the central C–H fragment of the π-allyl moiety. According to the geometrical parameters and the maximum electron density,18 this H-bond is moderately strong but not negligible compared to the two O⋯H–O hydrogen bonds. In [πS]-1, the rear acetate oxygen also interacts with the π-allyl moiety, yet this time with the internal CH fragment. Nevertheless, this bond is also quite strong. In both [πR]-1 and [πS]-1, these networks of H-bonds preorganize the complexes for cyclization. In the transition state [πR]-1‡, the O⋯H–C hydrogen bond remains but passes from the rear oxygen to the front one. It is also weaker than in [πR]-1 (O⋯H–C 2.38 Å; OHC 136.6°; ρmax = 0.012 e Å−3). On the other hand, the O⋯H–C interaction is unworkable in [πS]-1‡. Also in the products A-cis and A-trans, this extra H-bond does not exist anymore. Although [πS]-1 is more stable than [πR]-1 by 3.6 kcal mol−1, it is [πR]-1‡ that is the lowest lying transition state (ΔΔG‡298 = 2.6 kcal mol−1). This could be due, among other factors, to the fact that the extra O⋯H–C bond still exists in the transition state, conferring additional stability. Both transformations are moderately exergonic. Under kinetic control, the calculations predict A-cis type products to be the major ones, which is corroborated experimentally.
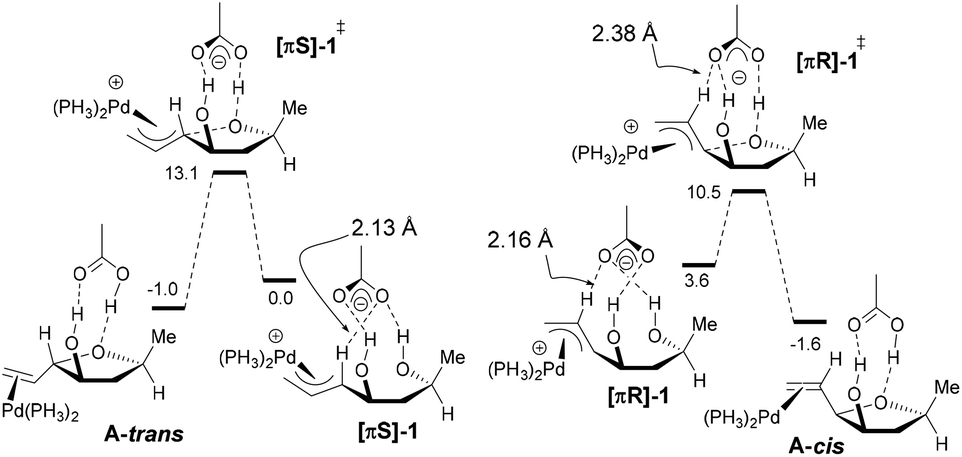 | ||
| Scheme 4 DFT predictions in the syn series. | ||
To confirm these DFT predictions, and in particular those regarding the intriguing occurrence of an H-bond involving the π-allylpalladium moiety, two deuterated derivatives of 7b were synthesized: 7b* labelled at the internal allylic position, and 7b• labeled at the central allylic position (Scheme 5). 7b* was obtained as follows: aldol condensation led to the known β-hydroxyester 11 and then to the corresponding Weinreb amide 12. The latter was transformed into propargylic ketone 13, the syn-selective reduction of which gave syn-alcohol 14. Deuteration was performed using the Lindlar catalyst leading to (Z)-allyl alcohol 14. Finally, the allyl acetate function was installed through cross-metathesis leading then to 7b*. Concerning deuteration at the central position of the π-allylpalladium (Scheme 5) we used 6-syn which through cross-metathesis with (Z)-but-2-ene-1,4-diyl-2,3-d2 diacetate19 led to 7b•. Cyclizations of 7b* into 9* and of 7b• into 9• were carried out under our standard conditions (Scheme 5). Compared to 7b → 9, 7b* cyclized into 9* with no dr difference which is consistent with the DFT calculations that predicted that the H at the internal position does not participate in the stabilization of the transition state. That is with delight that we observed that cyclization of 7b• into 9• occurred with a clear erosion of the dr (80.5/19.5 instead of 83/17 from 7b), a result that matches perfectly the DFT prediction regarding the formation of an H-bond at the internal position of the π-allylpalladium.
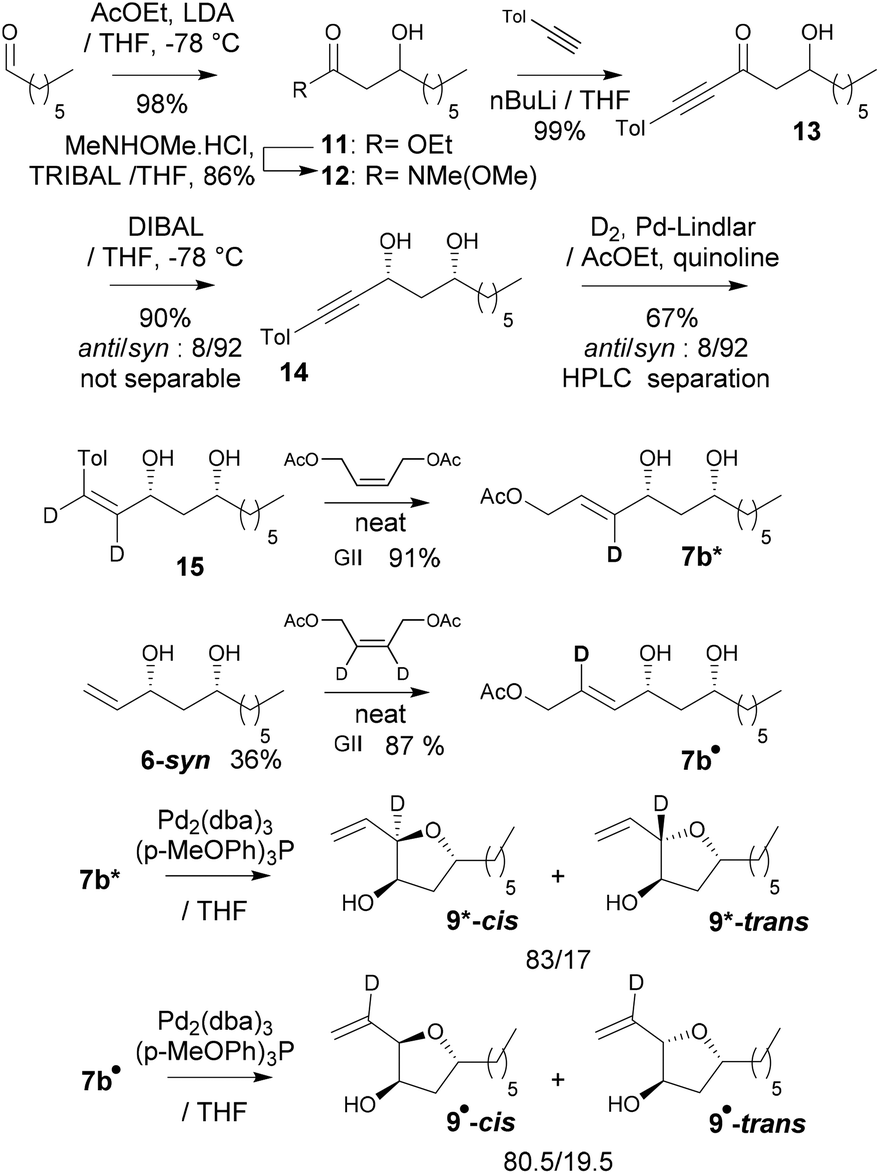 | ||
| Scheme 5 Synthesis of deuterated cyclization precursors and cyclization. | ||
Deuterium provides slightly weaker H-bonds than hydrogen in this case, explaining thus the erosion of the selectivity; this proves the existence of this predicted H-bond. Calculations have also been performed in the anti series predicting the formation of a similar network of H-bonds consistent with the experimentally observed dr and cis-selectivity.20
Conclusions
We have demonstrated that the synthesis of 5-alkyl-2-vinyl tetrahydrofuran-3-ol by the Tsuji–Trost reaction also occurs in a CDC mode with 4,6-dihydroxyalk-2-en-1-yl esters as substrates. This constitutes a valuable scope extension of our method that first worked with 5,6-dihydroxyalk-2-en-1-yl esters as described in our previous study.6 Interestingly, these two pathways are fully complementary since with [0.1]-THF the directing OH favors the installation of the vinyl group in cis while with [1.0]-THF the selectivity is trans. As a consequence, our approach now allows access to all possible enantioenriched 3-hydroxyl-2,5-disubstituted THF isomers with synthetically useful dr and yields rendering the use of this strategy particularly attractive in the context of the total synthesis of natural products. DFT predictions have been confirmed experimentally with deuterium labeling, providing in particular an unprecedented and reliable method of detection of the formation of H-bonds, a method that is particularly attractive considering the ever-growing interest in counterion directed catalysis.Notes and references
- T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277 CrossRef CAS PubMed.
- (a) V. Komanduri and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 16448 CrossRef CAS PubMed; (b) J. R. Zbieg, E. Yamaguchi, E. L. McInturff and M. J. Krische, Science, 2012, 336, 324 CrossRef CAS PubMed; (c) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed; (d) M. Rueping, R. M. Koenigs and I. Atodiresei, Chem. – Eur. J., 2010, 16, 9350 CrossRef CAS PubMed; (e) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed.
- G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496 CrossRef CAS PubMed.
- (a) S. Mukherjee and B. List, J. Am. Chem. Soc., 2007, 129, 11336 CrossRef CAS PubMed; (b) G. Jiang and B. List, Adv. Synth. Catal., 2011, 353, 1667 CrossRef CAS; (c) G. Jiang and B. List, Angew. Chem., Int. Ed., 2011, 50, 9471 CrossRef CAS PubMed; (d) K. Ohmatsu, M. Ito, T. Kunieda and T. Ooi, Nat. Chem., 2012, 4, 473 CrossRef CAS PubMed; (e) Z.-L. Tao, W.-Q. Zhang, D.-F. Chen, A. Adele and L.-Z. Gong, J. Am. Chem. Soc., 2013, 135, 9255 CrossRef CAS PubMed; (f) G. Pupo, R. Properzi and B. List, Angew. Chem., Int. Ed., 2016, 55, 6099 CrossRef CAS PubMed.
- G. Jindal and R. B. Sunoj, J. Org. Chem., 2014, 79, 7600 CrossRef CAS PubMed.
- M. Arthuis, R. Beaud, V. Gandon and E. Roulland, Angew. Chem., Int. Ed., 2012, 51, 10510 CrossRef CAS PubMed.
- O. Hara, K. Fujii, Y. Hamada and Y. Sakagami, Heterocycles, 2001, 54, 419 CrossRef CAS.
- E. Roulland, Angew. Chem., Int. Ed., 2008, 47, 3762 CrossRef CAS PubMed.
- (RR) or (S,S)-1,2-Diaminocyclohexane-N,N′-bis(2′-diphenylphosphinobenzoyl).
- A. Lorente, J. Lamariano-Merketegi, F. Albericio and M. Álvarez, Chem. Rev., 2013, 113, 4567 CrossRef CAS PubMed.
- T. Nakata, Chem. Rev., 2005, 105, 4314 CrossRef CAS PubMed.
- Y.-G. Liu, X.-Q. Gong, H.-Y. Tian and B.-G. Sun, J. Chem. Res., 2012, 36, 333 CrossRef CAS.
- L. Alcaraz, J. J. Harnett, C. Mioskowski, J. P. Martel, T. Le Gall, D.-S. Shin and J. R. Falck, Tetrahedron Lett., 1994, 35, 5449 CrossRef CAS.
- Relative configurations of the major cyclization products were unambiguously determined by 1H NMR NOESY experiments on the 4-bromobenzoyl ester of 9-cis and on the 4-methoxybenzoate ester of 10-cis (see the ESI†).
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS; (b) J. M. L. Martin and A. Sundermann, J. Chem. Phys., 2001, 114, 3408 CrossRef CAS.
- See the ESI† for details.
- B. P. Hay and V. S. Bryantsev, Chem. Commun., 2008, 2417 RSC.
- Obtained by deuteration of but-2-yne-1,4-diyl diacetate using the Lindlar catalyst under classical conditions.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00337k |
| This journal is © the Partner Organisations 2016 |