
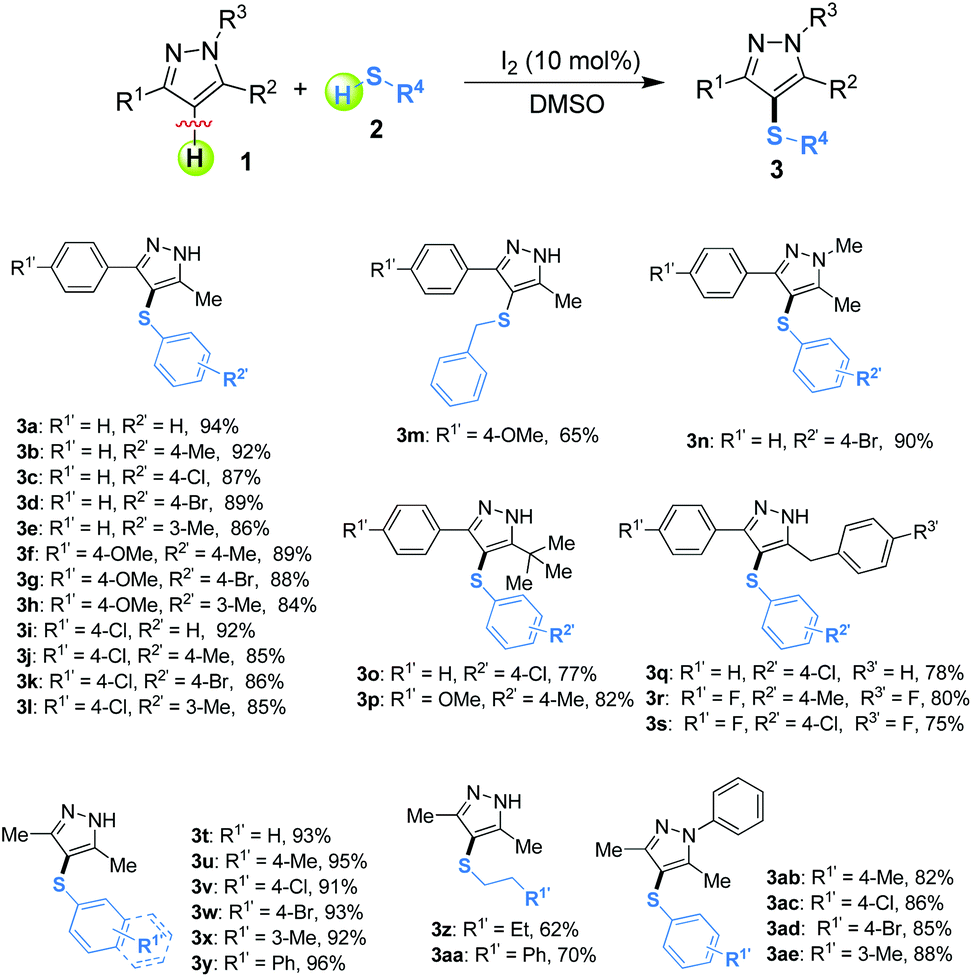
Metal-free iodine-catalyzed direct cross-dehydrogenative coupling (CDC) between pyrazoles and thiols†
Daoshan
Yang
*,
Pengfei
Sun
,
Wei
Wei
,
Lingduan
Meng
,
Lingchao
He
,
Baokai
Fang
,
Wei
Jiang
and
Hua
Wang
*
The Key Laboratory of Life-Organic Analysis and Key Laboratory of Pharmaceutical Intermediates and Analysis of Natural Medicine, School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, Shandong, P. R. China. E-mail: yangdaoshan@tsinghua.org.cn; huawang_qfnu@126.com
First published on 31st August 2016
Abstract
A green and efficient iodine-catalyzed cross-dehydrogenative C–S coupling method for the synthesis of C-4 sulfenylated pyrazoles has been initially developed under metal-free conditions. This new method is highly efficient and practical, and the starting materials are readily available. The present method should provide a useful strategy for the construction of different thiol-substituted pyrazole motifs.
Sulfur-containing compounds are of great importance in pharmaceuticals, bioactive products, agrochemicals, organic dyes, and materials chemistry.1 As a consequence, the development of novel, efficient, and practical methods for C–S bond formation is still a prime topic in organic chemistry. Transition-metal-catalyzed cross-couplings of thiols or disulfides with aryl halides, pseudo halides or arylboronic acids, have been demonstrated to be a versatile method for constructing C–S bonds.2 Meanwhile, the direct sulfenylation of inert C–H bonds using thiols, disulfides, sulfenyl halides, arylsulfonyl cyanides, sulfonyl hydrazides or 1-(substituted phenylthio)pyrrolidine-2,5-dione as the thiol source under Cu,3 Fe,4 Pd,5 and Ru6 catalytic conditions has also been reported as a powerful tool for C–S bond formation thus far. For example, Fu et al. developed an elegant work for the synthesis of diaryl sulfides via iron- or boron-catalyzed C–H arylthiation of phenols under mild conditions.4a Despite some great advantages, these methods could encounter certain limitations, including unavailability of precursors, harsh reaction conditions, and toxic metal salt catalysts. As a consequence, it remains a challenging, but very attractive, task to develop more efficient, economical, and practical synthesis methods for constructing C–S bonds.
With the increasing voice of green and sustainable chemistry, the development of more efficient and simple synthetic methodologies has still stimulated an impressive number of research groups all over the world. In this context, the cross-dehydrogenative coupling (CDC) reaction has emerged as a potentially powerful strategy for the construction of a diverse array of C–C and C–X (X = heteroatom) bonds7 because it provides intrinsic advantages such as shorter synthetic routes, higher atom-economy, and more economical materials, which leads to “benign by design” (Green Chemistry).8 However, research surveys of this synthetic strategy for the C–S bond-forming reaction is surprisingly limited when compared to the formation of C–C, C–O or C–N bonds.9
Recently, there has been growing demand to develop metal-free organic transformations owing to the fact that trace-metal impurities might be avoided in the target molecules.10 There is no doubt that a metal-free protocol for C–S bond formation via C–H bond functionalization appears desirable and synthetically attractive. In these protocols, various sulfenylating reagents such as arylsulfonyl chlorides,11 aryl sulfonyl hydrazides,12 diaryldisulfides13 sodium sulfinates,14 and sulfinic acids15 have been used. However, many of these sulfenylating reagents either need moisture-free conditions or need multiple steps for their synthesis. Consequently, directly using thiols as sulfenylating reagents appears desirable and synthetically attractive.16
The pyrazole skeleton is a key structural motif that appears in the core structure of an impressive number of biologically active molecules.17 For example, they can be used as antibacterial, antitumor, anti-inflammatory, antiobesity, and analgesic agents.18 Additionally, some commercially available drugs contain this core structure (Fig. 1), such as Fipronil, Crizotinib, Zaleplon, and Celebrex.19 Furthermore, many substituted pyrazole derivatives possess diverse pharmacological properties.20 Although some methods using thiols as the sulfur source have been developed for the synthesis of sulfenylated pyrazoles these routes are often troublesome and some of the starting materials are not readily available (Scheme 1).21 It is highly desirable to develop more convenient and efficient approaches. To the best of our knowledge, the synthesis of the combined motifs of pyrazole and thiol frameworks via cross-dehydrogenative C–S coupling has not been explored thus far (Scheme 2). Therefore, we wish to synthesize this fused sulfur-containing N-heterocycle which could possibly exhibit biological activities.21b With our growing interest in the synthesis of sulfur-containing organic compounds,22 we herein report a green and efficient strategy for the construction of thiol-substituted pyrazoles via iodine-catalyzed cross-dehydrogenative C–S coupling of pyrazoles with thiols under mild conditions.
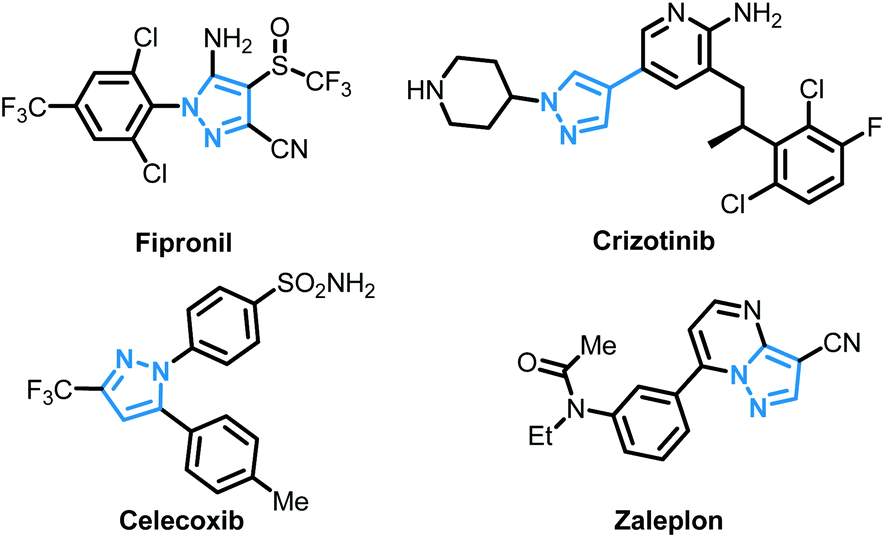 | ||
| Fig. 1 Representative examples of drugs containing a pyrazole framework. | ||
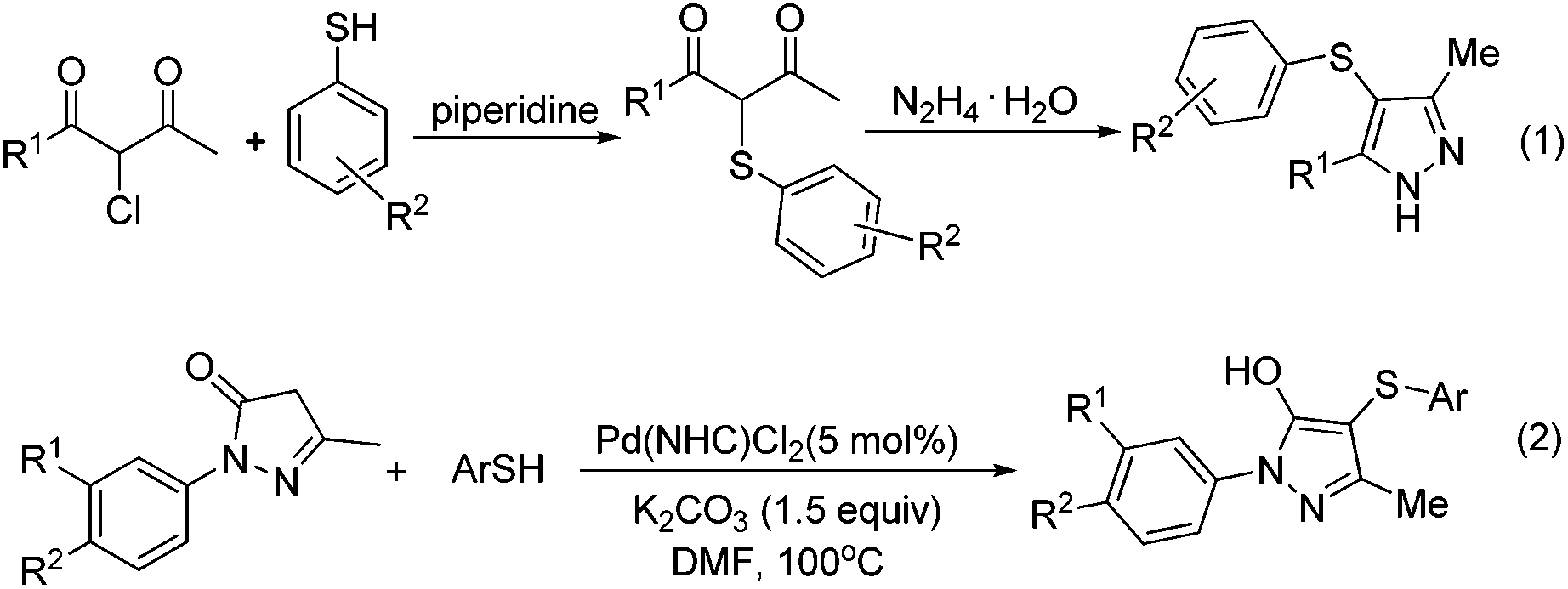 | ||
| Scheme 1 Examples of the synthesis of thiol-substituted pyrazoles. | ||
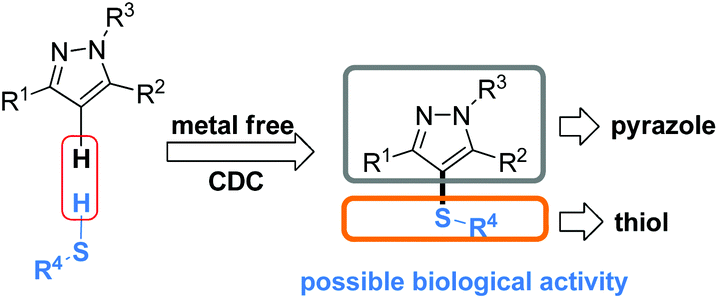 | ||
| Scheme 2 Strategy for thiol-substituted pyrazoles via the cross dehydrogenative coupling reaction of pyrazoles with thiols. | ||
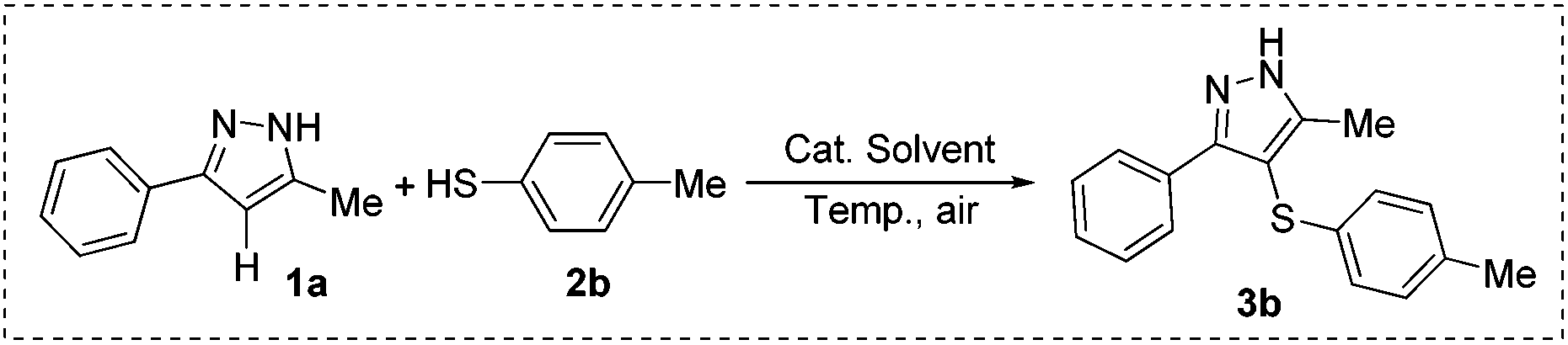
Initially, 5-methyl-3-phenyl-1H-pyrazole (1a) and 4-methylbenzenethiol (2b) were chosen as the model substrates to optimize reaction conditions including the catalysts, solvents, and reaction temperatures under ambient air conditions. As shown in Table 1, five catalysts such as nBu4NI, KI, NaI, I2O5 and I2 were investigated at 100 °C by using DMSO (2 mL) as the solvent, and I2 gave the highest yield (92%) (entries 1–5). Notably, no target product was observed in the absence of any catalysts (entry 6). Furthermore, different solvents including single and mixed ones were tested, showing that DMSO was superior to the others (compare entries 5, 7–12). Additionally, various reaction temperatures were attempted (entries 5, 13–16), and 100 °C was found to be suitable for this reaction (entry 5). Elevated temperature did not obviously enhance the yield (entry 13). After the optimization process of catalysts, solvents and temperature, various thiol-substituted pyrazole derivatives were synthesized under our standard conditions: 10 mol% of iodine as the catalyst, 2 mL DMSO as the solvent at 100 °C in air.
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Solvent | Temp. [°C] | Yieldb [%] |
| a Reaction conditions: 5-methyl-3-phenyl-1H-pyrazole (1a) (0.2 mmol), 4-methylbenzenethiol (2b) (0.24 mmol), catalyst (0.02 mmol), solvent (2 mL), reaction time (18 h) in air. b Isolated yield. | ||||
| 1 | n Bu4NI | DMSO | 100 | 15 |
| 2 | KI | DMSO | 100 | 60 |
| 3 | NaI | DMSO | 100 | 56 |
| 4 | I2O5 | DMSO | 100 | 20 |
| 5 | I 2 | DMSO | 100 | 92 |
| 6 | None | DMSO | 100 | 0 |
| 7 | I2 | DCE | 100 | Trace |
| 8 | I2 | CH3CN | 100 | Trace |
| 9 | I2 | H2O | 100 | 0 |
| 10 | I2 | DMF | 100 | 30 |
| 11 | I2 | DMSO/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) :3) |
100 | 80 |
| 12 | I2 | DMSO/CH3CN (1:3) |
100 | 85 |
| 13 | I2 | DMSO | 110 | 92 |
| 14 | I2 | DMSO | 90 | 80 |
| 15 | I2 | DMSO | 80 | 73 |
| 16 | I2 | DMSO | 60 | 65 |
After establishing the optimized reaction conditions for the I2-catalyzed cross-dehydrogenative C–S coupling of substituted pyrazoles (1) with thiols (2), we investigated the generality and the scope of the protocol by varying the range of pyrazoles and thiols. As shown in Table 2, a variety of pyrazoles, bearing either aryl groups or alkyl groups reacted smoothly with thiols, affording the corresponding C-4 sulfenylated pyrazoles 3a–3ae in good to excellent yields. To our delight, aryl thiols which have electron-donating or withdrawing groups could be converted to the desired sulfides in good to excellent yields. Several aliphatic thiols including phenylmethanethiol, butane-1-thiol, and 2-phenylethanethiol were also examined, which afforded the desired products in good yields (3m, 3z and 3aa). Also, naphthalene-2-thiol could be employed in the reaction to generate the desired product 3y in 96% yield. The steric hindrance in the thiols did not significantly affect the catalytic efficiency (3b, 3e and 3h). Additionally, pyrazoles bearing bulky substrates such as tertiary butyl, were able to react with thiols to give the corresponding products 3o and 3p in excellent yields. Although pyrazoles showed high reactivity, unfortunately, pyrazolones, such as 3-methyl-1-phenyl-1H-pyrazol-5(4H)-one 4, 3-methyl-1-p-tolyl-1H-pyrazol-5(4H)-one 5, and 1-(4-chlorophenyl)-3-methyl-1H-pyrazol-5(4H)-one 6 were poor substrates, and didn't give the desired products (Scheme 3). Thus, further investigations to explore more powerful catalytic conditions were required. The I2-catalyzed cross-dehydrogenative C–S coupling reactions could tolerate some functional groups such as alkyl and ether, and bonds such as C–Cl bonds, C–Br bonds and C–F bonds which could be used for further modifications at the substituted positions.
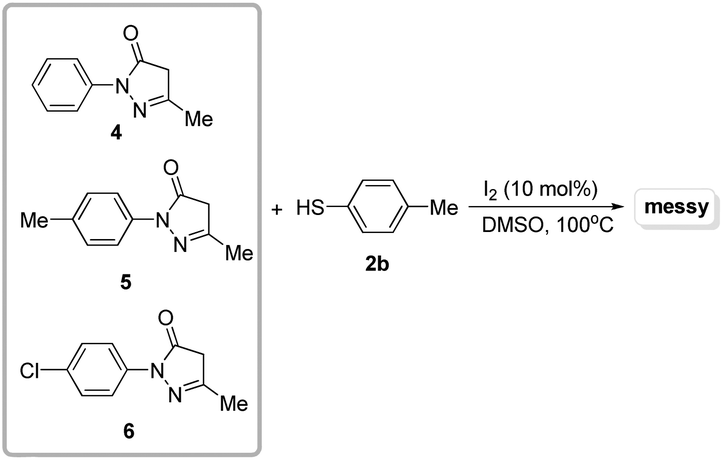 | ||
| Scheme 3 Investigations of the substrate scope. | ||
Gram-scale applications for the present method were also explored. As shown in Scheme 3, the proposed reaction between 1a and 2a was investigated under the standard conditions, which could give 1.26 g of 3a in 95% yield without any significant loss of reactive efficiency [eqn (1), Scheme 4]. Furthermore, the sulfonyl product 7 was directly synthesized in good yield from 3b through an oxidative reaction with m-chloroperbenzoic acid (m-CPBA) [eqn (2), Scheme 4]. Thus, this simple, metal-free protocol could be extended as an efficient and practical method to construct various potentially bioactive C-4 sulfenylated or sulfonylated pyrazoles.
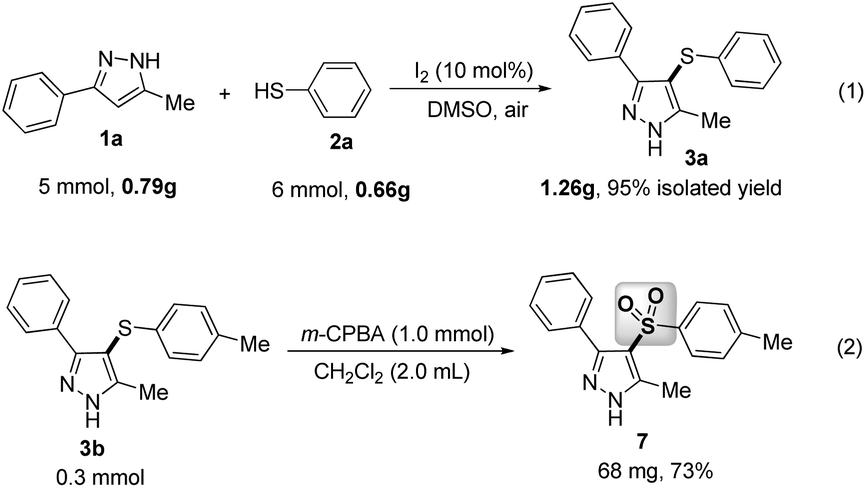 | ||
| Scheme 4 The applications of this method. | ||
To gain further insight into the reaction mechanism, several control experiments were performed. When the model reaction was performed under a nitrogen atmosphere, the sulfenylated product 3a was obtained in 93% yield, indicating that the dioxygen in air might not act as the oxidant to realize the catalytic cycle in the present reaction [eqn (1), Scheme 5]. Furthermore, we added the well-known radical scavenger TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) to the reaction system, but no significant difference was observed in the yield, ruling out the presence of radicals during the reaction [eqn (2), Scheme 5]. Additionally, the intermolecular kinetic isotope effects (KIE) were investigated, and no kinetic isotope effect (kH/kD = 1.0) was observed, which indicates that C–H bond cleavage might not be the rate-determining step [eqn (3), Scheme 5] (Fig. 1, see the ESI†). Besides, the reaction of benzenethiol 2a (0.24 mmol), only under the standard conditions, gave 1,2-diphenyldisulfane 8 in 98% yield, which implied that 1,2-diphenyldisulfane 8 might be the important intermediate in the present transformation [eqn (4), Scheme 5].
 | ||
| Scheme 5 Investigations of the reaction mechanism. | ||
On the basis of these preliminary results mentioned above together with the previous related literature,23 a proposed mechanism would be herein presented (Scheme 6). Initially, thiols 2 were transformed into disulfides 8 under the present reaction conditions. Next, disulfides 8 reacted with I2 to form the electrophilic intermediate Ar–SI A. Subsequently, the intermediate Ar–SI reacted with pyrazoles 1 to give carbocation B, which could be stabilized by the adjacent nitrogen. Then, intermediate B loses a proton to give the desired products 3 and HI. The HI reacted with DMSO to form intermediate C, which reacted with HI to give intermediate D. Finally, nucleophilic attack of I− on the iodide atom of D takes place to regenerate I2 releasing DMS and H2O. Further investigations on the more detailed mechanism are in progress in our laboratory.
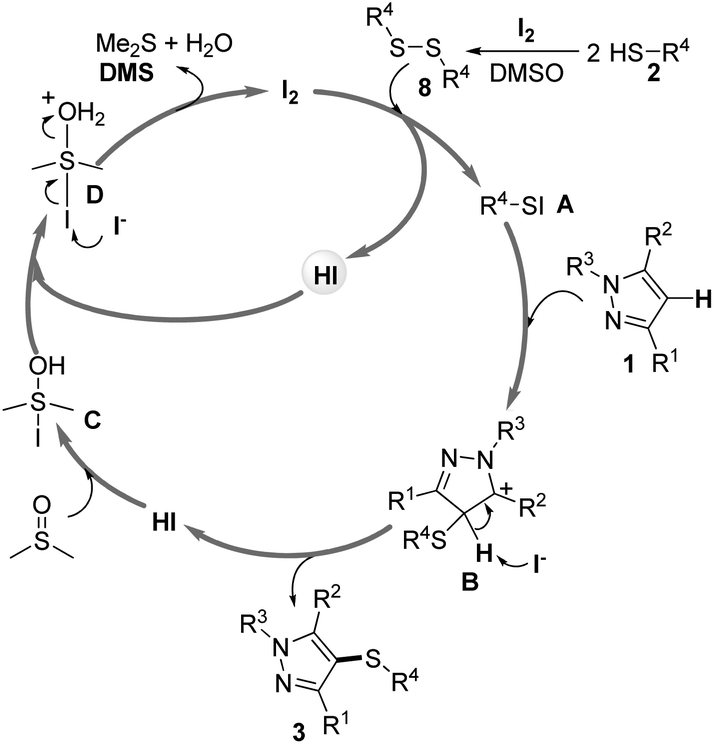 | ||
| Scheme 6 A proposed mechanism for the direct transformation. | ||
In summary, an efficient, green and environmentally friendly protocol has been initially developed for the synthesis of C-4 sulfenylated pyrazoles via I2-catalyzed cross-dehydrogenative coupling of pyrazoles with readily available thiols. A series of potential biological C-4 sulfenylated pyrazole frameworks could be conveniently obtained in good to excellent yields. This new method can enjoy the following advantages: (a) commercially available and non-toxic I2 as the catalyst; (b) DMSO as both a solvent and an oxidant; (c) simple and readily available materials; (d) easy workup procedure; (e) no addition of any base, ligand or additive; (f) outstanding tolerance of functional groups. The easy and efficient method for the synthesis of pyrazole compounds should attract much attention in synthetic and pharmaceutical chemistry.
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 21302110, 21302109 and 21375075), the Taishan Scholar Foundation of Shandong Province, the Natural Science Foundation of Shandong Province (ZR2013BQ017 and ZR2015JL004), and the Project of Shandong Province Higher Educational Science and Technology Program (J13LD14). We thank Jin Li in this group for reproducing the results of 3a, 3o and 3e.
Notes and references
- Selective reviews, see: (a) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (b) S. S. Mansy and J. A. Cowan, Acc. Chem. Res., 2004, 37, 719 CrossRef CAS PubMed; (c) T. Punniyamurthy, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed; (d) T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS PubMed; (e) S. Oida, Y. Tajima, T. Konosu, Y. Nakamura, A. Somada, T. Tanaka, S. Habuki, T. Harasaki, Y. Kamai, T. Fukuoka, S. Ohya and H. Yasuda, Chem. Pharm. Bull., 2000, 48, 694 CrossRef CAS PubMed.
- For selected examples, see: (a) G. Mann, D. Baranano, J. F. Hartwig, A. L. Rheingold and I. A. Guzei, J. Am. Chem. Soc., 1998, 120, 9205 CrossRef CAS; (b) D. Baranano and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 2937 CrossRef CAS; (c) D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450 CrossRef CAS PubMed; (d) F. Y. Kwong and S. L. Buchwald, Org. Lett., 2002, 4, 3517 CrossRef CAS PubMed; (e) A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 2880 CrossRef CAS PubMed; (f) L. L. Joyce, G. Evindar and R. A. Batey, Chem. Commun., 2004, 446 RSC; (g) L. Rout, T. K. Sen and T. Punniyamurthy, Angew. Chem., Int. Ed., 2007, 46, 5583 CrossRef CAS PubMed; (h) Y. C. Wong, T. T. Jayanth and C. H. Cheng, Org. Lett., 2006, 8, 5613 CrossRef CAS PubMed; (i) M. Arisawa, T. Suzuki, T. Ishikawa and M. Yamaguchi, J. Am. Chem. Soc., 2008, 130, 12214 CrossRef CAS PubMed.
- For selected examples, see: (a) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed; (b) L. Chu, X. Yue and F.-L. Qing, Org. Lett., 2010, 12, 1644 CrossRef CAS PubMed; (c) S. Ranjit, R. Lee, D. Heryadi, C. Shen, J. E. Wu, P. Zhang, K.-W. Huang and X. Liu, J. Org. Chem., 2011, 76, 8999 CrossRef CAS PubMed.
- (a) H. Tian, C. Zhu, H. Yang and H. Fu, Chem. Commun., 2014, 50, 8875 RSC; (b) A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 2880 CrossRef CAS PubMed; (c) W.-Y. Wu, J.-C. Wang and F.-Y. Tsai, Green Chem., 2009, 11, 326 RSC.
- (a) P. Anbarasan, H. Neumann and M. Beller, Chem. Commun., 2011, 47, 3233 RSC; (b) P. Saravanan and P. Anbarasan, Org. Lett., 2014, 16, 848 CrossRef CAS PubMed.
- (a) O. Saidi, J. Marae, A. E. W. Ledger, P. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298 CrossRef CAS PubMed; (b) M. Chen, Z.-T. Huang and Q.-Y. Zheng, Chem. Commun., 2012, 48, 11686 RSC.
- Selected reviews, see: (a) H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS PubMed; (b) B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154 CrossRef CAS; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (d) J. L. Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- (a) R. Shu, L. Hao, J. F. Harrod, H.-G. Woo and E. Samuel, J. Am. Chem. Soc., 1998, 120, 12988 CrossRef CAS; (b) H. Dorn, R. A. Singh, J. A. Massey, J. M. Nelson, C. A. Jaska, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2000, 122, 6669 CrossRef CAS; (c) L.-B. Han and T. D. Tilley, J. Am. Chem. Soc., 2006, 128, 13698 CrossRef CAS PubMed; (d) Y. Liu, Y. Zhang, C. Hu, J.-P. Wanand and C. Wen, RSC Adv., 2014, 4, 35528 RSC.
- C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed.
- Q. Wu, D.-B. Zhao, X.-R. Qin, J.-B. Lan and J.-S. You, Chem. Commun., 2011, 47, 9188 RSC.
- (a) F.-L. Yang and S.-K. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929 CrossRef CAS PubMed; (b) X. Zhao, L. Zhang, X. Lu, T. Li and K. Lu, J. Org. Chem., 2015, 80, 2918 CrossRef CAS PubMed; (c) J. Sun, J.-K. Qiu, Y.-L. Zhu, C. Guo, W.-J. Hao, B. Jiang and S.-J. Tu, J. Org. Chem., 2015, 80, 8217 CrossRef CAS PubMed; (d) X. Zhao, L. Zhang, T. Li, G. Liu, H. Wang and K. Lu, Chem. Commun., 2014, 50, 13121 RSC; (e) A. Hajra, A. K. Bagdi, S. Mitra and M. Ghosh, Org. Biomol. Chem., 2015, 13, 3314 RSC; (f) X. Kang, R. Yan, G. Yu, X. Pang, X. Liu, X. Li, L. Xiang and G. Huang, J. Org. Chem., 2014, 79, 10605 CrossRef CAS PubMed; (g) X. Zhao, T. Li, L. Zhang and K. Lu, Org. Biomol. Chem., 2016, 14, 1131 RSC.
- (a) W. Ge and Y. Wei, Green Chem., 2012, 14, 2066 RSC; (b) P. Sang, Z.-K. Chen, J.-W. Zou and Y.-H. Zhang, Green Chem., 2013, 15, 2096 RSC; (c) C. D. Prasad, S. J. Balkrishna, A. Kumar, B. S. Bhakuni, K. Shrimali, S. Biswas and S. Kumar, J. Org. Chem., 2013, 78, 1434 CrossRef CAS PubMed; (d) L. Zou, J. Reball, J. Mottweiler and C. Bolm, Chem. Commun., 2012, 48, 11307 RSC; (e) R. Tang, Y. Xie, Y. Xie, J. Xiang and J. Li, Chem. Commun., 2011, 47, 12867 RSC; (f) S.-R. Guo, Y.-Q. Yuan and J.-N. Xiang, Org. Lett., 2014, 15, 4654 CrossRef PubMed; (g) Y.-F. Liao, P.-C. Jiang, S.-P. Chen, H.-R. Qi and G.-J. Deng, Green Chem., 2013, 15, 3302 RSC.
- (a) F. Xiao, S. Chen, J. Tian, H. Huang, Y. Liu and G.-J. Deng, Green Chem., 2016, 18, 1538 RSC; (b) F.-H. Xiao, H. Xie, S.-W. Liu and G.-J. Deng, Adv. Synth. Catal., 2014, 356, 364 CrossRef CAS; (c) D. Wang, R. Zhang, S. Lin, Z. Yan and S. Guo, RSC Adv., 2015, 5, 108030 RSC; (d) H. Rao, P. Wang, J. Wang, Z. Li, X. Sun and S. Cao, RSC Adv., 2014, 4, 49165 RSC.
- C. Liu and L. Ding, Org. Biomol. Chem., 2015, 13, 2251 CAS.
- (a) J.-P. Wan, S. Zhong, L. Xie, X. Cao, Y. Liu and L. Wei, Org. Lett., 2016, 18, 584 CrossRef CAS PubMed; (b) Y. Liu, Y. Zhang, C. Hu, J.-P. Wan and C. Wen, RSC Adv., 2014, 4, 35528 RSC.
- For selected examples, see: (a) S. G. Kucukguzel and S. Şenkardeş, Eur. J. Med. Chem., 2015, 97, 786 CrossRef CAS PubMed; (b) P. Khloya, S. Kumar, P. Kaushik, P. Surain, D. Kaushik and P. K. Sharma, Bioorg. Med. Chem. Lett., 2015, 25, 1177 CrossRef CAS PubMed; (c) M. G. Ferlin, G. Chiarelotto, S. D. Acqua, E. Maciocco, M. P. Mascia, M. G. Pisu and G. Biggio, Bioorg. Med. Chem., 2005, 13, 3531 CrossRef CAS PubMed.
- (a) A. A. Bekhit, A. Hymete, A. E.-D. A. Bekhit, A. Damtew and H. Y. Aboul-Enein, Mini-Rev. Med. Chem., 2010, 10, 1014 CrossRef CAS PubMed; (b) A. A. Bekhit, H. T. Y. Fahmy, S. A. F. Rostom and A. E.-D. A. Bekhit, Eur. J. Med. Chem., 2010, 45, 6027 CrossRef CAS PubMed; (c) A. M. Vijesh, A. M. Isloor, P. Shetty, S. Sundershan and H. K. Fun, Eur. J. Med. Chem., 2013, 62, 410 CrossRef CAS PubMed.
- (a) J. J. Cui, M. Tran-Dube, H. Shen, M. Nambu, P.-P. Kung, M. Pairish, L. Jia, J. Meng, L. Funk, I. Botrous, M. McTigue, N. Grodsky, K. Ryan, E. Padrique, G. Alton, S. Timofeevski, S. Yamazaki, Q. Li, H. Zou, J. Christensen, B. Mroczkowski, S. Bender, R. S. Kania and M. P. Edwards, J. Med. Chem., 2011, 54, 6342 CrossRef CAS PubMed; (b) N. K. Terrett, A. S. Bell, D. Brown and P. Ellis, Bioorg. Med. Chem. Lett., 1996, 6, 1819 CrossRef; (c) T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. Collins, W. S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347 CrossRef CAS PubMed; (d) K. W. Weitzel, J. M. Wickman, S. G. Augustin and J. G. Strom, Clin. Ther., 2000, 22, 1254 CrossRef CAS PubMed; (e) K. Kısmet, M. T. Akay, O. Abbasoglu and A. Ercan, Cancer Detect. Prev., 2004, 28, 127 CrossRef PubMed; (f) T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, W. Collins, P. S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347 CrossRef CAS PubMed.
- (a) M. Su, M. Tomás-Gamasa, S. Serdjukow, P. Mayer and T. Carell, Chem. Commun., 2014, 50, 409 RSC; (b) M. Zora, A. Kivrak and C. Yazici, J. Org. Chem., 2011, 76, 6726 CrossRef CAS PubMed; (c) H. Batchu, S. Bhattacharyya, R. Kant and S. Batra, J. Org. Chem., 2015, 80, 7360 CrossRef CAS PubMed.
- (a) V. B. Purohit, S. C. Karad, K. H. Patel and D. K. Raval, Tetrahedron, 2016, 72, 1114 CrossRef CAS; (b) H. Y. Kim, V. B. Jadhav, D. Y. Jeong, W. K. Park, J.-H. Song, S. Lee and H. Cho, Arch. Pharm. Res., 2015, 38, 1019 CrossRef CAS PubMed.
- (a) D. Yang, K. Yan, W. Wei, G. Li, S. Lu, C. Zhao, L. Tian and H. Wang, J. Org. Chem., 2015, 80, 11073 CrossRef CAS PubMed; (b) D. Yang, K. Yan, W. Wei, J. Zhao, M. Zhang, X. Sheng, G. Li, S. Lu and H. Wang, J. Org. Chem., 2015, 80, 6083 CrossRef CAS PubMed; (c) K. Yan, D. Yang, W. Wei, S. Lu, G. Li, C. Zhao, Q. Zhang and H. Wang, Org. Chem. Front., 2016, 3, 66 RSC; (d) D. Yang, K. Yan, W. Wei, L. Tian, Q. Li, J. You and H. Wang, RSC Adv., 2014, 4, 48547 RSC; (e) N. Zhang, D. Yang, W. Wei, L. Yuan, Y. Cao and H. Wang, RSC Adv., 2015, 5, 37013 RSC; (f) K. Yan, D. Yang, P. Sun, W. Wei, Y. Liu, G. Li, S. Lu and H. Wang, Tetrahedron Lett., 2015, 56, 4792 CrossRef CAS; (g) K. Yan, D. Yang, W. Wei, J. Zhao, Y. Shuai, L. Tian and H. Wang, Org. Biomol. Chem., 2015, 13, 7323 RSC; (h) K. Yan, D. Yang, W. Wei, P. Sun, Y. Lu and H. Wang, Org. Chem. Front., 2016, 3, 556 RSC.
- (a) S. K. R. Parumala and R. K. Peddinti, Green Chem., 2015, 17, 4068 RSC; (b) Y. Liao, P. Jiang, S. Chen, H. Qi and G.-J. Deng, Green Chem., 2013, 15, 3302 RSC; (c) W. Ge and Y. Wei, Green Chem., 2012, 14, 2066 RSC; (d) J. B. Azeredo, M. Godoi, G. M. Martins, C. C. Silveira and A. L. Braga, J. Org. Chem., 2014, 79, 4125 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00407e |
| This journal is © the Partner Organisations 2016 |