
Stereo- and regio-selective synthesis of 3′-C-substituted-(N)-methanocarba adenosines as potential anticancer agents†
Siddhi D.
Naik
ab,
Girish
Chandra
c,
Pramod K.
Sahu
b,
Hong-Rae
Kim
b,
Shuhao
Qu
ab,
Ji-seong
Yoon
b and
Lak Shin
Jeong
*b
aCollege of Pharmacy, Ewha Womans University, Seoul 03760, Korea
bResearch Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea. E-mail: lakjeong@snu.ac.kr; Tel: +82-2-880-7850
cDepartment of Chemistry, School of Chemical and Physical Sciences, Central University of Bihar, Gaya, Bihar 823001, India
First published on 5th September 2016
Abstract
Stereo- and regio-selective synthesis of 3′-C-substituted-(N)-methanocarba adenosines 3a–c as potential anticancer agents was achieved by employing stereoselective cyclopropanation, stereoselective nucleophilic addition, regioselective isopropylidene cleavage and regioselective base condensation of cyclic sulfate as key steps.
Introduction
Modified nucleosides have long been developed as antimetabolite anticancer agents. They show anticancer activity by inhibiting normal metabolic pathways essential for the synthesis of both DNA and RNA in tumor cells.1 Among a number of antimetabolite nucleosides, 3′-C-ethynyl-cytidine (1a) and -uridine (1b) have been reported to exhibit potent antitumor activities against mouse leukemic L1210 (IC50 = 0.016 and 0.13 μM respectively) and human KB cells (IC50 = 0.028 and 0.029 μM respectively).2 Compound 1a is initially phosphorylated by uridine–cytidine kinase (UCK) to its monophosphate, and further phosphorylated to its di- and triphosphate, thus inhibiting RNA polymerase competitively.3 On the other hand, 3′-C-methyladenosine (2) was also reported to exhibit the most potent anticancer activity among 1′-, 2′-, and 3′-C-substituted compounds.4 Compound 2 showed anticancer activity against human myelogenous leukemia K562, multidrug resistant human leukemia K562IU, human promyelocytic leukemia HL-60, human colon carcinoma HT-29 and human breast carcinoma MCF-7 cell lines.4 The anticancer activity of 2 is attributed to the inhibition of ribonucleotide reductase after being converted to the corresponding diphosphate.5Conformational studies by NMR indicate that in general the ribonucleosides are engaged in a rapid dynamic north (2′-exo/3′-endo) and south (2′-endo/3′-exo) equilibrium and only a small preference towards either one of the hemispheres is detectable.6 Marquez and co-workers hypothesized that DNA/RNA polymerase and cellular kinases prefer either conformation on binding to nucleoside analogues, which was proved by nucleoside analogues built on a conformationally locked bicyclo[3.1.0]hexane template, locked at the north or the south conformation.7 As judged from the X-ray crystal structures, most of the ribonucleosides prefer to adopt the north conformation. On the basis that compounds 1 and 2 might be adopting the north conformation as well, we designed and synthesized their conformationally locked bicyclo[3.1.0]hexane nucleoside analogs and evaluated their anticancer activities (Fig. 1).
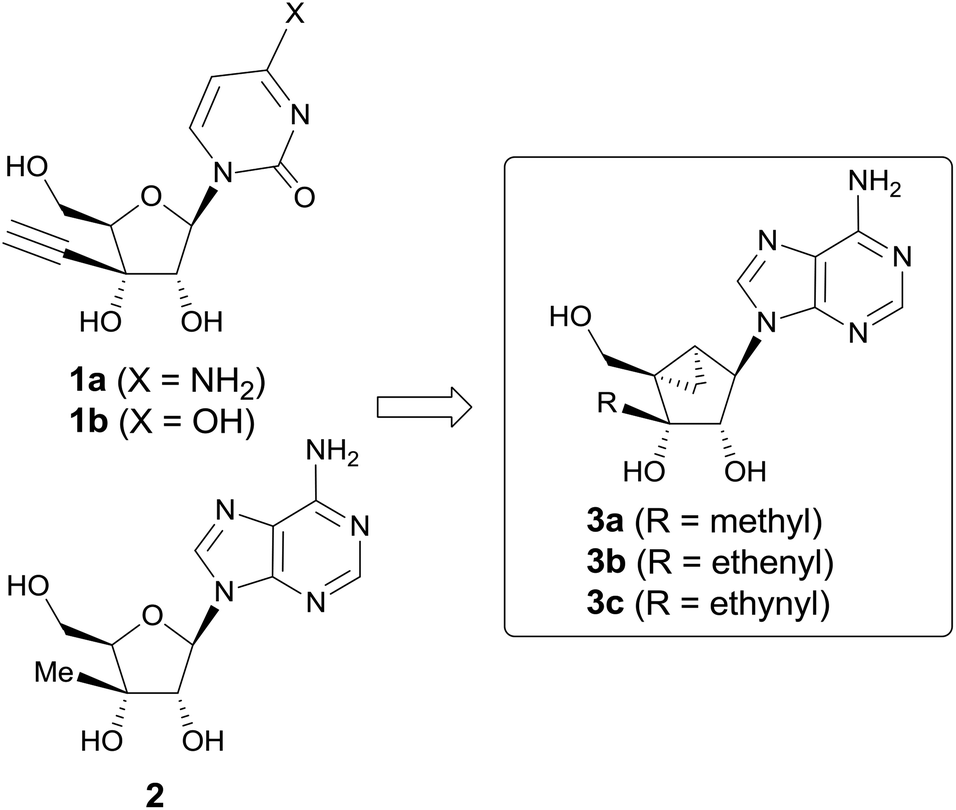 | ||
| Fig. 1 The rationale for design of the final nucleosides 3a–c. | ||
Herein, we report the stereo- and regioselective synthesis of 3′-C-substituted (N)-methanocarba nucleosides 3a–c, using stereoselective nucleophilic addition, regioselective cyclic sulfate nucleobase condensation and regioselective isopropylidene cleavage as key steps and their anticancer activities.
Results and discussion
It was envisaged to utilize the stereoselective addition of organometallic nucleophilic reagents to ketones at the sugar stage, via route A or route B, which uses the same reaction at the nucleoside level (Scheme 1).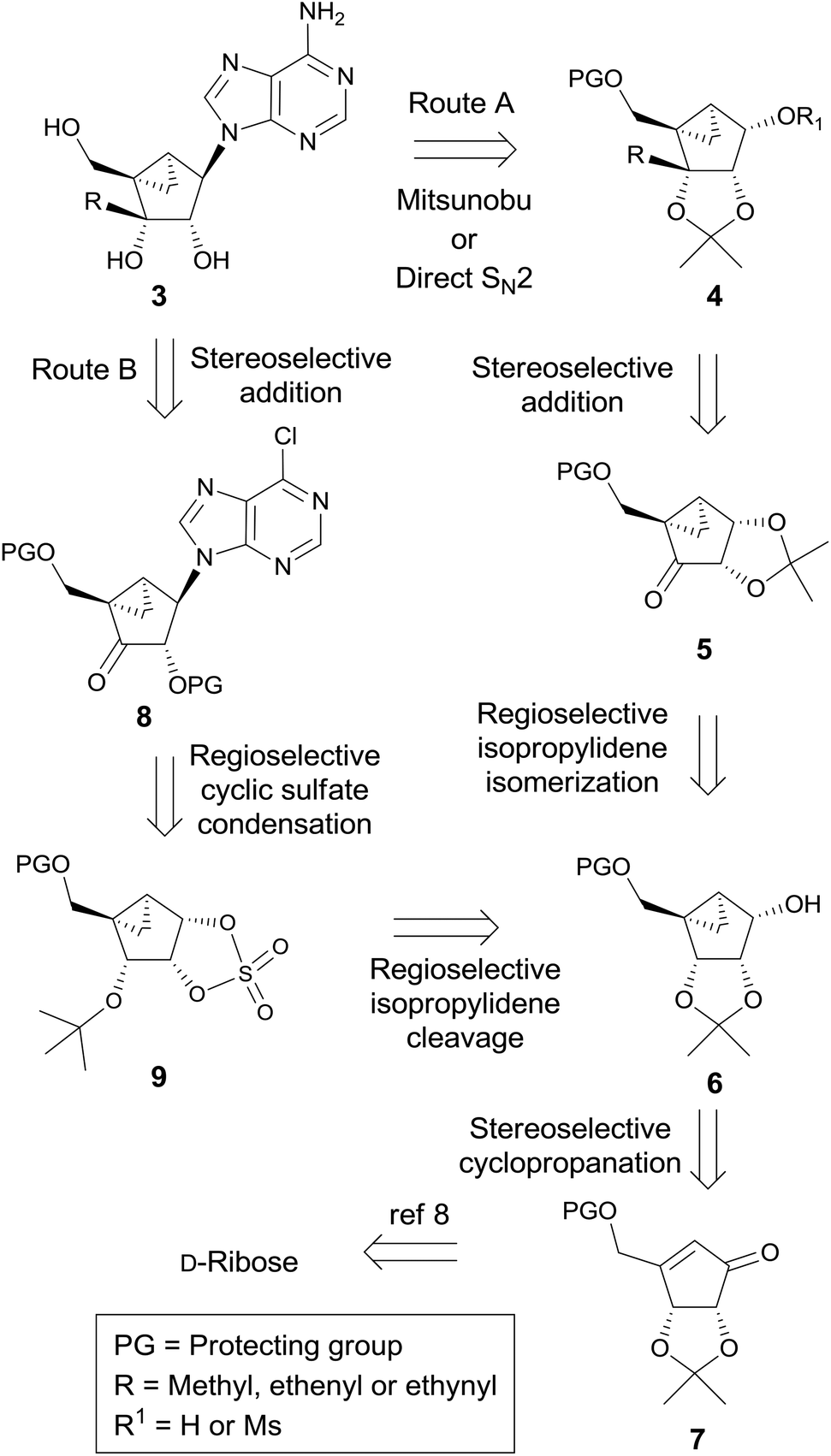 | ||
| Scheme 1 Retrosynthetic analysis of the final nucleosides 3a–c. | ||
The final nucleosides 3 could be synthesized from 4 using the Mitsunobu or direct SN2 reaction (route A). The key intermediate 4 could be derived from ketone 5via stereoselective addition of organometallic reagents followed by the isomerization of the acetonide. Ketone 5 could be derived from the isomerization of 6 followed by oxidation. Compound 6 could be easily synthesized from 7via stereoselective cyclopropanation as the key step, which could be synthesized from D-ribose according to our previously published procedure.8
On the other hand, the final nucleosides 3a–c could be derived from the 3′-ketonucleoside 8, using the stereoselective addition of organometallic reagents as the key step (route B). Intermediate 8 could be synthesized from the cyclic sulfate 9, using the regioselective cyclic sulfate base condensation as the key step, which could be derived from 6via the regioselective cleavage of the isopropylidene group.
Scheme 2 depicts the synthesis of the glycosyl donors 15 and 16, employing the synthetic strategy of route A. Compound 7, synthesized from D-ribose by following a known method8 was reduced to allylic alcohol 109 under Luche conditions,10 which was converted to cyclopropyl-fused alcohol 119 under the standard cyclopropanation conditions. Isomerization of 2,3-acetonide 11 to 1,2-acetonide 12 was achieved by treatment with acetone in the presence of p-TsOH. TPAP/NMO oxidation11 of 12 afforded ketone 13 quantitatively. Treatment of ketone 13 with vinylmagnesium bromide at 0 °C afforded the tertiary alcohol 14 as the single stereoisomer due to the steric effect induced by 1,2-acetonide and the cyclopropane ring. Another isomerization of 1,2-acetonide 14 to 2,3-acetonide 15 smoothly proceeded under the same acidic conditions. The glycosyl donor 15 was also converted to another glycosyl donor 16 by mesylation.
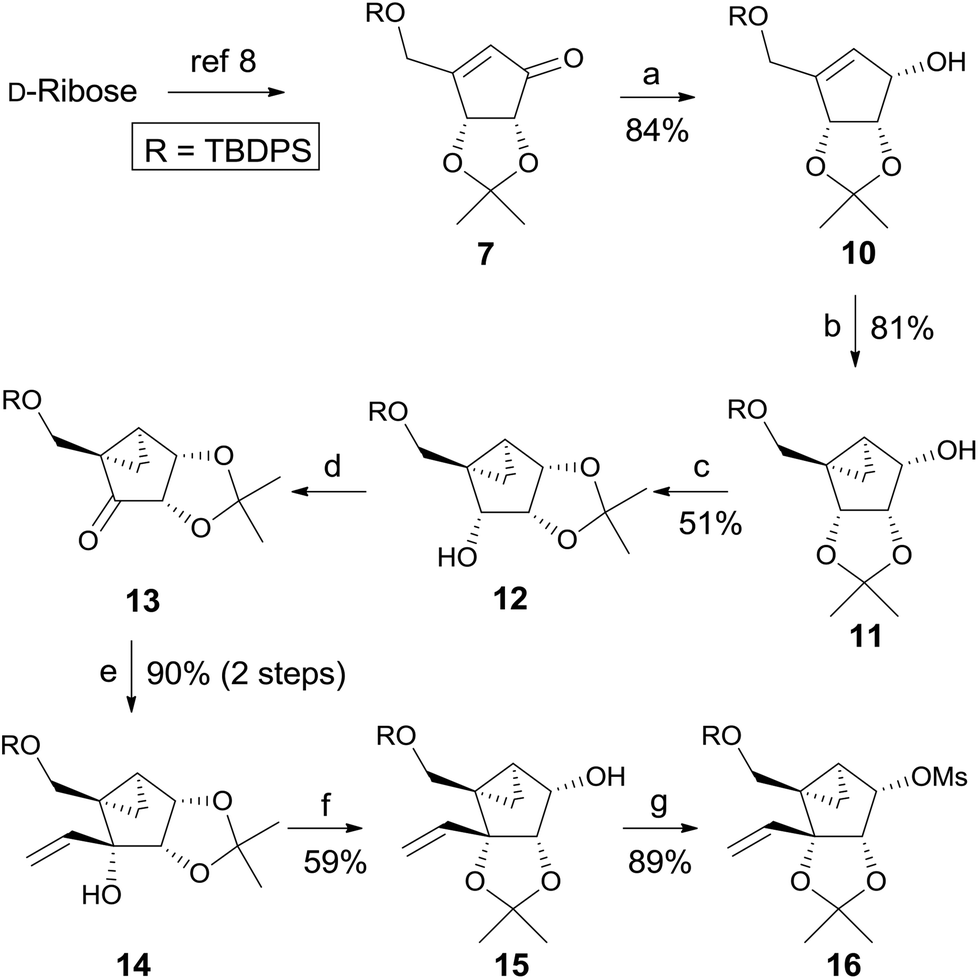 | ||
Scheme 2 Synthesis of the glycosyl donors 15 and 16 using route A. Reagents and conditions: (a) NaBH4, CeCl3·7H2O, MeOH, 0 °C, 30 min; (b) Et2Zn, CH2I2, CH2Cl2, rt, 5 h; (c) p-TsOH, acetone, rt, 4 h; (d) TPAP, NMO, mol. sieves, CH2Cl2, rt, 15 h; (e) CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHMgBr, THF, 0 °C, 30 min; (f) p-TsOH, acetone, rt, 4 h; (g) MsCl, Et3N, CH2Cl2, 40 min. CHMgBr, THF, 0 °C, 30 min; (f) p-TsOH, acetone, rt, 4 h; (g) MsCl, Et3N, CH2Cl2, 40 min. | ||
However, condensation of 15 with 6-chloropurine or adenine under the standard Mitsunobu conditions did not give the condensed product 17 (Scheme 3). Condensation of 16 with 6-chloropurine or an adenine anion under the direct SN2 conditions also failed to afford the desired product 17.
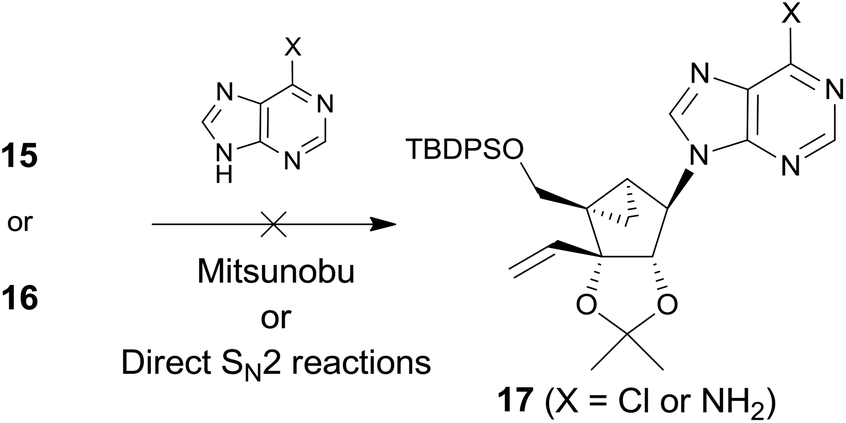 | ||
| Scheme 3 Synthetic approach towards 17 using the Mitsunobu or direct SN2 base condensation. | ||
It was speculated that no formation of 17 is attributed to the 1,3-diaxial repulsion between the incoming nucleophile (6-chloropurine or adenine anion) and the 3-vinyl group, as illustrated in Fig. 2 because the substrate without the vinyl group afforded base condensed products without any problem.12
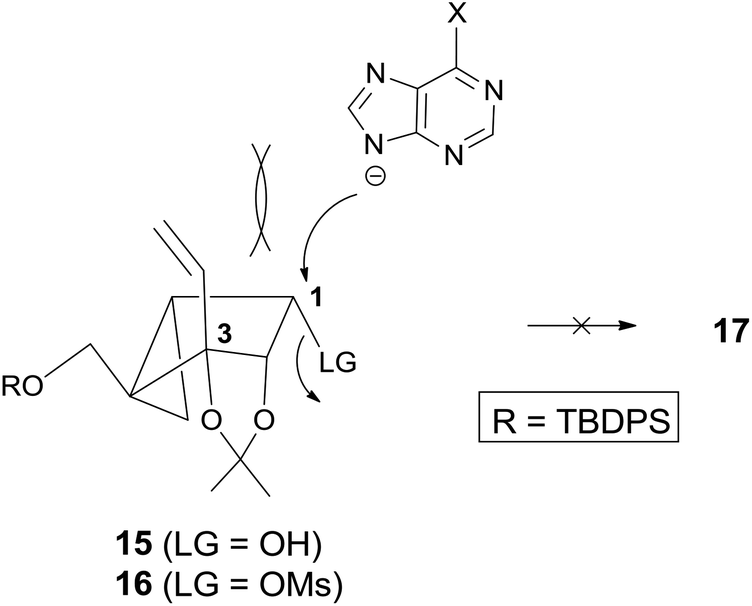 | ||
| Fig. 2 Plausible mechanism for no formation of 17 due to 1,3-diaxial repulsion. | ||
Thus, we decided to utilize the synthetic route B, which avoids 1,3-diaxial repulsion. We also employed cyclic sulfate chemistry13 since cyclic sulfate is very reactive as well as cleaved regioselectively at a position with low electron density.14 Thus, another glycosyl donor, cyclic sulfate 20 was synthesized, as shown in Scheme 4. Regioselective cleavage of 2,3-acetonide 11 was achieved by treating with trimethyl aluminum15 at room temperature to afford diol 189 as a single regioisomer. Treatment of 18 with thionyl chloride yielded cyclic sulfite 19 which was oxidized to cyclic sulfate 20 by treating with excess NaIO4 in the presence of catalytic amounts of RuCl3·3H2O.13
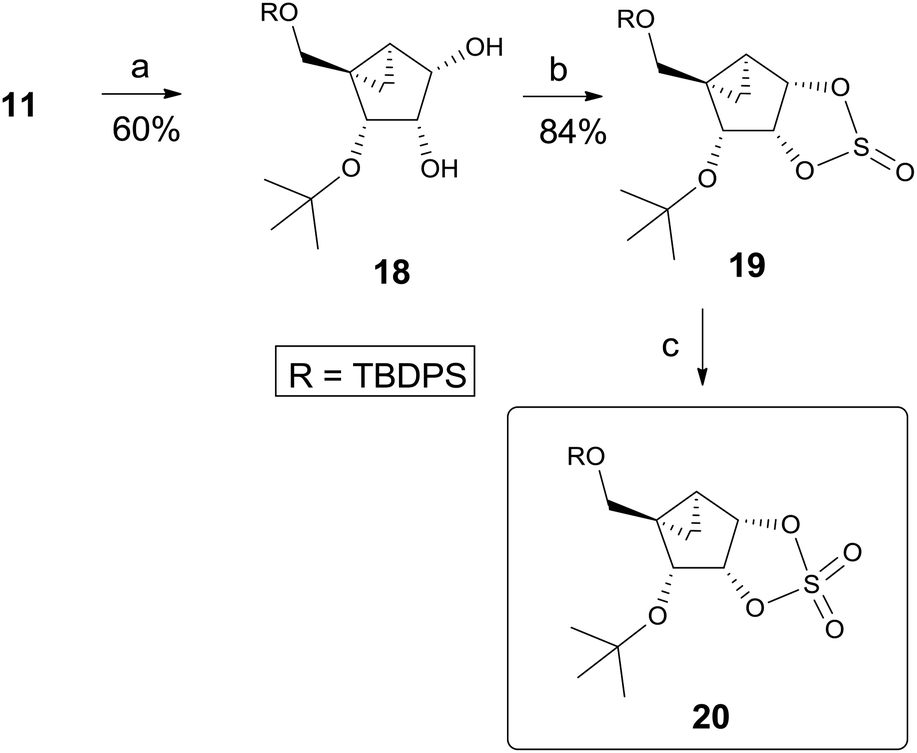 | ||
| Scheme 4 Synthesis of the cyclic sulfate 20 as another glycosyl donor. Reagents and conditions: (a) AlMe3, CH2Cl2, rt, 4 d; (b) SOCl2, Et3N, CH2Cl2, 0 °C, 10 min; (c) NaIO4, RuCl3·3H2O, CCl4, CH3CN, H2O, 0 °C, 10 min. | ||
Synthesis of the final products 3a–c using cyclic sulfate chemistry is illustrated in Scheme 5. Condensation of cyclic sulfate 20 with a 6-chloropurine anion at 70 °C smoothly proceeded to give the desired regioisomer 21 as the single regioisomer after acidic hydrolysis of the resulting sulfate.9 Based on the previous reports involving cyclic sulfate chemistry14, the excellent regioselectivity was attributed to the differences in the electron density between the C-1 and C-2 carbons. According to computational calculations, it seems to be a trend that the cyclic sulfate carbon atom with more electron-withdrawing substituents at the vicinal carbon atom displays a higher electron density than the other cyclic sulfate carbon.14b Thus, the C-1 carbon of 20 should display a lower electron density than the C-2 carbon because of the electron-withdrawing 3′-alkoxy group, giving 21 as the single regioisomer. The hydroxyl group of 21 was protected as the TBS group to give 22, whose t-butyl group was removed with TiCl4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 at −78 °C for 30 min to afford 23 in good yield. Oxidation of 23 with DMP yielded ketone 24, which can readily undergo stereoselective addition. Nucleophilic addition of 24 with strong organometallic carbon nucleophiles such as MeMgI, CH2CHMgBr, and Li–C
16 at −78 °C for 30 min to afford 23 in good yield. Oxidation of 23 with DMP yielded ketone 24, which can readily undergo stereoselective addition. Nucleophilic addition of 24 with strong organometallic carbon nucleophiles such as MeMgI, CH2CHMgBr, and Li–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–TMS produced the desired addition products 25a–c as the single stereoisomers, respectively. Removal of the silyl protecting groups in 25a–c with n-Bu4NF afforded the 6-chloropurine derivatives 26a–c, respectively. Treatment of 26a–c with t-butanolic ammonia yielded the final nucleosides 3a–c, respectively.
C–TMS produced the desired addition products 25a–c as the single stereoisomers, respectively. Removal of the silyl protecting groups in 25a–c with n-Bu4NF afforded the 6-chloropurine derivatives 26a–c, respectively. Treatment of 26a–c with t-butanolic ammonia yielded the final nucleosides 3a–c, respectively.
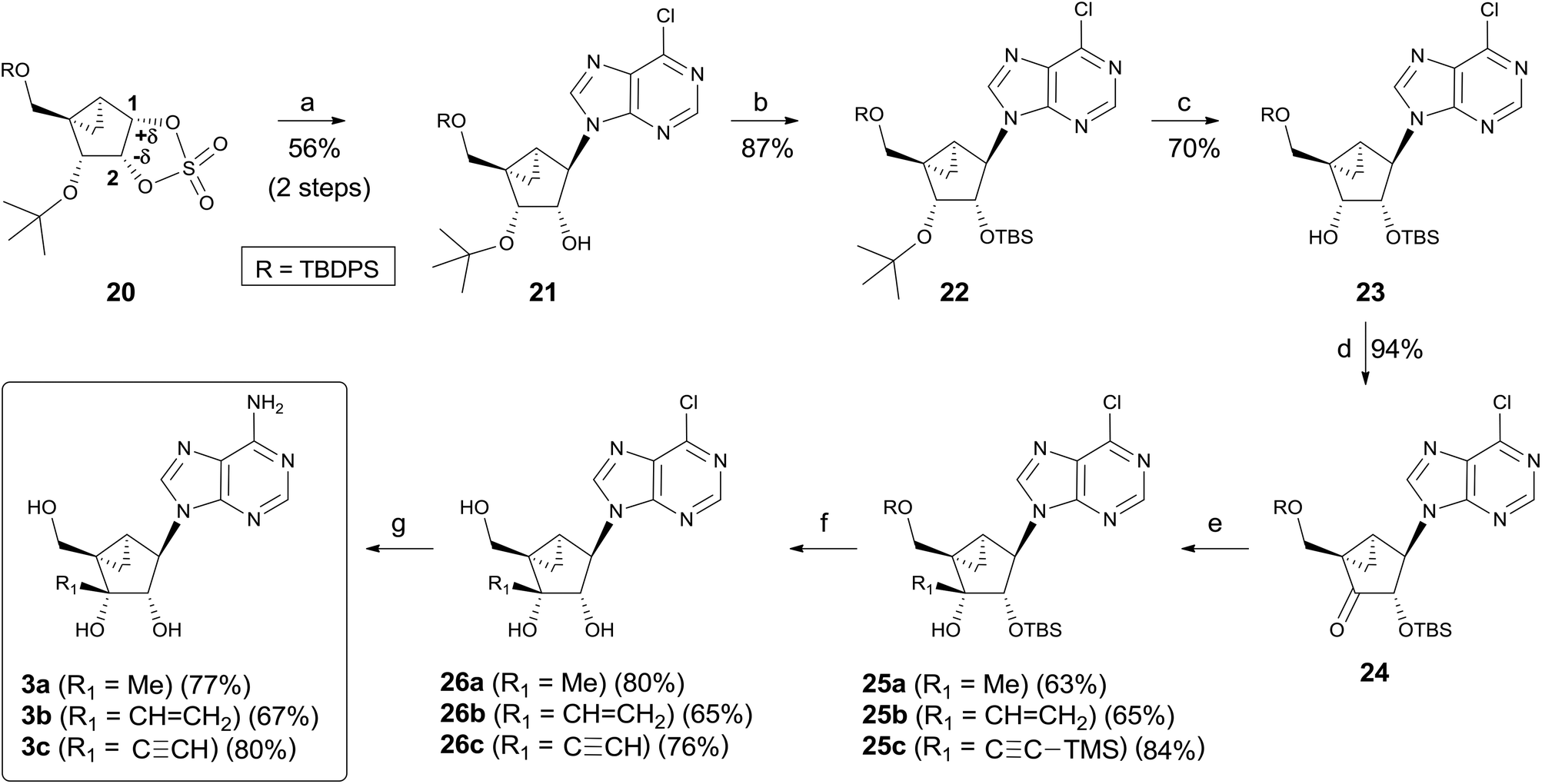 | ||
| Scheme 5 Synthesis of the final nucleosides 3a–c using cyclic sulfate chemistry. Reagents and conditions: (a) (i) 6-chloropurine, NaH, DMF, 70 °C, 8 h; (ii) c-HCl, 70 °C, 2 h; (b) TBSCl, imidazole, DMF, 70 °C, 15 h; (c) TiCl4, CH2Cl2, −78 °C, 30 min; (d) DMP, NaHCO3, CH2Cl2, rt, 2 h; (e) MeMgI, THF, −78 °C, 30 min for 25a; CH2CHMgBr, THF, 0 °C, 30 min for 25b; TMS-acetylene, n-BuLi, THF, −78 °C, 1.5 h for 25c; (f) n-Bu4NF, THF, 0 °C – rt, 4 h; (g) NH3/t-BuOH, 80 °C, 18 h. | ||
Stereochemistry of 3c was confirmed by X-ray crystallography (vide infra) (Fig. 3).17 As expected, the X-ray crystal structure of 3c with a bicyclo [3.1.0] system revealed a preferred north (N) conformation with a P (pseudorotation angle) value of −19.5°, which corresponds to a more characteristic C2′-exo/C3′-endo ring puckering found in conventional ribonucleosides.6
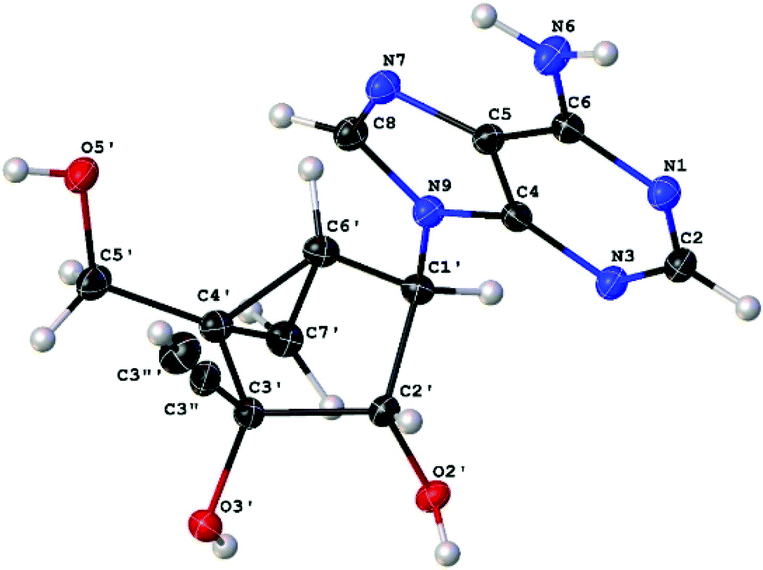 | ||
| Fig. 3 X-ray crystal structure of 3c. | ||
All final compounds 3a–c were assayed for anticancer activity in several human cancer cell lines such as A549 (lung cancer), HCT (colon cancer), SNU (stomach cancer), and PC (prostate cancer) cell lines.18 To our disappointment, none of the compounds showed any significant anticancer activity up to 100 μM. No biological activity of the north conformers 3a–c may be explained in several ways. There might be no phosphorylation by cellular kinases as cellular kinases may prefer the south conformation, indicating that 13 and 25 are phosphorylated with the south conformation in the solution state and/or because of the steric effects induced by the fused cyclopropyl ring.7 Another possible speculation is that RNA polymerase may also prefer the south conformation for anticancer activity even though they are phosphorylated with the north conformation in cells.7 Cellular phosphorylation and RNA polymerase assay should be further studied to elucidate these speculations.
Conclusions
In summary, we have accomplished the stereo- and regioselective synthesis of 3′-C-substituted-(N)-methanocarba-adenosines 3a–c, starting from D-ribose. Regioselective cleavages of the isopropylidene group and cyclic sulfate were employed for the regioselective synthesis of the final products, while stereoselective nucleophilic addition was utilized for the introduction of the 3′-C-substituents. Although we could not discover promising anticancer agents, we expect that all chemistries employed here will be extensively utilized for the synthesis of modified nucleosides. The cellular metabolism study is in progress and will be reported elsewhere.Experimental section
General procedures
Proton (1H) and carbon (13C) NMR spectra were obtained on a Bruker AV 400 (400/100 MHz), Varian Unity Invoa 400 (400/100 MHz), or Jeol JNM-ECA600 (600/150 MHz) spectrometer. Chemical shifts are reported in ppm with Me4Si as the internal standard. Optical rotations were determined on a Jasco III in an appropriate solvent. UV spectra were recorded on U-3000 made by Hitachi in methanol or water. Melting points were determined on a Buchan B-540 instrument and are uncorrected. All reactions were routinely carried out under an inert atmosphere of dry nitrogen. Reactions were checked by thin layer chromatography (Kieselgel 60 F254, Merck). Spots were detected by viewing under UV light, and by colorizing with charring after dipping in a p-anisaldehyde solution or a phosphomolybdic acid solution. In aqueous work-up, all organic solutions were dried over anhydrous magnesium sulfate and filtered prior to rotary evaporation at water pump pressure. The crude compounds were purified by column chromatography on silica gel (Kieselgel 60, 70–230 mesh, Merck). All the anhydrous solvents used were redistilled over CaH, P2O5, or sodium/benzophenone prior to the reaction.(3aS,3bS,4aR,5R,5aR)-4a-(((tert-Butyldiphenylsilyl)oxy)-methyl)-2,2-dimethylhexahydrocyclopropa-[3,4]cyclopenta-[1,2-d][1,3]dioxol-5-ol (12)
To a stirred solution of 119 (20.36 g, 46.4 mmol) in acetone (300 mL) was added p-TsOH·H2O (4.41 g, 23.2 mmol) and the reaction mixture was stirred at room temperature under a nitrogen atmosphere for 4 h. Triethylamine was added to neutralize the reaction mixture until a pH of 7–8 was achieved. The reaction mixture was evaporated to dryness under reduced pressure; the residue was dissolved in ethyl acetate and washed with water. The organic layer was dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 17:3) to give 12 (10.31 g, 51%, brsm 97%) as a colorless syrup. 1H NMR (600 MHz, CDCl3): δ 7.63–7.60 (m, 4H), 7.41–7.24 (m, 6H), 4.77 (t, J = 5.9 Hz, 1H), 4.64 (t, J = 7.7 Hz, 1H), 4.52 (t, J = 6.9 Hz, 1H), 4.03 (d, J = 11.0 Hz, 1H), 3.30 (d, J = 11.0 Hz, 1H), 2.25 (d, J = 9.2 Hz, 1H), 1.38 (m, 1H), 1.27 (s, 3H), 1.07 (t, J = 4.1 Hz, 1H), 1.03 (s, 9H), 0.53 (m, 1H); 13C NMR (150 MHz, CDCl3): δ 135.56, 135.55, 133.6, 133.4, 129.74, 129.71, 127.69, 127.67, 112.3, 80.2, 79.3, 71.4, 65.3, 41.3, 26.8, 26.2, 26.0, 24.6, 19.2, 10.2; [α]25D = −22.88 (c 0.09, MeOH); HRMS (FAB+): m/z calculated for C26H35O4Si (M + H)+ 439.2305, found: 439.2292.
(3aS,3bS,4aR,5R,5aS)-4a-(((tert-Butyldiphenylsilyl)oxy)-methyl)-2,2-dimethyl-5-vinylhexahydrocyclopropa[3,4]-cyclopenta[1,2-d][1,3]dioxol-5-ol (14)
To a stirred solution of 12 (5.86 g, 13.38 mmol) in anhydrous methylene chloride (150 mL) were added N-methylmorpholine N-oxide (2.35 g, 20.07 mmol) and tetrapropylammonium perruthenate (0.235 g, 0.70 mmol). After being stirred at room temperature for 15 h under a nitrogen atmosphere the reaction mixture was filtered through a Celite pad. The filtrate was evaporated under reduced pressure and the residue was filtered through a short silica pad using hexane–ethyl acetate (9:1) as an eluent. The filtrate was evaporated to give 13 (crude) as a colorless syrup, which was used for the next reaction without any further purification. To a stirred solution of crude 13 in dry THF (100 mL) under a nitrogen atmosphere was added a solution of vinyl magnesium bromide (1 M in THF) (67 mL, 67 mmol) at 0 °C and the mixture was stirred at 0 °C for 30 min. The reaction was quenched with a saturated aqueous ammonium chloride solution and the aqueous phase was extracted with ethyl acetate. The organic layer was washed with brine, dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 9:1) to give 14 (5.40 g, 90%) as a colorless syrup: 1H NMR (600 MHz, CDCl3): δ 7.61–7.59 (m, 4H), 7.41–7.33 (m, 6H), 5.86 (m, 1H), 5.46 (dd, J = 1.4, 17.2 Hz, 1H), 5.16 (dd, J = 1.8, 10.8 Hz, 1H), 4.81 (t, J = 6.0 Hz, 1H), 4.16 (d, J = 6.8 Hz, 1H), 3.78 (d, J = 10.5 Hz, 1H), 3.50 (d, J = 10.5 Hz, 1H), 2.78 (s, 1H), 1.67 (m, 1H), 1.57 (s, 3H), 1.31 (t, J = 4.6 Hz, 1H), 1.26 (s, 3H), 1.01 (s, 9H), 0.82 (s, 1H); 13C NMR (150 MHz, CDCl3): δ 140.2, 135.5, 133.3, 133.2, 129.6, 127.6, 113.1, 112.5, 84.8, 79.8, 79.7, 64.0, 42.2, 26.7, 26.1, 25.9, 24.7, 19.1, 11.8; [α]25D = 4.60 (c 0.15, MeOH); HRMS (FAB+): m/z calculated for C28H37O4Si (M + H)+ 465.2461, found: 465.2452.
(3aR,3bR,4aS,5S,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)-methyl)-2,2-dimethyl-3a-vinylhexahydrocyclopropa[3,4]-cyclopenta[1,2-d][1,3]dioxol-5-ol (15)
To a stirred solution of 14 (7.39 g, 3.00 mmol) in acetone (25 mL) was added p-TsOH·H2O (0.285 g, 1.5 mmol) and the reaction mixture was stirred at room temperature under a nitrogen atmosphere for 4 h. Triethylamine was added to neutralize the reaction mixture until a pH of 7–8 was achieved, the mixture was evaporated to dryness under reduced pressure and the residue was dissolved in ethyl acetate and washed with water. The organic layer was dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 17:3) to give 15 (4.35 g, 59%, brsm 78%) as a colorless syrup: 1H NMR (600 MHz, CDCl3): δ 7.62–7.58 (m, 4H), 7.40–7.33 (m, 6H), 6.04 (m, 1H), 5.37 (dd, J = 1.8, 17.1 Hz, 1H), 5.10 (dd, J = 1.8, 10.8 Hz, 1H), 4.50 (m, 1H), 4.20 (d, J = 6.9 Hz, 1H), 3.70 (d, J = 11.0 Hz, 1H), 3.57 (d, J = 11.0 Hz, 1H), 2.28 (d, J = 10.5 Hz, 1H), 1.58 (s, 3H), 1.88 (m, 1H), 1.26 (s, 3H), 1.24 (t, J = 4.5 Hz, 1H), 1.00 (s, 9H), 0.74 (s, 1H); 13C NMR (150 MHz, CDCl3): δ 138.2, 135.6, 135.5, 133.59, 133.55, 129.5, 127.58, 127.57, 113.1, 112.9, 92.3, 84.4, 70.6, 63.3, 37.5, 31.3, 26.7, 26.4, 25.2, 19.2, 11.8; [α]25D = 28.00 (c 0.10, MeOH); HRMS (FAB+): m/z calculated for C28H37O4Si (M + H)+ 465.2461, found: 465.2456.
(3aR,3bR,4aS,5S,5aS)-3b-(((tert-Butyldiphenylsilyl)oxy)-methyl)-2,2-dimethyl-3a-vinylhexahydrocyclopropa[3,4]-cyclopenta[1,2-d][1,3]dioxol-5-yl methanesulfonate (16)
To a stirred solution of 15 (117 mg, 0.2619 mmol) in methylene chloride (10 mL) under a nitrogen atmosphere were added triethylamine (0.167 mL, 0.328 mmol) and methanesulfonyl chloride (0.030 mL, 0.7857 mmol) at 0 °C and the reaction mixture was stirred at room temperature for 40 min. Water was added to the mixture and the aqueous phase was extracted with methylene chloride. The combined organic extracts were dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 9:1) to give 16 (126 mg, 89%) as a colorless foam: 1H NMR (400 MHz, CDCl3): δ 7.61–7.56 (m, 4H), 7.41–7.33 (m, 6H), 5.98 (m, 1H), 5.49 (d, J = 17.1 Hz, 1H), 5.25 (t, J = 5.7 Hz, 1H), 5.12 (d, J = 10.9 Hz, 1H), 4.33 (d, J = 6.3 Hz, 1H), 3.69 (d, J = 11.0 Hz, 1H), 3.59 (d, J = 11.0 Hz, 1H), 3.20 (s, 3H), 1.92 (m, 1H), 1.57 (s, 3H), 1.25 (s, 3H), 1.24 (m, 1H), 0.99 (s, 9H), 0.91 (s, 1H).
(3aS,3bS,4aR,5R,5aR)-5-(tert-Butoxy)-4a-(((tert-butyl-diphenylsilyl)oxy)methyl)hexahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3,2]dioxathiole 2-oxide (19)
To a stirred solution of 18 (14.17 g, 31.16 mmol) in methylene chloride (200 mL) under a nitrogen atmosphere were added triethylamine (15.65 mL, 112.19 mmol) and thionyl chloride (3.39 mL, 46.47 mmol) at 0 °C and the reaction mixture was stirred at 0 °C for 10 min. The reaction was quenched with water and the aqueous phase was extracted with methylene chloride. The organic layer was washed with brine, dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 9:1) to give 19 (12.78 g, 84%) as a diasteriomeric mixture in the form of a colorless syrup: 1H NMR (600 MHz, CDCl3): (for major isomer) δ 7.44–7.41 (m, 4H), 7.38–7.35 (m, 7H), 5.40 (t, J = 5.8 Hz, 1H), 5.06 (t, J = 6.4 Hz, 1H), 4.76 (d, J = 6.4 Hz, 1H), 4.12 (d, J = 11.4 Hz, 1H), 3.02 (d, J = 11.4 Hz, 1H), 1.25 (s, 9H), 1.22 (m, 1H), 1.04 (s, 9H), 0.97 (m, 1H), 0.49 (m, 1H); HRMS (FAB+): m/z calculated for C27H37O5SSi (M + H)+ 501.2131, found: 501.2133.
(1R,2R,3S,4R,5S)-2-(tert-Butoxy)-1-(((tert-butyldiphenyl-silyl)oxy)methyl)-4-(6-chloro-9H-purin-9-yl)bicyclo[3.1.0]-hexan-3-ol (21)
To a stirred solution of 19 (3.40 g, 6.79 mmol) in carbon tetrachloride, acetonitrile, and water (1:1:1.5, 52.5 mL) were added ruthenium chloride trihydrate (0.098 g, 0.475 mmol) and sodium metaperiodate (2.17 g, 10.18 mmol) and the reaction mixture was stirred at room temperature for 10 min. The reaction was diluted with water and the aqueous phase was extracted with diethyl ether. The organic layer was washed with brine and a saturated aqueous sodium bicarbonate solution. The combined organic extracts were dried over MgSO4, filtered, concentrated and filtered through a short silica gel pad using hexane–ethyl acetate (1:2) as the eluent which on evaporation afforded 20 as a colorless syrup which was immediately subjected to the next reaction without any further purification. To a stirred suspension of 6-chloropurine (2.09 g, 13.54 mmol) in DMF (20 mL) under a nitrogen atmosphere was added sodium hydride (0.487 g, 20.31 mmol) and the reaction mixture was stirred at 80 °C for 3 h. To this reaction mixture was added 20 in DMF (20 mL) at 60 °C and the reaction mixture was stirred at 70 °C for 5 h. Conc. HCl was added slowly at 0 °C to attain a pH of 1–2 and the mixture was stirred at 60 °C for 2 h. The reaction mixture was neutralized with a saturated NaHCO3 solution at 0 °C and the aqueous phase was extracted with ethyl acetate. The organic layer was dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 9:1) to give 21 (2.27 g, 56%) as a white foam: UV (MeOH) λmax 265 nm; 1H NMR (600 MHz, CDCl3): δ 8.96 (s, 1H), 8.54 (s, 1H), 7.61–7.55 (m, 4H), 7.42–7.29 (m, 6H), 5.02 (s, 1H), 4.80 (d, J = 6.4 Hz, 1H), 4.19 (d, J = 10.9 Hz, 1H), 3.87 (d, J = 6.6 Hz, 1H), 3.32 (d, J = 10.9 Hz, 1H), 3.16 (bs, 1H), 1.42 (m, 1H), 1.31 (m, 1H), 1.17 (s, 9H), 1.11 (s, 9H), 0.71 (m, 1H); 13C NMR (150 MHz, CDCl3): δ 151.9, 151.4, 151.0, 143.7, 135.7, 135.4, 132.7, 132.6, 131.9, 130.0, 129.9, 127.8, 127.7, 75.1, 71.1, 64.4, 61.7, 36.3, 28.1, 27.1, 23.3, 19.1, 11.5; [α]20D = 61.0 (c 0.14, MeOH); HRMS (FAB+): m/z calculated for C32H40ClN4O3Si (M + H)+ 591.2558, found: 591.2551.
9-((1S,2R,3S,4R,5R)-4-(tert-Butoxy)-3-((tert-butyldimethyl-silyl)oxy)-5-(((tert-butyldiphenylsilyl)oxy)methyl)bicyclo-[3.1.0]hexan-2-yl)-6-chloro-9H-purine (22)
To a stirred solution of 21 (2.58 g, 4.36 mmol) in DMF (30 mL) under a nitrogen atmosphere were added imidazole (1.73 g, 26.20 mmol), tert-butyldimethylchlorosilane (2.63 g, 17.46 mmol) and 4-dimethylaminopyridine (0.266 g, 2.18 mmol) at 0 °C and the reaction mixture was stirred for 15 h at 70 °C. The reaction was cooled to room temperature, quenched with water and the aqueous phase was extracted with ethyl acetate. The organic layer was dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 15:1) to give 22 (2.68 g, 87%) as a colorless foam: UV (MeOH) λmax 265 nm; 1H NMR (600 MHz, CDCl3): δ 8.70 (s, 1H), 8.67 (s, 1H), 7.67–7.61 (m, 4H), 7.43–7.32 (m, 6H), 4.82 (s, 1H), 4.49 (d, J = 6.0 Hz, 1H), 4.24 (d, J = 11.5 Hz, 1H), 3.97 (m, 1H), 3.24 (d, J = 11.0 Hz, 1H), 1.64 (m, 1H), 1.49 (m, 1H), 1.15 (s, 9H), 1.10 (s, 9H), 0.85 (s, 9H), 0.67 (m, 1H), 0.01 (s, 3H), −0.02 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 151.7, 151.6, 150.9, 144.2, 135.8, 133.5, 132.9, 132.7, 132.0, 129.9, 129.8, 127.8, 127.7, 78.1, 73.8, 70.9, 65.4, 62.4, 34.8, 28.3, 27.1, 25.7, 21.4, 19.1, 17.8, 13.0, −4.4, −5.0; [α]20D = −121.36 (c 0.11, MeOH); HRMS (FAB+): m/z calculated for C38H54ClN4O3Si2 (M + H)+ 705.3423, found: 705.3429.
(1R,2R,3S,4R,5S)-3-((tert-Butyldimethylsilyl)oxy)-1-(((tert-butyldiphenylsilyl)oxy)methyl)-4-(6-chloro-9H-purin-9-yl)-bicyclo[3.1.0]hexan-2-ol (23)
To a stirred solution of 22 (2.60 g, 4.00 mmol) in anhydrous methylene chloride (150 mL) under a nitrogen atmosphere was added a solution of TiCl4 (1 M in methylene chloride) (100 mL, 100.9 mmol) at −78 °C and the reaction mixture was stirred at −78 °C for 30 min. The reaction was quenched with saturated aqueous ammonium chloride and the precipitate formed was dissolved by adding a saturated aqueous sodium bicarbonate solution. The aqueous phase was extracted with ethyl acetate, the organic layer was dried over MgSO4, filtered, and evaporated and the residue was purified by silica gel column chromatography (hexane:ethyl acetate = 22:3) to give 23 (1.73 g, 70%) as a glassy solid: UV (MeOH) λmax 265 nm; 1H NMR (600 MHz, CDCl3): δ 8.74 (s, 1H), 8.72 (s, 1H), 7.70–7.65 (m, 4H), 7.42–7.35 (m, 6H), 5.02 (s, 1H), 4.90 (m, 1H), 4.37 (d, J = 11.0 Hz, 1H), 3.93 (d, J = 5.9 Hz, 1H), 3.32 (d, J = 10.9 Hz, 1H), 2.26 (d, J = 10.5 Hz, 1H), 1.60 (m, 1H), 1.42 (m, 1H), 1.13 (s, 9H), 0.92 (s, 9H), 0.65 (m, 1H), 0.21(s, 3H), 0.04 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 151.8, 151.3, 151.1, 143.6, 135.6, 135.5, 133.0, 132.5, 131.8, 129.9, 129.8, 127.8, 77.2, 70.8, 64.9, 61.6, 36.7, 26.9, 25.7, 24.0, 19.2, 17.9, 10.5, −4.6, −5.3; [α]20D = 47.68 (c 0.16, MeOH); HRMS (FAB+): m/z calculated for C34H46ClN4O3Si2 (M + H)+ 649.2797, found: 649.2788.
(1R,3S,4R,5S)-3-((tert-Butyldimethylsilyl)oxy)-1-(((tert-butyl-diphenylsilyl)oxy)methyl)-4-(6-chloro-9H-purin-9-yl)bicyclo-[3.1.0]hexan-2-one (24)
To a stirred solution of 23 (2.67 g, 4.11 mmol) in dry methylene chloride (30 mL) under a nitrogen atmosphere were added solid sodium bicarbonate (1.03 g, 12.33 mmol) and Dess–Martin periodinane (8.70 g, 20.55 mmol) at room temperature and the reaction mixture was stirred at room temperature for 2 h. The reaction was quenched with a saturated aqueous sodium bicarbonate solution at 0 °C and the aqueous phase was extracted with methylene chloride. The organic layer was dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 9:1) to give 24 (2.53 g, 94%) as a colorless foam: UV (MeOH) λmax 265 nm; 1H NMR (600 MHz, CDCl3): δ 8.59 (s, 1H), 8.37 (s, 1H), 7.60–7.58 (m, 6H), 7.41–7.28 (m, 6H), 4.9 (s, 1H), 4.44 (d, J = 10.9 Hz, 1H), 3.78 (s, 1H), 3.76 (d, J = 11.0 Hz, 1H), 2.21 (m, 1H), 1.71 (m, 1H), 1.56 (m, 1H), 1.03 (s, 9H), 0.85 (s, 9H), 0.05 (s, 3H), 0.02 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 205.9, 152.0, 151.3, 151.1, 143.1, 135.5, 135.4, 132.7, 132.4, 131.9, 129.93, 129.91, 127.8, 127.7, 79.0, 60.1, 59.5, 39.0, 26.8, 26.6, 25.5, 19.1, 17.9, 14.3, −4.9, −5.1; [α]20D = −44.78 (c 0.14, MeOH); HRMS (FAB+): m/z calculated for C34H43ClN4O3Si2 (M + H)+ 647.2641, found: 647.2643.
(1R,2R,3S,4R,5S)-3-((tert-Butyldimethylsilyl)oxy)-1-(((tert-butyldiphenylsilyl)oxy)methyl)-4-(6-chloro-9H-purin-9-yl)-2-methylbicyclo[3.1.0]hexan-2-ol (25a)
To a stirred solution of 24 (500 mg, 0.77 mmol) in anhydrous THF (8 mL) under a nitrogen atmosphere was added a solution of methyl magnesium iodide (3 M in ether) (1.28 mL, 3.85 mmol) at −78 °C and the mixture was stirred at −78 °C for 30 min and then at room temperature for 30 minutes. The reaction was quenched with a saturated aqueous ammonium chloride solution at 0 °C and the aqueous phase was extracted with ethyl acetate. The organic layer was washed with brine, dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 9:1) to give 25a (320 mg, 63%) as a colorless foam: UV (MeOH) λmax 265 nm; 1H NMR (600 MHz, CDCl3): δ 8.69 (s, 1H), 8.42 (s, 1H), 7.68–7.36 (m, 4H), 7.45–7.36 (m, 6H), 4.78 (s, 1H), 4.24 (d, J = 11.4 Hz, 1H), 3.61 (d, J = 10.9 Hz, 1H), 2.68 (s, 1H), 1.72 (m, 1H), 1.34 (pseudo t, J = 3.3 Hz, 1H), 1.28 (s, 3H), 1.11 (s, 9H), 0.90 (s, 9H), 0.85 (m, 1H), 0.12 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 151.6, 151.5, 151.1, 143.6, 135.6, 135.4, 133.0, 132.7, 132.0, 130.0, 129.9, 127.8, 84.3, 63.9, 62.7, 38.8, 29.6, 29.2, 26.9, 25.7, 25.4, 22.3, 19.1, 17.8, 13.0, −4.6, −5.2; [α]20D = 15.48 (c 0.52, MeOH); HRMS (FAB+): m/z calculated for C35H48ClN4O3Si2 (M + H)+ 663.2954, found: 663.2938.
(1R,2R,3S,4R,5S)-3-((tert-Butyldimethylsilyl)oxy)-1-(((tert-butyldiphenylsilyl)oxy)methyl)-4-(6-chloro-9H-purin-9-yl)-2-vinylbicyclo[3.1.0]hexan-2-ol (25b)
To a stirred solution of 24 (500 mg, 0.76 mmol) in anhydrous THF (8 mL) under a nitrogen atmosphere was added a solution of vinyl magnesium bromide (1 M in THF) (3.84 mL, 3.84 mmol) at 0 °C and the mixture was stirred at 0 °C for 30 min. The reaction was quenched with a saturated aqueous ammonium chloride solution and the aqueous phase was extracted with ethyl acetate. The organic layer was washed with brine, dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 9:1) to give 25b (340 mg, 65%) as a colorless foam: UV (MeOH) λmax 265 nm; 1H NMR (600 MHz, CDCl3): δ 8.65 (s, 1H), 8.30 (s, 1H), 7.66–7.62 (m, 4H), 7.44–7.35 (m, 6H), 6.01 (m, 1H), 5.42 (dd, J = 1.3, 16.9 Hz, 1H), 5.08 (dd, J = 1.8, 10.5 Hz, 1H), 4.71 (d, J = 2.7 Hz, 1H), 4.16 (d, J = 11.0 Hz, 1H), 4.11 (d, J = 3.2 Hz, 1H), 3.46 (d, J = 11.4 Hz, 1H), 2.66 (bs, 1H), 1.73 (m, 1H), 1.43 (t, J = 4.14 Hz, 1H), 1.09 (s, 9H), 0.97 (m, 1H), 0.81 (s, 9H), −0.05 (s, 3H), −0.15 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 151.8, 151.6, 151.2, 143.8, 139.1, 135.6, 135.5, 133.1, 132.9, 132.0, 129.9, 129.0, 127.8, 115.0, 84.7, 79.3, 64.2, 63.3, 38.9, 26.9, 25.6, 22.1, 19.2, 17.8, 14.3, −4.8, −4.9; [α]20D = −50.86 (c 0.15, MeOH); HRMS (FAB+): m/z calculated for C36H48ClN4O3Si2 (M + H)+ 675.2954, found: 675.2967.
(1R,2S,3S,4R,5S)-3-((tert-Butyldimethylsilyl)oxy)-1-(((tert-butyldiphenylsilyl)oxy)methyl)-4-(6-chloro-9H-purin-9-yl)-2-((trimethylsilyl)ethynyl)bicyclo[3.1.0]hexan-2-ol (25c)
To a stirred solution of trimethylsilylacetylene (0.326 mL, 2.31 mmol) in anhydrous THF (3 mL) under a nitrogen atmosphere was added an n-butyllithium solution (1.6 M in hexane) (1.44 mL, 2.31 mmol) at −78 °C and the mixture was stirred at −78 °C for 1 h. To this reaction mixture, a solution of 24 (500 mg, 0.77 mmol) in anhydrous THF (7 mL) was added dropwise at −78 °C and the reaction mixture was stirred at −78 °C for 30 min. A saturated aqueous ammonium chloride solution was added at −78 °C to quench the reaction and the aqueous phase was extracted with ethyl acetate. The organic layer was washed with brine, dried over MgSO4, filtered and evaporated. The residue was purified by silica gel column chromatography (hexane:ethyl acetate = 19:1) to give 25c (482 mg, 84%) as a colorless foam: UV (MeOH) λmax 265 nm; 1H NMR (600 MHz, CDCl3): δ 8.71 (s, 1H), 8.20 (s, 1H), 7.65–7.63 (m, 4H), 7.44–7.37 (m, 6H), 4.94 (s, 1H), 4.28 (merged dd, J = 10.9, 8.7 Hz, 2H), 4.04 (s, 1H), 2.95 (bs, 1H), 1.83 (m, 1H), 1.42 (m, 1H), 1.23 (m, 1H), 1.05 (s, 9H), 0.93 (s, 9H), 0.24 (s, 3H), 0.10 (s, 3H), −0.22 (s, 9H); 13C NMR (150 MHz, CDCl3): δ 151.6, 151.5, 150.9, 144.2, 135.5, 135.4, 133.08, 133.02, 131.8, 130.0, 129.9, 127.8, 103.9, 94.1, 84.0, 74.7, 62.2, 61.6, 39.5, 26.9, 25.7, 22.4, 19.3, 17.9, 10.1, −0.4, −4.6, −5.1; [α]20D = 9.69 (c 0.13, MeOH); HRMS (FAB+): m/z calculated for C39H54ClN4O3Si3 (M + H)+ 745.3192, found: 745.3198.
(1R,2R,3S,4R,5S)-4-(6-Chloro-9H-purin-9-yl)-1-(hydroxyl-methyl)-2-methylbicyclo[3.1.0]hexane-2,3-diol (26a)
To a stirred solution of 25a (250 mg, 0.376 mmol) in anhydrous THF (8 mL) under a nitrogen atmosphere was added a solution of tetrabutyl ammonium fluoride (1 M in THF) (0.56 mL, 0.56 mmol) at 0 °C and the reaction mixture was stirred at room temperature for 4 h. The reaction mixture was evaporated and the residue was purified by silica gel column chromatography (methylene chloride:methanol = 19:1) to give 26a (97 mg, 80%) as a white solid (contaminated with a trace amount of TBAF impurity): mp 116–118 °C; UV (MeOH) λmax 265 nm; 1H NMR (600 MHz, CD3OD): δ 8.90 (s, 1H), 8.72 (s, 1H), 4.76 (d, J = 3.2 Hz, 1H), 4.01 (d, J = 11.8 Hz, 1H), 3.86 (d, J = 3.6 Hz, 1H), 3.60 (d, J = 11.9 Hz, 1H), 1.86 (m, 1H), 1.47 (t, J = 4.5 Hz, 1H), 1.38 (s, 3H), 0.93 (m 1H); 13C NMR (150 MHz, CD3OD): δ 153.9, 153.6, 152.1, 147.5, 133.5, 86.1, 78.5, 65.3, 65.2, 40.6, 26.3, 24.0, 16.5; [α]20D = −82.30 (c 0.10, MeOH); HRMS (FAB+): m/z calculated for C13H16ClN4O3 (M + H)+ 311.0911, found: 311.0903.
(1R,2R,3S,4R,5S)-4-(6-Chloro-9H-purin-9-yl)-1-(hydroxyl-methyl)-2-vinylbicyclo[3.1.0]hexane-2,3-diol (26b)
Compound 25b (321 mg, 0.47 mmol) was converted to 26b (100 mg, 65%) as a white solid (contaminated with a trace amount of TBAF impurity) according to the same procedure used in the preparation of 26a: mp 107–109 °C; UV (MeOH) λmax 265 nm; 1H NMR (600 MHz, CD3OD): δ 8.85 (s, 1H), 8.73 (s, 1H), 6.14 (m, 1H), 5.39 (dd, J = 1.8, 16.9 Hz, 1H), 5.14 (dd, J = 1.8, 10.8 Hz, 1H), 4.76 (d, J = 5.0 Hz, 1H), 4.16 (d, J = 5.0 Hz, 1H), 3.91 (d, J = 11.4 Hz, 1H), 3.54 (d, J = 11.4 Hz, 1H), 1.88 (m, 1H), 1.53 (t, J = 4.6 Hz, 1H), 1.01 (m, 1H); 13C NMR (150 MHz, CD3OD): δ 153.9, 153.6, 152.1, 147.7, 142.2, 133.6, 115.3, 85.5, 81.0, 65.5, 65.3, 41.1, 23.9, 16.9; [α]20D = −47.00 (c 0.09, MeOH); HRMS (FAB+): m/z calculated for C14H16ClN4O3 (M + H)+ 323.0911, found: 323.0909.(1R,2R,3S,4R,5S)-4-(6-Chloro-9H-purin-9-yl)-2-ethynyl-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (26c)
Compound 25c (449 mg, 0.602 mmol) was converted to 26c (164 mg, 76%) as a white solid (contaminated with a trace amount of TBAF impurity) according to the same procedure used in the preparation of 26a: mp 183–185 °C; UV (MeOH) λmax 265 nm; 1H NMR (600 MHz, DMSO-d6): δ 8.80 (d, J = 1.8 Hz, 2H), 5.92 (d, J = 5.4 Hz, 1H), 5.29 (s, 1H), 4.79 (s, 1H), 4.61 (d, J = 5.7 Hz, 1H), 4.07 (m, 1H), 3.94 (d, J = 5.0 Hz, 1H), 3.79 (m, 1H), 3.39 (s, 1H), 1.88 (m, 1H), 1.39 (m, 1H), 0.92 (m, 1H); 13C NMR (150 MHz, DMSO-d6): δ 151.5, 151.4, 148.9, 145.9, 131.1, 85.4, 82.8, 77.4, 73.5, 62.0, 59.7, 38.8, 21.3, 10.5; [α]20D = 129.0 (c 0.10, MeOH); HRMS (FAB+): m/z calculated for C14H13ClN4O3 (M + H)+ 321.0754, found: 321.0761.(1R,2R,3S,4R,5S)-4-(6-Amino-9H-purin-9-yl)-1-(hydroxyl-methyl)-2-methylbicyclo[3.1.0]hexane-2,3-diol (3a)
A stirred solution of 26a (60 mg, 0.185 mmol) in ammonia/t-butanol (8 mL) was heated at 80 °C in a steel bomb for 18 hours. The reaction mixture was cooled to room temperature, evaporated to dryness and the residue was purified by silica gel column chromatography (methylene chloride:methanol = 11:1) to give 3a (43 mg, 77%) as a white solid: mp 273–275 °C; UV (MeOH) λmax 261 nm; 1H NMR (600 MHz, CD3OD): δ 8.43 (s, 1H), 8.19 (s, 1H), 4.62 (d, J = 2.7 Hz, 1H), 4.05 (d, J = 11.9 Hz, 1H), 3.78 (d, J = 3.2 Hz, 1H), 3.56 (d, J = 11.9 Hz, 1H), 1.83 (dd, J = 4.1, 8.7 Hz, 1H), 1.48 (t, J = 4.6 Hz, 1H), 1.37 (s, 3H), 0.90 (m, 1H); 13C NMR (150 MHz, CD3OD): δ 158.2, 154.3, 151.3, 141.9, 121.4, 86.2, 78.7, 65.4, 64.8, 40.7, 26.5, 24.2, 16.3; [α]20D = −86.18 (c 0.11, MeOH); HRMS (FAB+): m/z calculated for C13H18N5O3 (M + H)+ 292.1410, found: 292.1407. Anal. Calcd for C13H17N5O3: C, 53.60; H, 5.88; N, 24.04. Found: C, 53.34; H, 5.90; N, 24.15.
(1R,2R,3S,4R,5S)-4-(6-Amino-9H-purin-9-yl)-1-(hydroxyl-methyl)-2-vinylbicyclo[3.1.0]hexane-2,3-diol (3b)
Compound 26b (64 mg, 1.98 mmol) was converted to 3b (40 mg, 67%) as a white solid according to the same procedure used in the preparation of 3a: mp 250–252 °C; UV (MeOH) λmax 261 nm; 1H NMR (600 MHz, CD3OD): δ 8.39 (s, 1H), 8.19 (s, 1H), 6.12 (m, 1H), 5.40 (dd, J = 1.8, 16.9 Hz, 1H), 5.12 (dd, J = 1.8, 10.5 Hz, 1H), 4.62 (d, J = 4.1 Hz, 1H), 4.06 (d, J = 4.6 Hz, 1H), 3.94 (d, J = 11.9 Hz, 1H), 3.49 (d, J = 11.8 Hz, 1H), 1.85 (m, 1H), 1.54 (t, J = 4.6 Hz, 1H), 0.99 (m, 1H); 13C NMR (150 MHz, CD3OD): δ 158.2, 154.3, 151.3, 142.4, 142.1, 121.4, 115.0, 85.7, 81.3, 65.5, 65.2, 41.4, 24.1, 16.7; [α]20D = −123.12 (c 0.08, MeOH); HRMS (FAB+): m/z calculated for C14H18N5O3 (M + H)+ 304.1410, found: 304.1406. Anal. Calcd for C14H17N5O3: C, 55.44; H, 5.65; N, 23.09. Found: C, 55.52; H, 5.82; N, 23.21.(1R,2R,3S,4R,5S)-4-(6-Amino-9H-purin-9-yl)-2-ethynyl-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (3c)
Compound 26c (84 mg, 0.261 mmol) was converted to 3c (63 mg, 80%) as a white solid according to the same procedure used in the preparation of 3a: mp 267–268 °C; UV (MeOH) λmax 261 nm; 1H NMR (600 MHz, DMSO-d6): δ 8.21 (s, 1H), 8.15 (s, 1H), 7.19 (s, 2H), 5.82 (d, J = 5.5 Hz, 1H), 5.17 (s, 1H), 4.64 (s, 1H), 4.56 (pseudo t, J = 5.9 Hz, 1H), 4.03 (m, 1H), 3.91 (d, J = 5.0 Hz, 1H), 3.81 (m, 1H), 3.37 (s, 1H), 1.78 (m, 1H), 1.32 (t, J = 4.0 Hz, 1H), 0.89 (m, 1H); 13C NMR (150 MHz, DMSO-d6): δ 155.9, 152.2, 149.2, 139.3, 118.9, 85.4, 83.2, 77.1, 73.5, 61.2, 59.8, 38.8, 21.7, 10.4; [α]20D = −33.21 (c 0.11, H2O); HRMS (FAB+): m/z calculated for C14H16N5O3 (M + H)+ 302.1253, found: 302.1256. Anal. Calcd for C14H15N5O3: C, 55.81; H, 5.02; N, 23.24. Found: C, 55.53; H, 5.14; N, 23.54.Acknowledgements
This work was supported by a grant from the Mid-career Research program (370C-20160046) of the National Research Foundation (NRF), Korea.References
- Modified Nucleosides: in Biochemistry, Biotechnology and Medicine, ed. P. Herdewijn, Wiley-VCH, Weinheim, Germany, 2008, p. 1 Search PubMed.
- (a) H. Hattori, M. Tanaka, M. Fukushima, T. Sasaki and A. Matsuda, J. Med. Chem., 1996, 39, 5005 CrossRef CAS PubMed; (b) H. Hattori, E. Nozawa, T. Iino, Y. Yoshimura, S. Shuto, Y. Shimamoto, M. Nomura, M. Fukushima, M. Tanaka, T. Sasaki and A. Matsuda, J. Med. Chem., 1998, 41, 2892 CrossRef CAS PubMed.
- A. Matsuda and T. Sasaki, Cancer Sci., 2004, 95, 105 CrossRef CAS PubMed.
- P. Franchetti, L. Cappellacci, M. Pasqualini, R. Petrelli, P. Vita, H. N. Jayaram, Z. Horvath, T. Szekeres and M. Grifantini, J. Med. Chem., 2005, 48, 4983 CrossRef CAS PubMed.
- S. P. Ong, L. S. Nelson and H. P. Hogenkamp, Biochemistry, 1992, 31, 11210 CrossRef CAS PubMed.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1973, 95, 2333 CrossRef CAS PubMed.
- (a) V. E. Marquez, M. A. Siddiqui, A. Ezzitouni, P. Russ, J. Wang, R. W. Wagner and M. D. Matteucci, J. Med. Chem., 1996, 39, 3739 CrossRef CAS PubMed; (b) V. E. Marquez, Y. Choi, M. J. Comin, P. Russ, C. George, M. Huleihel, T. Ben-Kasus and R. Agbaria, J. Am. Chem. Soc., 2005, 127, 15145 CrossRef CAS PubMed.
- W. J. Choi, H. R. Moon, H. O. Kim, B. N. Yoo, J. A. Lee, D. H. Shin and L. S. Jeong, J. Org. Chem., 2004, 69, 2634 CrossRef CAS PubMed.
- J. A. Lee, H. O. Kim, D. K. Tosh, H. R. Moon, S. Kim and L. S. Jeong, Org. Lett., 2006, 8, 5081 CrossRef CAS PubMed.
- R. M. Wilson and F. Geiser, J. Am. Chem. Soc., 1978, 100, 2226 CrossRef.
- For a review on TPAP oxidation, see: S. V. Ley, J. Norman, W. P. Griffith and S. T. Marsden, Synthesis, 1994, 639 CrossRef.
- L. S. Jeong, V. E. Marquez, C.-S. Yuan and R. T. Borchardt, Heterocycles, 1995, 41, 2651 CrossRef CAS.
- For a review on cyclic sulfates, see: (a) B. B. Lohray, Synthesis, 1992, 1035 CrossRef CAS; (b) B. B. Lohray and V. Bhushan, Adv. Heterocycl. Chem., 1997, 68, 89 CrossRef CAS; (c) H.-S. Byun, L. He and R. Bittman, Tetrahedron, 2000, 56, 7051 CrossRef CAS.
- (a) T. K. M. Shing and V. W.-F. Tai, J. Chem. Soc. Perkin Trans 1, 1994, 2017 RSC; (b) S. Lee, S. J. Lee, H. J. Park, S. K. Lee and S. Kim, Org. Biomol. Chem., 2011, 9, 4580 RSC.
- S. Takano, T. Ohkawa and K. Ogasawara, Tetrahedron Lett., 1994, 35, 2331 CrossRef.
- R. H. Schlessinger and R. A. Nugent, J. Am. Chem. Soc., 1982, 104, 1116 CrossRef CAS.
- Compound 3c: C14H15N5O3·H2O, Mr = 319.32, monoclinic, space group P212121 (no. 8), a = 7.51038(10) Å, b = 17.3551(2) Å, c = 23.2172(4) Å, V = 3026.21(7) Å3, T = 100.0(2) K, Z = 8, ρcalc = 1.402 g cm−3, F(000) = 1344.0, crystal dimension 0.148 × 0.081 × 0.035 mm3, μ(Mo Kα) = 0.886 mm−1, CuKα radiation (λ = 1.54184 Å). Of 25009 reflections collected in the 2θ range from 7.616 to 153.096° using an ω scan on a Rigaku Rapid R-axis diffractometer, 6304 were unique reflections (Rint = 0.0270). The structure was solved and refined against F2 using SHELXS97 and SHELXL97, wR2 = 0.0680, R1 = 0.0280, GOF = 1.027, and max/min residual electron density 0.18/−0.20 e Å−3. Flack x parameter = 0.07(5). CCDC 1472958.
- S. K. Lee, Y. H. Heo, V. E. Steelee and J. M. Pezzuto, Anticancer Res., 2002, 22, 97 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1472958. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00358c |
| This journal is © the Partner Organisations 2016 |