
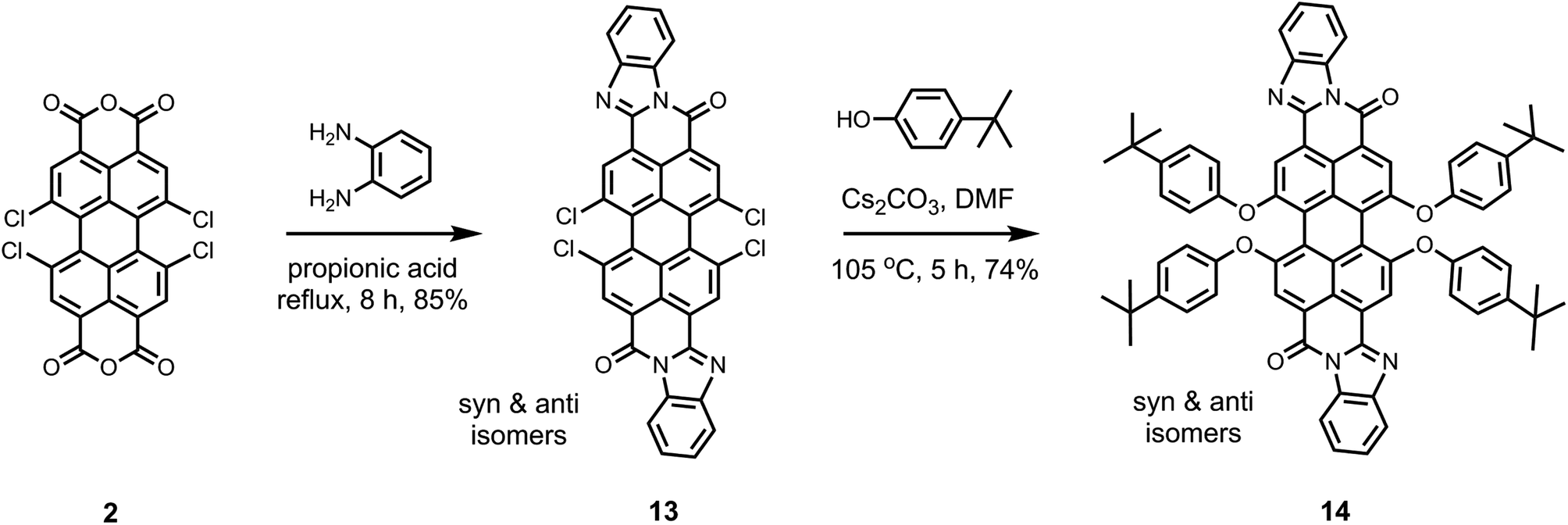
Novel derivatives of 1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxylic acid: synthesis, electrochemical and optical properties†
Rajeev K.
Dubey
*ab,
Nick
Westerveld
a,
Ernst J. R.
Sudhölter
a,
Ferdinand C.
Grozema
b and
Wolter F.
Jager
*a
aLaboratory of Organic Materials & Interfaces, Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: R.K.Dubey@tudelft.nl; W.F.Jager@tudelft.nl
bLaboratory of Optoelectronic Materials, Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
First published on 6th September 2016
Abstract
A family of novel unsymmetrical “peri”-substituted perylene-3,4,9,10-tetracarboxylic acid derivatives (5–10), with 1,6,7,12-tetrachloro-substituents at the bay-positions, has been synthesized. Subsequently, their redox and optical properties have been explored with the intent of unveiling opto-electronic characteristics of these newly synthesized compounds. To synthesize these new compounds, pure 1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxylic tetra-n-butylester (4) has been employed as the precursor and the structural modifications have been carried out exclusively at the “peri” positions in an efficient manner. The two synthons prepared in this work, 1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxylic di-n-butylester monoanhydride (5) and 1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxylic monoimide monoanhydride (8), are extremely valuable and versatile starting materials as they possess free anhydride functionality at the “peri” position in addition to the 1,6,7,12-tetrachloro-bay-substituents. Finally, the conventional methodology for the synthesis of 1,6,7,12-tetraphenoxy-bay-functionalized perylene bisimides and perylene bisbenzimidazoles has been modified to make it faster and more convenient.
Introduction
The chemistry of perylene-based dyes has been thoroughly investigated and developed in both the academic and the industrial domains.1–3 Over the course of time, a number of important synthetic routes towards perylene-3,4,9,10-tetracarboxylic acid derivatives have been developed. Some representative classes of compounds that have been synthesized are presented in Fig. 1, such as perylene-3,4,9,10-tetracarboxy tetraesters (A1–A3),4–7 perylene-3,4,9,10-tetracarboxy monoimide diesters (B1–B2),5,8,9 perylene-3,4,9,10-tetracarboxy bisimides (C1–C3),1,3 and perylene-3,4,9,10-tetracarboxy bisbenzimidazoles (D1–D3).8,10 In addition, methods to obtain perylene-3,4-dicarboxylic acid derivatives have also been reported.11,12 These perylene derivatives have gained a lot of attention because of their inherently beneficial properties, e.g. chemical robustness, photo and thermal stability, strong absorption and emission in the visible region, as well as high electron affinities and charge carrier mobilities. These compounds have been extensively employed as the photo-functional materials for the construction of efficient and tunable electron donor–acceptor systems13–17 and light-harvesting arrays.18–21 These dyes have also been used in other photo-physical processes of current interest, such as photocatalysis,22,23 singlet exciton fission,24,25 triplet–triplet annihilation,26 organic photovoltaics,27–29 lasing,30 fluorescence probing,31–33 and bio-labelling.34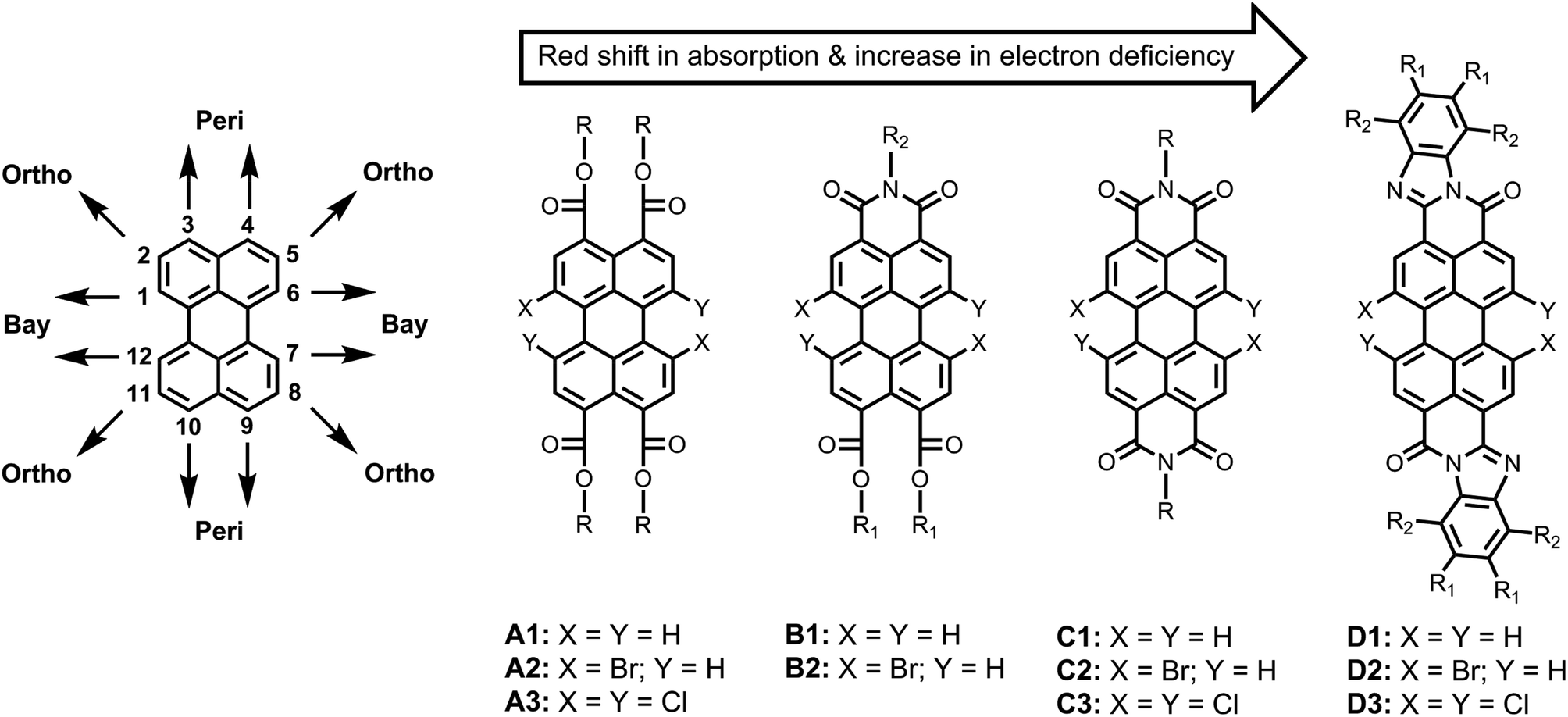 | ||
| Fig. 1 Previously prepared perylene-tetracarboxylic acid derivatives with tunable optical and electronic properties. | ||
As compared to other organic dyes, perylene dyes provide a wealth of opportunities for the tuning of their properties via structural modification. These structural modifications can be achieved at three distinct positions around the perylene core; namely, the “peri” (3,4,9,10) positions, the “bay” (1,6,7,12) positions, and the “ortho” (2,5,8,11) positions (Fig. 1). Among these, the functionalization of peri and bay positions have been the preferred choices so far. At the peri positions, mostly imide, ester, and benzimidazole groups have been substituted (A1–D1; Fig. 1). The absorption and emission spectra shift bathochromically upon going from A1 to D1.5,20 These peri-substituents influence other properties as well. Generally, the ester functionalized derivatives are the least electron-deficient and exhibit the best solubility.9,32 On the other hand, the imide and benzimidazole groups significantly increase the electron deficiency of the perylene core but decrease the solubility.5,20 In particular, the bisbenzimidazole derivatives have extremely low solubility in common organic solvents.35
Once the peri positions are functionalized, the bay positions (1,6,7,12) provide an additional “handle” to control the opto-electronic properties of the dye. Simultaneously, the bay substituents significantly increase the solubility of the dye by twisting the otherwise planer perylene core.1,36,37 For the bay functionalization, 1,7-disubstitution and 1,6,7,12-tetrasubstitution have been used as the two major approaches, for which 1,7-dibromo- and 1,6,7,12-tetrachloro-perylene derivatives have been used as precursors, respectively.3
Since the halogen atoms can undergo a large variety of substitution and carbon–carbon coupling reactions, the bay-halogenated derivatives (A2–D2 and A3–D3) are highly valuable and versatile synthons. For attaching substituents to bay area, various methods, such as Suzuki21 and Sonogashira38 couplings, and nucleophilic aromatic substitution reactions, mostly with sulfides,16,39,40 cyclic amines,14,33,41 and phenols,5,15,20,41,42 have been reported.
The ortho substitution is an additional and recently developed method to prepare perylene derivatives with altered properties.43–45 Contrary to the bay-substitution, the ortho-substitution does not twist the perylene core. Therefore, this substitution is especially important for applications where planarity of the perylene core is an important requirement. This approach is still in the phase of development and has exclusively been applied on the perylene bisimide derivatives, so far.
We present, herein, the very first synthesis of unsymmetrical peri-substituted 1,6,7,12-tetrachloro-perylene-3,4,9,10-tetracarboxilic acid derivatives 5–10. These bay-chlorinated compounds are excellent starting materials for the synthesis of a large range of perylene derivatives due to the presence of four chlorine atoms that can be easily substituted. In addition, we have modified the conventional synthetic methodology for the tetraphenoxy-bay-substitution to achieve high chemical yield in significantly less reaction time as demonstrated with the synthesis of previously reported tetraphenoxy-bay-area-substituted compounds 12 and 14.
Results and discussion
Synthesis and characterization
The synthesis of the pure perylene precursor 1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxylic tetra-n-butylester 4 was carried out from commercially available perylene-3,4,9,10-tetracarboxylic bisanhydride (PBA, 1) and is outlined in Scheme 1.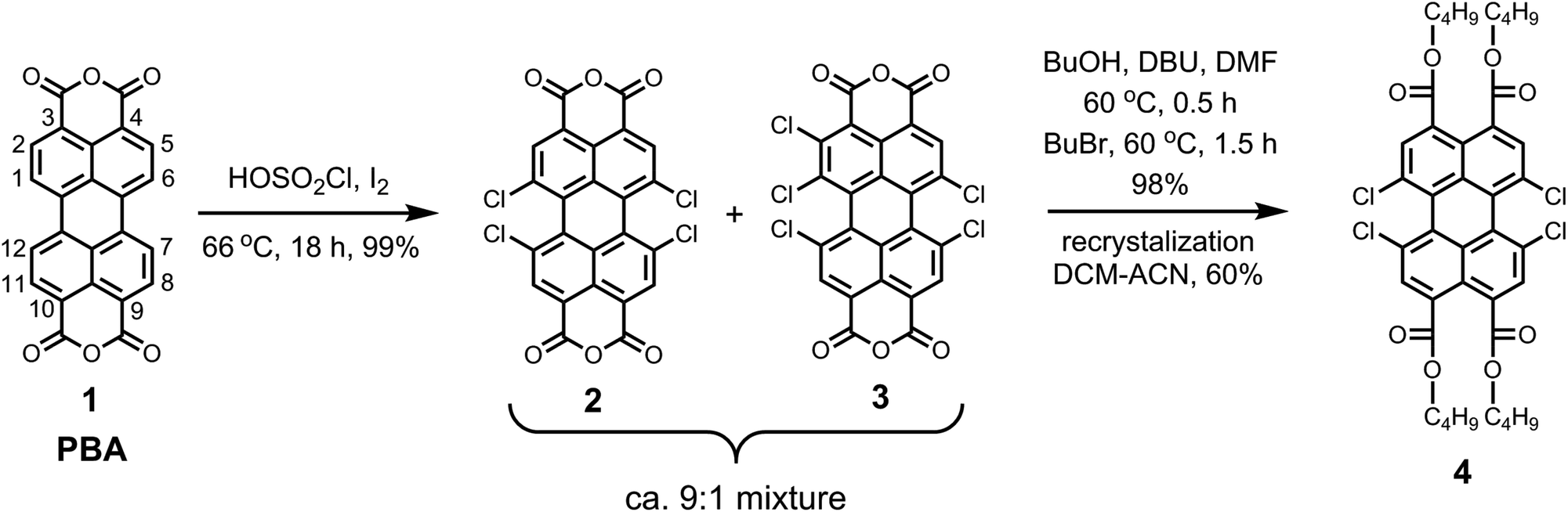 | ||
| Scheme 1 Synthesis of pure 1,6,7,12-tetrachloroperylene tetra-n-butylester 4. | ||
The first step involved the tetra-chlorination of PBA 1 using chlorosulfonic acid. In our recent studies, we showed that the conventional reaction conditions (70 °C, 20 h) to obtain 1,6,7,12-tetrachloro-PBA 2 produces ca. 20% of 1,2,6,7,12-pentachloro-PBA 3 as the side product.7 In this study, we observed that the amount of the side product 1,2,6,7,12-pentachloro-PBA 3 highly depends on the temperature of the reaction. By keeping the reaction mixture at 66 °C for 16 h, we could reduce the amount of pentachloro impurity to less than 10%. The reaction produced trichloro-derivative at reaction temperatures lower than 66 °C along with the pentachloro-derivative. Hence, a further decrease in the reaction temperature was not possible. In view of the sensitivity of this reaction towards temperature, it is recommended not to use an oil-bath, which often results in large temperature fluctuations. For this reaction, we specifically used an aluminum based dry heating block to attain a good temperature control.
In the second step, the crude mixture of 2 and 3 was converted into the corresponding perylene tetra-n-butylester derivatives, because these are highly soluble and crystalline compounds. The esterification reaction with n-butanol and n-bromobutane, in dimethylformamide46 at 60 °C, was highly efficient and gave a mixture of corresponding tetra- and pentachloroperylene tetra-n-butylesters in the expected ratio of ca. 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as revealed by the 1H NMR spectroscopy.7 Eventually, the desired tetrachloro-derivative 4 was obtained in the pure form by recrystallization from acetonitrile/dichloromethane mixtures in overall 60% yield. It is important to emphasize that the pure compound 4 can also be obtained by silica gel column chromatography, using toluene as the eluent.
:1 as revealed by the 1H NMR spectroscopy.7 Eventually, the desired tetrachloro-derivative 4 was obtained in the pure form by recrystallization from acetonitrile/dichloromethane mixtures in overall 60% yield. It is important to emphasize that the pure compound 4 can also be obtained by silica gel column chromatography, using toluene as the eluent.
Scheme 2 depicts the route to the new peri-substituted perylene derivatives that we have synthesized from pure perylene tetra-n-butylester 4. In the first step, the pure perylene tetra-n-butylester 4 was converted to perylene monoanhydride di-n-butylester 5 by an acid catalyzed removal of two ester moieties in refluxing n-heptane.5,9 The synthesis of compound 5 was the most challenging step in the whole synthetic scheme because of the simultaneous formation of corresponding tetrachloro-bisanhydride 2. The yield of the product 5 was found to be highly sensitive to various parameters, such as temperature, amount and type of the solvent, and the amount of p-TsOH·H2O (Table S1, ESI†). Noticeably, the selectivity of this reaction towards yielding 5 was significantly reduced at temperatures higher than 99 °C, in particular when using higher amount of p-TsOH·H2O, and when solvents that are more polar than n-heptane were used. This was because of an enhanced formation of the side product perylene bisanhydride 2. Herein, we maximized the yield of 5 by carrying out the reaction with ca. 1.1 equivalents of p-TsOH·H2O using a limited amount of n-heptane at its reflux temperature.
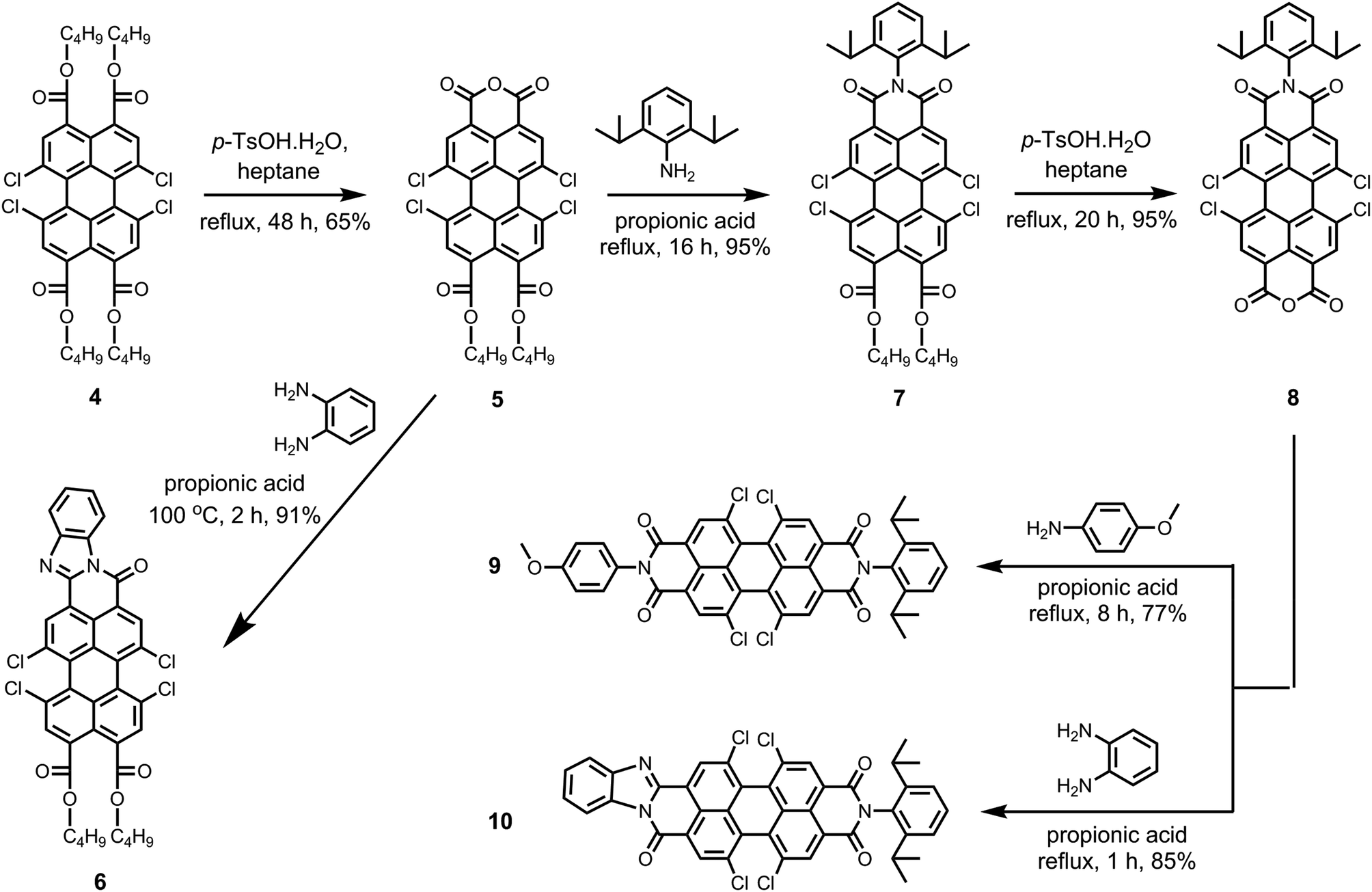 | ||
| Scheme 2 Synthesis of novel 1,6,7,12-tetrachloro substituted perylene derivatives 5–10. | ||
The isolation of compound 5 in pure form was also challenging. Column chromatography could not be employed because the anhydride group of compound 5 strongly adheres to the silica. The further purification attempts based on solubility difference between 5 and 2 also failed. This was mainly because of the moderate solubility of side product 2 in common organic solvents, such as dichloromethane, chloroform, and toluene. The solubility of side product 2 can be explained by the presence of four chlorine atoms at the bay positions. We experienced that alcohols, in particular methanol and ethanol, could not be used for the isolation of compound 5 because alcohols react with compounds 2 and 5 at higher temperatures. Refluxing mixtures of mono anhydride 5 and bisanhydride 2 in alcohols, resulted in the formation of red and highly fluorescent water-soluble compounds. These compounds are most likely formed by the opening of anhydride functionalities by a nucleophilic attack of an alcohol, thus forming ester carboxylate functionalities on a peri-position. This reaction is known for perylene anhydrides, but only occurs when the strong base DBU is added.46 The reason why this reaction takes place for tetrachloro perylene anhydrides 2 and 5 without DBU activation, is the higher reactivity of these chlorinated compound. We ascribe this high reactivity to the higher electron deficiency of the perylene core induced by the strongly electron withdrawing bay-chlorines, that is clearly reflected by the lower reduction potential of these compounds. Finally, compound 5 was purified by recrystallization using refluxing acetonitrile in ca. 65% overall yield. The product has a purity of 97% as 3% contamination of bisanhydride still persisted.
Perylene monoanhydride di-n-butylester 5 is an important intermediate because of the presence of free monoanhydride group, which can be utilized to generate new functionalities at the peri-positions. Herein, compound 5 was first reacted with 1,2-diaminobenzene in propionic acid at 100 °C to get tetrachloroperylene monobenzimidazole di-n-butylester 6 in 91% yield.6 It has to be emphasized that the di-n-butylester moieties present on compound 6 can further be converted to monoanhydride group, which can be used as active site to prepare unsymmetrically substituted bisbenzimidazoles.
The imidization of the anhydride 5 with 2,6-diisopropylaniline was performed in refluxing propionic acid to achieve tetrachloroperylene monoimide di-n-butylester 7 in an extremely high yield (95%). Clear enough, the ester functionalities in 5, as well as the chlorine atoms at the bay area, were stable under these harsh reaction conditions. Subsequently, the di-n-butylester moieties of compound 7 were converted to monoanhydride functionality by the treatment with an excess of p-TsOH·H2O in refluxing n-heptane. This reaction resulted in an hitherto unknown compound 1,6,7,12-tetrachloroperylene monoimide monoanhydride 8 in 95% yield. Compound 8 is an important intermediate, which is capable of providing an easy access to unsymmetrical 1,6,7,12-tetrachloroperylene bisimides and 1,6,7,12-tetrachloroperylene monoimide monobenzimidazoles. To prove this point, compound 8 was converted to unsymmetrical perylene bisimide 9 by the reaction of 4-methoxyaniline in high yield. Furthermore, the synthesis of the unsymmetrical perylene monoimide monobenzimidazole derivative 10 was also carried out in 85% yield by treating 8 with 1,2-diaminobenzene in propionic acid at reflux.
The starting compound 1,6,7,12-tetrachloroperyle tetraester 4 can also be used for the synthesis of pure 1,6,7,12-perylene bisanhydride 2, which is a highly important intermediate to obtain perylene bisimides (like 11). This is done by using an excess of p-TsOH·H2O at higher dilution in n-heptane and reflux conditions for 24 h (Scheme S1, ESI†). Subsequently, the imidization of bisanhydride 2 with 2,6-diisopropylaniline was performed in refluxing propionic acid and corresponding perylene bisimide 11 was obtained in 91% yield.
In view of the issues associated with low solubility of perylene bisimides, the substitution of the four bay-chlorine atoms by the phenoxy groups to give 1,6,7,12-tetraphenoxyperylene bisimides (like 12) is a highly desirable process (Scheme 3). The presence of four phenoxy groups at the bay-area generally results in dramatic increase in solubility. This nucleophilic aromatic substitution reaction has been extensively performed with various phenols in the presence of K2CO3 in NMP. The literature shows that a wide range of reaction times (8–48 h)47,48 and reaction temperatures (80–140 °C)47,49 have been employed to perform this reaction, for which low to extremely high yields (30–95%)42,49 have been reported.
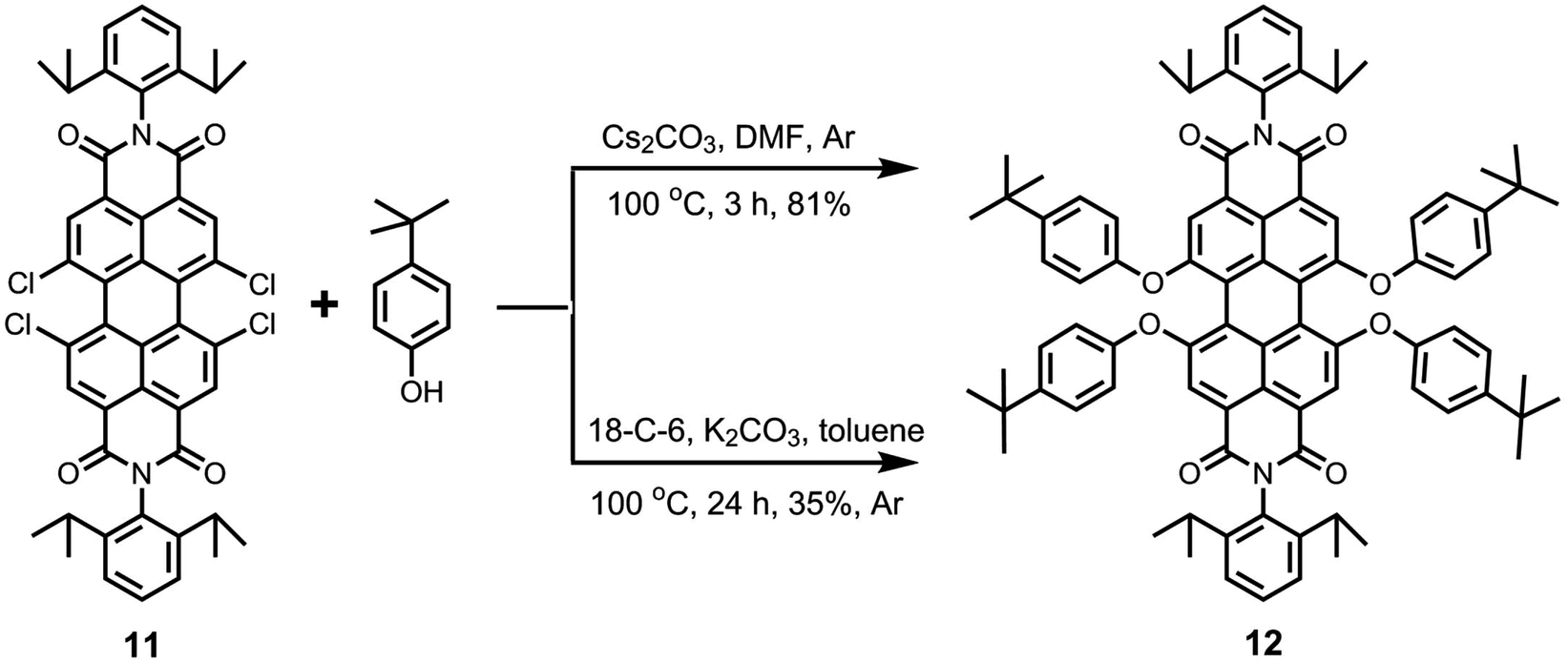 | ||
| Scheme 3 Synthesis of 1,6,7,12-tetraphenoxyperylene bisimide 12. | ||
In this study, we examined the efficacy of two other reactions to produce the tetraphenoxy derivative 12 (Scheme 3). In the first method, tetrachloroperylene bisimide 11 was treated with 4-tert-butylphenol in the presence of Cs2CO3 in DMF at 100 °C.5 Interestingly, the reaction was completed in only 3 hours and a high yield (81%) was achieved. Because of the short reaction time, this methodology provides a promising alternative to the exclusively used K2CO3/NMP reaction, which requires longer reaction times. In the second method, 4-tert-butylphenol was reacted with compound 11 in the presence of K2CO3 and 18-crown-6 was used as the phase transfer catalyst in toluene at 100 °C. This reaction has been previously employed on 1,7-dibromoperylene bisimides and was found to be quite fast and efficient.15,41 However, in this case, a maximum yield of only 35% could be obtained for compound 12, even after long reaction time (24 h). Interestingly, the reaction also yielded the triphenoxy-substituted product in a noticeable amount (ca. 30%).
As depicted in Scheme 2, the perylene derivatives with one free monoanhydride group (5 and 8) are important intermediates for the synthesis of corresponding derivatives with one benzimidazole group at the peri-position (6 and 10). The presence of benzimidazole group results in an extension of π-system of the perylene core.35 This effect significantly reduces the solubility of the dye. The solubility is still manageable for the monobenzimidazole derivatives (e.g.6 and 10) due to the presence of better soluble groups on the other side of perylene core.8 However, for the bisbenzimidazole perylene derivatives, tetraphenoxy-bay-substituents are essential to achieve moderate solubility in common organic solvents.10,35 Previously, the synthesis of tetraphenoxy-bay-substituted bisbenzimidazoles was performed in an indirect manner from 1,6,7,12-tetraphenoxyperylene bisanhydrides, which were obtained by the saponification of 1,6,7,12-tetraphenoxyperylene bisimides as described in Scheme S2 (ESI†).10,35
In this study, we followed a direct route for an efficient synthesis of bisbenzimidazoles with solubilization improving tetraphenoxy groups at the bay area (Scheme 4). First, 1,6,7,12-tetrachloroperylene bisanhydride 2 was converted into the corresponding 1,6,7,12-tetrachloroperylene bisbenzimidazole 13 by treatment with 1,2-diamonobenzene in refluxing propionic acid, resulting in 85% yield. As expected, compound 13 exhibited poor solubility, so that further reaction for phenoxy-substitution in NMP could not be realized. Therefore, similar to perylene bisimides, the coupling reaction of compound 13 with 4-tert-butylphenol was performed in DMF and it proceeded smoothly at 105 °C to afford the desired product 14 in 74% yield. In the aromatic region of the 1H NMR spectrum of 14, the presence of syn- and anti-isomers was evident by the appearance of four singlets (between 8.4 to 8.6 ppm) originating from perylene core protons (Fig. 2).10 The integration of these singlets revealed the presence of these isomers in a ratio of ca. 3:2. The unambiguous assignment of these singlets to particular isomers could not be done due the extensive overlapping of NMR signals. Therefore, it could not be determined which is the prevailing isomer. Noticeably, the two isomers were reported in 1:1 ratio, when the tetraphenoxy-bisbenzimidazoles were prepared from 1,6,7,12-tetraphenoxy-perylene bisanhydrides in quinolone at 220 °C.10
 | ||
| Scheme 4 New direct approach to synthesize 1,6,7,12-tetraphenoxyperylene bisbenzimidazole 14. | ||
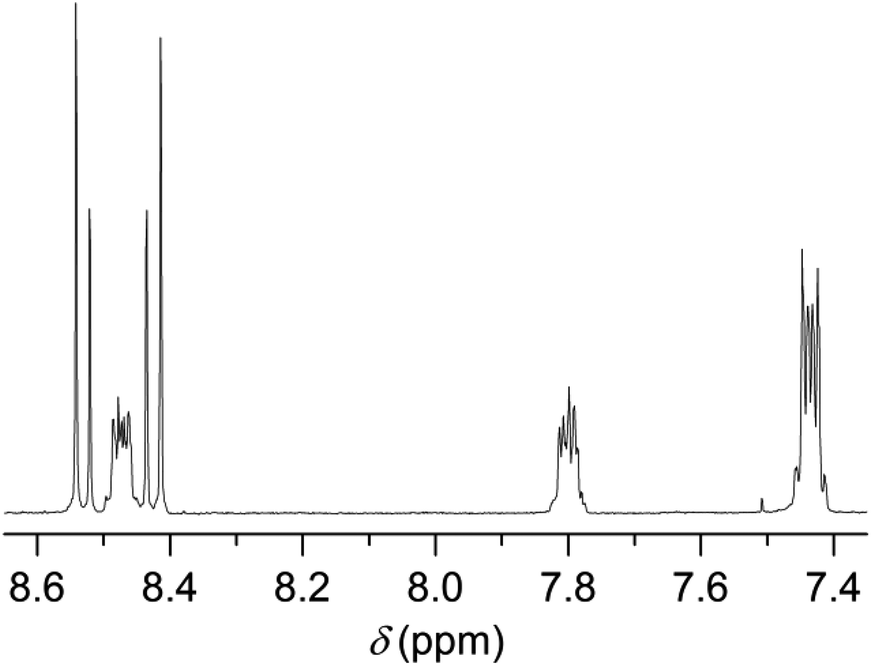 | ||
| Fig. 2 The 1H NMR spectrum (aromatic region) of compound 14. | ||
Electrochemical properties
The electrochemical characterization has been carried out using cyclic voltammetry (CV) to understand the electron-accepting nature of the synthesized chromophores 4, 6, 7, and 10–14. The obtained redox potentials in dichloromethane versus Fc/Fc+, together with calculated HOMO and LUMO energy levels versus vacuum, are listed in Table 1.| Compd | E 1red | E 2red | E 1ox |
E
gf (eV) |
E
LUMOg (eV) |
E
HOMOh (eV) |
|---|---|---|---|---|---|---|
| a The redox potentials (V vs. Fc/Fc+) measured by cyclic voltammetry in dichloromethane (scan rate = 0.10 V s−1). The potentials are reported as E1/2 (= (Eap + Ecp)/2) and quoted to the nearest 0.01 V. b Not observed. c Quasi-reversible. d Not visible very clearly. e Could not be measured because of poor solubility. f Optical band gap calculated from equation Eg = hc/λa.e. ≈ 1240/λa.e. (nm); where λa.e. denotes the absorption edge wavelength in nm, obtained from offset wavelength derived from the low energy absorption band.50 g Estimated vs. vacuum level from ELUMO = −(E1red + 4.8 eV). h Estimated from EHOMO = ELUMO − Eg. | ||||||
| 4 | −1.30 | −1.52 | —b | 2.53 | −3.50 | −6.03 |
| 6 | −0.95 | −1.17d | —b | 2.16 | −3.85 | −6.01 |
| 7 | −0.95 | −1.31c | —b | 2.35 | −3.85 | −6.20 |
| 10 | −0.78 | −0.98 | —b | 2.06 | −4.02 | −6.08 |
| 11 | −0.78 | −1.00 | —b | 2.25 | −4.02 | −6.27 |
| 12 | −1.17 | −1.39c | +0.84 | 2.00 | −3.63 | −5.63 |
| 13 | —e | —e | —e | 1.94 | —e | —e |
| 14 | −1.15 | −1.31 | +0.72 | 1.83 | −3.65 | −5.48 |
The perylene derivatives mostly used for applications are the archetypal perylene bisimides (e.g.11). In general, the perylene bisimides are good n-type semiconductors due to the presence of two electron-withdrawing imide groups, that exhibit two reversible reduction waves at low negative potentials corresponding to the formation of the radical anion and dianion.1
In this series of compounds, the perylene tetraester 4 exhibits first reduction potential at ca. −1.30 V, i.e. the most negative value in this series. This means, it is the least electron deficient compound. Upon moving to compounds 6 and 7, the electron deficiency increases because of the presence of either a benzimidazole or an imide group, respectively, that are more electron-withdrawing than the ester groups. Notably, both compounds 6 and 7 exhibit a first reduction at same potential. It means that the benzimidazole and imide groups exert electron-withdrawing effect of the same magnitude on the perylene core. Compound 10, with one imide and one benzimidazole groups, and perylene bisimide 11 exhibit the least negative values of first and second reduction potentials. Therefore, they are the most electron-deficient compounds in the series. It is worth noting that four electron-withdrawing bay-chlorine atoms make these derivatives comparatively more electron deficient than the corresponding derivatives with no bay-substituents.9 In the case of compounds 12 and 14, the presence of four phenoxy substituents at the bay-area clearly exerts a negative impact on the electron deficiency of the perylene core. For these compounds, the first reduction occurs at significantly more negative potential (ca. −1.15 V) compared to that (ca. −0.78 V) observed for similar perylene derivatives with four chlorine atoms (10 and 11).
Absorption and emission properties
The normalized absorption and emission spectra of the compounds 4, 6, 7, and 10–14 in chloroform are shown in Fig. 3, and the relevant optical data of these compounds are summarized in Table 2.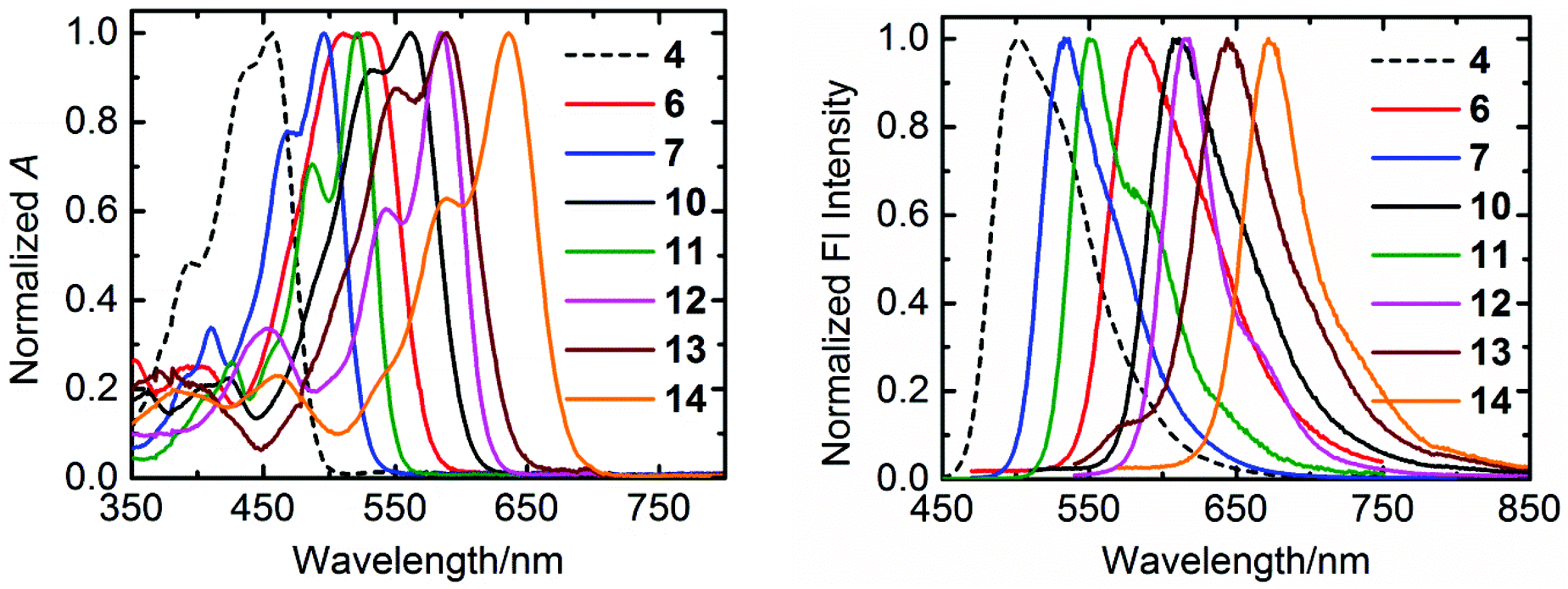 | ||
| Fig. 3 The normalized UV/Vis absorption (left) and emission (right) spectra of synthesized compounds in chloroform. | ||
| Compd | λ abs (nm) | ε (M−1 cm−1) | λ em (nm) | Stokes shift (cm−1) |
Φ
fa |
τ
fb (ns) |
k
Fc (108 s−1) |
k
Qd (108 s−1) |
|---|---|---|---|---|---|---|---|---|
| a Fluorescence quantum yield. b Fluorescence life-time. c Rate of fluorescence. d Rate of fluorescence quenching. e Could not be measured because of poor solubility. | ||||||||
| 4 | 457 | 25400 |
503 | 2001 | 0.26 | 1.6 | 1.63 | 4.63 |
| 6 | 530 | 34200 |
584 | 1745 | 0.36 | 4.8 | 0.75 | 1.33 |
| 7 | 496 | 38800 |
533 | 1400 | 0.98 | 5.4 | 1.81 | 0.04 |
| 10 | 561 | 44000 |
611 | 1459 | 0.38 | 4.7 | 0.81 | 1.32 |
| 11 | 521 | 46900 |
550 | 1012 | 0.91 | 4.9 | 1.86 | 0.18 |
| 12 | 585 | 53600 |
618 | 913 | 0.72 | 6.3 | 1.14 | 0.44 |
| 13 | 589 | —e | 644 | 1450 | 0.28 | 4.3 | 0.65 | 1.67 |
| 14 | 636 | 50700 |
672 | 842 | 0.20 | 5.0 | 0.40 | 1.60 |
All the compounds exhibit well-defined S0–S1 absorption and emission bands in the visible region, which is a characteristic feature of the aromatic perylene core. However, a systematic trend has been displayed by these compounds in their optical profile depending on the substituents either at “peri” or “bay” positions. The perylene tetraester 4, which carries four chlorine atoms at bay-positions, has the most blue-shifted absorption (λmax = 457 nm) and emission (λmax = 503 nm) spectra. Surprisingly, compound 4 shows a low fluorescence quantum yield (φf = 0.26) and life-time (τf = 1.6 ns). This is striking because the “bare” perylene tetraester, with no substituents at bay-area, is highly fluorescent with a fluorescence quantum yield of almost unity and a long life-time of 4.0 ns.51 The perylene monoimide diester 7, in which two ester groups are replaced by one imide moiety, gives bathochromically shifted absorption (λmax = 496 nm) and emission (λmax = 533 nm) spectra. In addition, an extremely high fluorescence quantum yield (ϕf = 0.98) and a long life-time (τf = 5.4 ns) is observed for this derivative. A high fluorescence (ϕf = 0.91) and a further red-shifted absorption (λmax = 521 nm) and emission (λmax = 550 nm) were observed for perylene bisimide 11.
For the compounds 6, 10, and 13, which contain either one or two benzimidazole groups at “peri” positions, a larger red-shift of the absorption and emission spectra is observed as compared to that of corresponding derivatives with imide groups (Table 2). This larger red-shift in benzimidazole based derivatives can be explained by an effective extension of π-system of the perylene core.35 Therefore, attaching a benzimidazole moiety as compared to imide group, works better to achieve bathochromic shifts in absorption and emission spectra. However, the presence of benzimidazole moiety clearly reduces the fluorescence quantum yield as compared to imide groups. Surprisingly, the benzimidazoles 6, 10, 13, and 14 possess long fluorescence life-times, which identifies low rates of fluorescence (kF) along with higher quenching rates (kQ), as the cause for the low fluorescence quantum yields of these compounds (Table 2).
Along with “peri” substitutions, the “bay” substitution also influences the optical properties of perylene dye. The tetraphenoxy-bay-substituents present in the derivatives 12 and 14 further shift the absorption band to longer wavelengths by 47–64 nm. This significant bathochromic shift was observed in emission spectra as well. These observations verify the importance of both “peri” and “bay”-functionalizations to fine-tune opto-electronic properties of perylene chromophores.
The HOMO and LUMO levels of synthesized perylene derivatives are shown in Fig. 4, which depicts the clear effect of peri and bay substitution on the electronic structure of perylene core. A noticeable feature is that the derivatives with benzimidazole group (e.g.6) and the corresponding derivatives with imide group (e.g.7) exhibit almost same LUMO energy levels. However, their HOMO levels are significantly different.
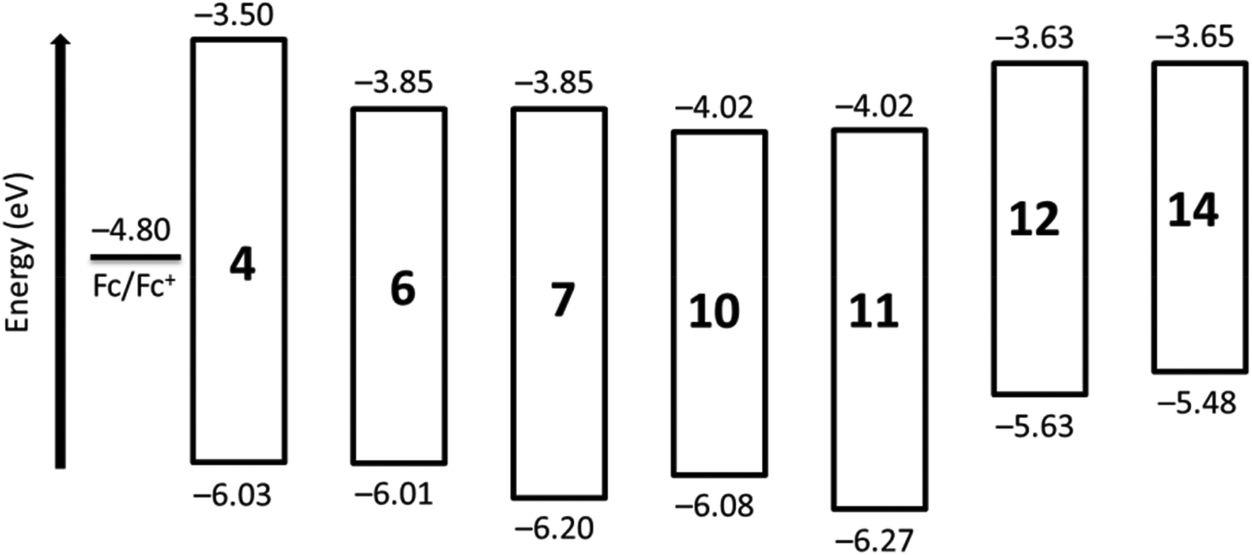 | ||
| Fig. 4 The HOMO and LUMO levels of synthesized perylene derivatives against vacuum. | ||
Conclusions
We have devised an efficient and convenient synthetic methodology for the preparation of novel unsymmetrically “peri”-substituted perylene-3,4,9,10-tetracarboxylic acid derivatives 5–10 starting from pure tetrachloroperylene tetra-n-butylester 4. The efficient preparation of unsymmetric tetrachloroperylene monoanhydride di-n-butylester 5 and the inertness of the bay-chlorine atoms and ester functionalities under the applied reaction conditions are the major factors contributing to the success of our method. The newly synthesized derivatives have four chlorine atoms at the 1,6,7,12-bay-positions of perylene core and therefore they are excellent starting materials for the synthesis of a large variety of perylene derivatives bearing bay-area substituents. We have achieved tetraphenoxy-bay-substitution on 1,6,7,12-tetrachloro-perylene bisimide and perylene bisbenzimidazole compounds with high yields in remarkably shorter reaction times using modified reaction conditions. The synthesized compounds displayed systematic trends in optical and electrochemical properties as a function of structure modification at “peri” positions. In this way, the synthetic modifications described in this manuscript provide excellent control over the opto-electronic characteristics of the obtained materials. The studied derivatives altogether are capable of absorbing light in a broad range of visible region of solar spectrum (i.e. 350–700 nm), which makes them highly desirable compounds as photo-functional materials. Further studies focusing on the selective bay-functionalization of the perylene core to produce promising materials for optoelectronic and photovoltaic applications are currently underway and will be reported in due course.Experimental section
Materials
All the reagents utilized in the synthesis were purchased from commercial suppliers and used as received unless otherwise stated. The DMF used in the synthesis was of anhydrous grade. Toluene was dried over sodium under an argon atmosphere prior to use. The purification of the products were performed by column chromatography. The TLC plates and the sorbent for the column chromatography (silica gel 40–63, mesh size 0.230–0.400 mm) were purchased from commercial suppliers.Instrumentation and characterization
The NMR spectra were recorded with 400 MHz pulsed Fourier transform NMR spectrometer in CDCl3 at room temperature. The 1H NMR spectra of compounds 2 and 13 were measured in D2SO4 and CF3COOD, respectively. The chemical shifts are quoted relative to CDCl3 [δ = 7.26 ppm (1H, singlet); 77.00 (13C, triplet)]. The chemical shift values are given in ppm and J values in Hz. High-resolution mass spectra were collected on an AccuTOF GCv 4G, JMS-T100GCV, mass spectrometer (JEOL, Japan). The FD/FI probe (FD/FI) was equipped with an FD Emitter, Carbotec (Germany), FD 10 μm. Typical measurement conditions were as follow: current rate 51.2 mA min−1 over 1.2 min; counter electrode −10 kV; ion source 37 V. The samples were prepared in dichloromethane.Electrochemical behavior of the compounds was studied using cyclic voltammetry (CHI 600D electrochemical analyzer) in a three-electrode single-compartment cell consisting of a platinum wire in glass as the working electrode, silver wire as the reference electrode, and a platinum sheet as the counter electrode. The cell was connected to the computer controlled potentiostat (CH Instruments Inc. 600D). Pre-dried CH2Cl2 containing 0.1 M tetrabutylammonium hexafluorophosphate was used as solvent. The measurements were done under continuous flow of nitrogen. The concentration of the prepared samples was ca. 0.5 mM. Under these experimental conditions, the ferrocene oxidation was observed at 0.52 V. The potentials of all the reversible peaks are reported as E1/2 (= (Eap + Ecp)/2) in V vs. Fc/Fc+ and quoted to the nearest 0.01 V. The measurements were carried out at 0.10 V s−1 scan rate.
All the spectroscopic measurements were carried out at room temperature. The absorption spectra were recorded with a double beam UV/vis spectrophotometer. The emission spectra were corrected for the wavelength response of the detection system. Fluorescence quantum yields were determined by the comparative method using following compounds as reference: perylene-3,4,9,10-tetracarboxylic tetramethylester (Φf = 0.95 in CH2Cl2) and N,N′-bis(1-hexylheptyl)-perylene-3,4,9,10-tetracarboxy bisimide (Φf = 0.99 in CHCl3).51 Fluorescence lifetime measurements were performed on a Lifespec-ps Fluorescence spectrometer from Edinburgh Instruments. Samples were placed in 1 cm quartz cuvettes and degassed prior to the measurement. The time correlated fluorescence was analyzed by exponential tail fit with F900 Lifespec software. The rates of fluorescence (kF) and rates of fluorescence quenching (kQ) were obtained from steady state and time-resolved optical measurements using eqn (1a)–(d).
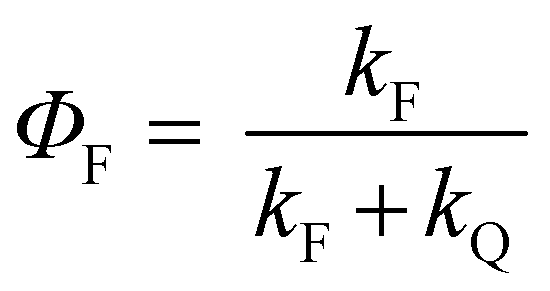 | (1a) |
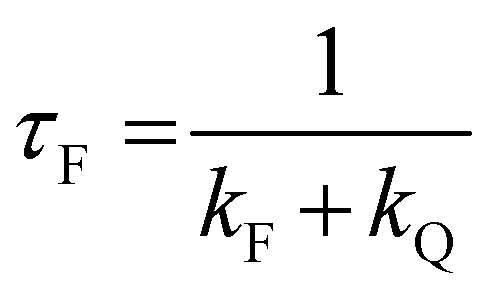 | (1b) |
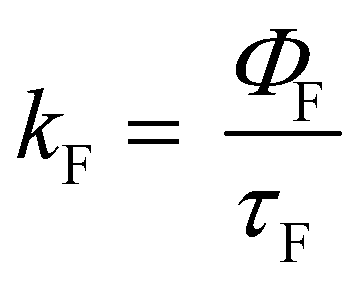 | (1c) |
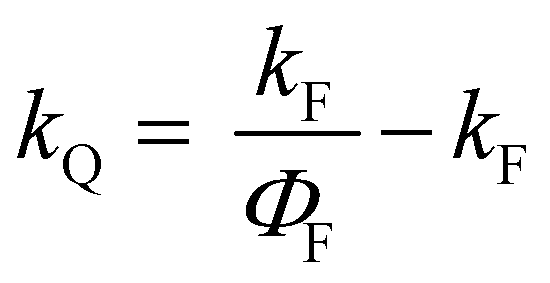 | (1d) |
:1 CH2Cl2–hexane, to afford the desired product 7 (1.12 g, 95%). Mp = 220 °C. 1H NMR (400 MHz, CDCl3): δ = 8.69 (s, 2H), 8.15 (s, 2H), 7.51 (t, J = 7.9 Hz, 1H), 7.36 (d, J = 7.9 Hz, 2H), 4.45–4.32 (m, 4H), 2.79–2.68 (m, 2H), 1.89–1.79 (m, 4H), 1.59–1.46 (m, 4H), 1.22–1.14 (m, 12H), 1.02 ppm (t, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ = 166.8, 162.5, 145.6, 134.7, 134.5, 133.8, 133.2, 132.1, 131.7, 131.6, 130.0, 129.9, 129.6, 126.5, 124.2, 123.6, 123.4, 122.4, 66.2, 30.5, 29.2, 24.0, 19.2, 13.8 ppm. MS (ESI-TOF): [M]+ calculated for C44H39Cl4NO6, 817.1532; found, 817.1518.
(i) In a 25 mL round-bottomed flask, weighed amounts of 1,6,7,12-tetrachloroperylene bisimide 11 (100 mg, 0.12 mmol), 4-tert-butylphenol (124 mg, 0.83 mmol), and Cs2CO3 (308 mg, 0.95 mmol) were taken. Subsequently, anhydrous DMF (6 mL) was added and the reaction mixture was stirred at 100 °C for 3 h under argon atmosphere. Afterwards, the reaction mixture was poured in water and the precipitate was collected by filtration. The solid residue was washed with water and ethanol. The dried crude product was chromatographed (silica/DCM) to afford the desired product 12 (125 mg, 81%). Mp > 350 °C. 1H NMR (400 MHz, CDCl3): δ = 8.28 (s, 4H), 7.42 (t, J = 8.0 Hz, 2H), 7.26 (d, J = 8.0 Hz, 4H), 7.23 (d, J = 8.4 Hz, 8H), 6.87 (d, J = 8.4 Hz, 8H), 2.75–2.66 (m, 4H), 1.27 (s, 36H), 1.12 ppm (d, J = 6.4 Hz, 24H). 13C NMR (100 MHz, CDCl3): δ = 163.3, 155.9, 152.8, 147.3, 145.6, 133.2, 130.7, 129.4, 126.6, 123.8, 122.7, 120.7, 120.2, 120.1, 119.2, 34.3, 31.4, 29.1, 24.0 ppm.
(ii) 1,6,7,12-Tetrachloroperylene bisimide 11 (100 mg, 0.12 mmol), 4-tert-butylphenol (124 mg, 0.83 mmol), and K2CO3 (114 mg, 0.83 mmol) were stirred in NMP (5 mL) at 100 °C for 24 h under argon atmosphere. The work-up and purification was carried out according to above mentioned procedure to afford the desired product 12 (112 mg, 73%).
(iii) A mixture of 4-tert-butylphenol (248 mg, 1.65 mmol), K2CO3 (326 mg, 2.36 mmol) and 18-crown-6 (624 mg, 2.36 mmol), in dry toluene (30 mL), was stirred for 30 min. at room temperature under argon atmosphere. Subsequently, 1,6,7,12-tetrachloroperylene bisimide 11 (200 mg, 0.24 mmol) was added. The reaction mixture was stirred for 24 h at 100 °C and allowed to cool to room temperature. The solvent was removed by rotary evaporation and the residue was washed with water and dried. The crude product was subjected to silica gel column chromatography (DCM–hexane, 1:1) to afford the product 12 (108 mg, 35%).
Acknowledgements
The research leading to these results has received funding from the European Research Council Horizon 2020 ERC Grant Agreement no. 648433.Notes and references
- F. Wurthner, Chem. Commun., 2004, 1564–1579, 10.1039/B401630K.
- H. Langhals, Helv. Chim. Acta, 2005, 88, 1309–1343 CrossRef CAS.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- T. Hassheider, S. A. Benning, H.-S. Kitzerow, M.-F. Achard and H. Bock, Angew. Chem., Int. Ed., 2001, 40, 2060–2063 CrossRef CAS.
- S. Sengupta, R. K. Dubey, R. W. M. Hoek, S. P. P. van Eeden, D. D. Gunbaş, F. C. Grozema, E. J. R. Sudhölter and W. F. Jager, J. Org. Chem., 2014, 79, 6655–6662 CrossRef CAS PubMed.
- Z. Yuan, Y. Xiao, Z. Li and X. Qian, Org. Lett., 2009, 11, 2808–2811 CrossRef CAS PubMed.
- R. K. Dubey, N. Westerveld, F. C. Grozema, E. J. R. Sudhölter and W. F. Jager, Org. Lett., 2015, 17, 1882–1885 CrossRef CAS PubMed.
- H. Langhals, S. Sprenger and M.-T. Brandherm, Liebigs Ann., 1995, 1995, 481–486 CrossRef.
- C. Xue, R. Sun, R. Annab, D. Abadi and S. Jin, Tetrahedron Lett., 2009, 50, 853–856 CrossRef CAS.
- L. Perrin and P. Hudhomme, Eur. J. Org. Chem., 2011, 2011, 5427–5440 CrossRef CAS.
- L. Feiler, H. Langhals and K. Polborn, Liebigs Ann., 1995, 1995, 1229–1244 CrossRef.
- Y. Zagranyarski, L. Chen, Y. Zhao, H. Wonneberger, C. Li and K. Müllen, Org. Lett., 2012, 14, 5444–5447 CrossRef CAS PubMed.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- R. K. Dubey, M. Niemi, K. Kaunisto, A. Efimov, N. V. Tkachenko and H. Lemmetyinen, Chem. – Eur. J., 2013, 19, 6791–6806 CrossRef CAS PubMed.
- R. K. Dubey, M. Niemi, K. Kaunisto, K. Stranius, A. Efimov, N. V. Tkachenko and H. Lemmetyinen, Inorg. Chem., 2013, 52, 9761–9773 CrossRef CAS PubMed.
- V. M. Blas-Ferrando, J. Ortiz, L. Bouissane, K. Ohkubo, S. Fukuzumi, F. Fernandez-Lazaro and A. Sastre-Santos, Chem. Commun., 2012, 48, 6241–6243 RSC.
- V. M. Blas-Ferrando, J. Ortiz, K. Ohkubo, S. Fukuzumi, F. Fernandez-Lazaro and A. Sastre-Santos, Chem. Sci., 2014, 5, 4785–4793 RSC.
- P. D. Frischmann, K. Mahata and F. Wurthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- F. C. De Schryver, T. Vosch, M. Cotlet, M. Van der Auweraer, K. Müllen and J. Hofkens, Acc. Chem. Res., 2005, 38, 514–522 CrossRef CAS PubMed.
- R. K. Dubey, D. Inan, S. Sengupta, E. J. R. Sudholter, F. C. Grozema and W. F. Jager, Chem. Sci., 2016, 7, 3517–3532 RSC.
- J. M. Serin, D. W. Brousmiche and J. M. J. Frechet, Chem. Commun., 2002, 2605–2607, 10.1039/B207905D.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, P. S. SamuelAmanda, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- C. Ramanan, A. L. Smeigh, J. E. Anthony, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2012, 134, 386–397 CrossRef CAS PubMed.
- N. Renaud and F. C. Grozema, J. Phys. Chem. Lett., 2015, 6, 360–365 CrossRef CAS PubMed.
- Z. Mahmood and J. Zhao, J. Org. Chem., 2016, 81, 587–594 CrossRef CAS PubMed.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- F. Fernandez-Lazaro, N. Zink-Lorre and A. Sastre-Santos, J. Mater. Chem. A, 2016, 4, 9336–9346 CAS.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C.-Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y.-L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6 Search PubMed.
- M. G. Ramírez, S. Pla, P. G. Boj, J. M. Villalvilla, J. A. Quintana, M. A. Díaz-García, F. Fernández-Lázaro and Á. Sastre-Santos, Adv. Opt. Mater., 2013, 1, 933–938 CrossRef.
- P. Spenst and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 10165–10168 CrossRef CAS PubMed.
- R. K. Dubey, G. Knorr, N. Westerveld and W. F. Jager, Org. Biomol. Chem., 2016, 14, 1564–1568 CAS.
- D. Aigner, S. M. Borisov, P. Petritsch and I. Klimant, Chem. Commun., 2013, 49, 2139–2141 RSC.
- K. Peneva, G. Mihov, A. Herrmann, N. Zarrabi, M. Börsch, T. M. Duncan and K. Müllen, J. Am. Chem. Soc., 2008, 130, 5398–5399 CrossRef CAS PubMed.
- H. Quante, Y. Geerts and K. Müllen, Chem. Mater., 1997, 9, 495–500 CrossRef CAS.
- K. Nagarajan, A. R. Mallia, V. S. Reddy and M. Hariharan, J. Phys. Chem. C, 2016, 120, 8443–8450 CAS.
- A. G. Slater, E. S. Davies, S. P. Argent, W. Lewis, A. J. Blake, J. McMaster and N. R. Champness, J. Org. Chem., 2013, 78, 2853–2862 CrossRef CAS PubMed.
- R. Mishra, J. M. Lim, M. Son, P. Panini, D. Kim and J. Sankar, Chem. – Eur. J., 2014, 20, 5776–5786 CrossRef CAS PubMed.
- C. Zhao, Y. Zhang, R. Li, X. Li and J. Jiang, J. Org. Chem., 2007, 72, 2402–2410 CrossRef CAS PubMed.
- M.-J. Lin, M. Schulze, K. Radacki and F. Wurthner, Chem. Commun., 2013, 49, 9107–9109 RSC.
- R. K. Dubey, A. Efimov and H. Lemmetyinen, Chem. Mater., 2011, 23, 778–788 CrossRef CAS.
- F. J. Cespedes-Guirao, A. B. Ropero, E. Font-Sanchis, A. Nadal, F. Fernandez-Lazaro and A. Sastre-Santos, Chem. Commun., 2011, 47, 8307–8309 RSC.
- G. Battagliarin, Y. Zhao, C. Li and K. Müllen, Org. Lett., 2011, 13, 3399–3401 CrossRef CAS PubMed.
- S. Nakazono, Y. Imazaki, H. Yoo, J. Yang, T. Sasamori, N. Tokitoh, T. Cédric, H. Kageyama, D. Kim, H. Shinokubo and A. Osuka, Chem. – Eur. J., 2009, 15, 7530–7533 CrossRef CAS PubMed.
- T. Teraoka, S. Hiroto and H. Shinokubo, Org. Lett., 2011, 13, 2532–2535 CrossRef CAS PubMed.
- J. Kelber, H. Bock, O. Thiebaut, E. Grelet and H. Langhals, Eur. J. Org. Chem., 2011, 2011, 707–712 CrossRef.
- F. Würthner, A. Sautter and J. Schilling, J. Org. Chem., 2002, 67, 3037–3044 CrossRef.
- S. Mathew and M. R. Johnston, Chem. – Eur. J., 2009, 15, 248–253 CrossRef CAS PubMed.
- C. Ego, D. Marsitzky, S. Becker, J. Zhang, A. C. Grimsdale, K. Müllen, J. D. MacKenzie, C. Silva and R. H. Friend, J. Am. Chem. Soc., 2003, 125, 437–443 CrossRef CAS PubMed.
- J. C. S. Costa, R. J. S. Taveira, C. F. R. A. C. Lima, A. Mendes and L. M. N. B. F. Santos, Opt. Mater., 2016, 58, 51–60 CrossRef CAS.
- H. Langhals, J. Karolin and L. B.-A. Johansson, J. Chem. Soc., Faraday Trans., 1998, 94, 2919–2922 RSC.
- Z. B. Hill, D. B. Rodovsky, J. M. Leger and G. P. Bartholomew, Chem. Commun., 2008, 6594–6596 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammograms of the all the compounds (Fig. S1 and S2); 1H and 13C NMR spectra of all synthesized compounds (Fig. S4–S15); mass spectra of all the new compounds (Fig. S16–S21). See DOI: 10.1039/c6qo00374e |
| This journal is © the Partner Organisations 2016 |