
LDA-mediated synthesis of ortho-cyanated diarylmethanes by reaction of fluoroarene with arylacetonitrile at room temperature†
Fang
Liang
,
Xinfei
Ji
,
Juan
Zhang
and
Song
Cao
*
Shanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology (ECUST), Shanghai 200237, China. E-mail: scao@ecust.edu.cn
First published on 26th August 2016
Abstract
An efficient and facile method for the synthesis of ortho-cyanated diarylmethane by reaction of fluoroarene with arylacetonitrile in the presence of LDA (lithium diisopropylamide) at room temperature was developed. The reaction of arylacetonitrile with electron-rich fluoroarene involves the formation of an aryne intermediate.
Aryl nitriles are important structural motifs found in pharmaceuticals, agrochemicals, and natural products.1 Furthermore, aromatic nitriles are versatile synthetic precursors and useful intermediates because they can be easily transformed into a diverse class of functionalities, such as amides, esters, primary amines, aldehydes, and carboxylic acids.2 Consequently, the development of efficient methods for the synthesis of aryl nitriles has drawn much attention from the organic synthetic community.3 The Sandmeyer and Rosenmund–von Braun reactions are two typical methods for the preparation of aromatic nitriles.4 The transition metal-catalyzed cyanations of aryl halides, aryl boronic acids and arenes with various cyanating agents have emerged as an alternative route to aryl nitriles.5 However, these protocols suffer from drawbacks, such as the use of an expensive transition-metal catalyst or stoichiometric amounts of mostly toxic metal cyanides and harsh reaction conditions.6 Recently, the search for organonitriles as nonmetallic cyano-group sources such as acetone cyanohydrin,7 CH3CN,8 ethyl cyanoacetate,9 trimethylsilyl cyanide (TMSCN),10 and malononitrile,11 has aroused much interest and significant progress has been made.12
Benzyl cyanide is a cheap and commercially available nonmetallic cyanide source. Wang and Lu have successfully achieved the cyanation of various substrates such as aryl halides, indole, aryl boronic acids, and arenes by using benzyl cyanide as the cyanation reagent in the presence of a transition metal catalyst.13 However, stoichiometric amounts of copper salts, a longer reaction time and higher reaction temperature were needed to complete the reaction. Furthermore, their results also indicated that the benzyl group in benzyl cyanide was converted into the benzoyl group and thus a substantial amount of by-products, benzaldehyde or benzoic acid was formed. Therefore, the methodology is less efficient from the viewpoint of atom economy. In 2009, Nakao and Hiyama reported nickel/AlMe2Cl-catalysed carbocyanation of alkynes using arylacetonitriles and two groups (cyano and arylmethyl) are added simultaneously to alkynes, affording a range of triply substituted acrylonitriles.14
Diarylmethane derivatives have received a great deal of attention due to their growing importance in pharmaceuticals, dye precursors and supramolecules.15 Diarylmethane and their derivatives are also very useful building blocks for organic synthesis.16 As a consequence, over the past few decades, many chemists have devoted their efforts to the development of novel methods for the synthesis of diarylmethanes.17 Two most powerful and conventional methods for the preparation of diarylmethanes are the Friedel–Crafts alkylation of aromatic compounds with benzylic halides18 and the transition metal-catalysed cross-coupling between aryl halides and benzylic metals.19 However, the disadvantages of these methods are the use of expensive transition-metal catalysts, the limited availability of the starting materials, and the prefunctionalization or pre-activation of substrates. Therefore, the development of an efficient alternative method for the synthesis of diarylmethanes is highly desirable.
In recent years, the cleavage and functionalization of the inert carbon–fluorine bonds have been a challenging topic in modern organic chemistry.20 The most commonly employed method for the functionalization of the carbon–fluorine bond of fluoroarenes is nucleophilic aromatic substitution (SNAr).21 However, the scope of this nucleophilic substitution remains somewhat limited to highly electron-deficient fluoroaromatics. For SNAr reactions of non-activated fluoroarenes such as electron-rich aryl fluorides, although the reaction could occur, a rather higher reaction temperature or prolonged reaction time are required.22 Recently, the transition-metal-catalyzed cross-coupling of fluoroarenes with organometallics has emerged as an alternative method for the functionalization of fluoroarenes.23 Although defluorination of fluoroarenes with a strong base via benzyne intermediates has been known for many years, it is seldom used for the functionalization of the carbon–fluorine bond.24
Biehl et al. have previously reported the reaction of electron-rich bromobenzene with arylacetonitrile to afford 2-arylmethylbenzonitrile in the presence of LITMP (lithium 2,2,6,6-tetramethylpiperidide) or LDA. However, the drawbacks of this reaction are the use of very low temperatures (−78 °C) and CH3O or OH as the ortho-directing group, which greatly limits their widespread applications.25
In continuation of our research on the cleavage and functionalization of carbon–fluorine bonds,26 we herein report an efficient and mild method for the synthesis of ortho-cyanated diarylmethanes by the reaction of various non-activated fluoroarenes with arylacetonitriles in the presence of LDA at room temperature (Scheme 1).
 | ||
| Scheme 1 Synthesis of ortho-cyanated diarylmethanes. | ||
We began our experiment with the model reaction of fluorobenzene 1a with benzyl cyanide 2a at room temperature under an argon atmosphere (Table 1). No desired product 3aa was detected when LiHMDS, NaHMDS, t-BuOLi and NaH were used as the base (entries 1–4). LITMP and n-BuLi could provide 3aa in moderate yields (entries 5 and 6) and LDA gave the best result (entry 7). Increasing the amount of fluorobenzene 1a from 1.2 equivalents to 1.5 equivalents slightly improved the yield of 3aa (entry 8). Further increasing the amounts of 1a led to a lower yield of 3aa (entry 9). It was found that 4.0 equivalents of LDA were sufficient to carry out this reaction smoothly (entry 8). However, increasing or decreasing the amounts of LDA would result in diminished yields (entries 10 and 11). Moreover, the choice of the solvent was vitally important. Only THF could afford the expected product 3aa in good yield (entry 8). If the reaction was carried out in Et2O, hexane, DMF, DMSO or DMA, almost no desired product was found (entries 12–16). Noticeably, the reaction proceeded rapidly and reached completion in 0.5 h.
|
|
||||
|---|---|---|---|---|
| Entry | 1a (equiv.) | Base (equiv.) | Solvent | 3aa (%) |
| a Reaction conditions: 2a (0.2 mmol), solvent (2 mL), 0.5 h, r.t., Ar. b Yields determined by GC analysis and based on 2a. | ||||
| 1 | 1.2 | LiHMDS (4.0) | THF | 0 |
| 2 | 1.2 | NaHMDS (4.0) | THF | 0 |
| 3 | 1.2 | t-BuOLi (4.0) | THF | 0 |
| 4 | 1.2 | NaH (4.0) | THF | 0 |
| 5 | 1.2 | LITMP (4.0) | THF | 45 |
| 6 | 1.2 | n-BuLi (4.0) | THF | 59 |
| 7 | 1.2 | LDA (4.0) | THF | 69 |
| 8 | 1.5 | LDA (4.0) | THF | 76 |
| 9 | 2.0 | LDA (4.0) | THF | 64 |
| 10 | 1.5 | LDA (3.5) | THF | 69 |
| 11 | 1.5 | LDA (4.5) | THF | 46 |
| 12 | 1.5 | LDA (4.0) | Et2O | Trace |
| 13 | 1.5 | LDA (4.0) | Hexane | 0 |
| 14 | 1.5 | LDA (4.0) | DMF | 0 |
| 15 | 1.5 | LDA (4.0) | DMSO | 0 |
| 16 | 1.5 | LDA (4.0) | DMA | 0 |

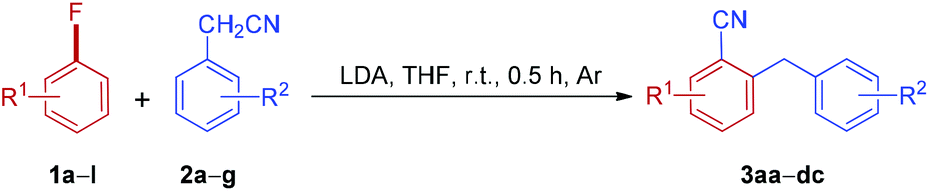
With the optimized reaction conditions in hand, the scope of arylacetonitrile was investigated, as shown in Table 2. The substituent effect on the benzene ring of arylacetonitrile was not apparent. Both electron-donating groups, such as Me and OMe (2b and 2d), and electron-withdrawing groups, such as CF3 (2f), located in the aromatic rings of arylacetonitriles were found to be tolerated in this reaction and the desired products were obtained in moderate to good yields. Interestingly, the sterically hindered substrate, 2-phenylpropanenitrile 2g, also afforded the expected product in a good yield (3ag). To our delight, the reaction of 1a with 2c could be scaled up from 1.0 to 15.0 mmol (2c, 1.97 g) without any loss in effectiveness and 3ac was obtained in 68% yield.
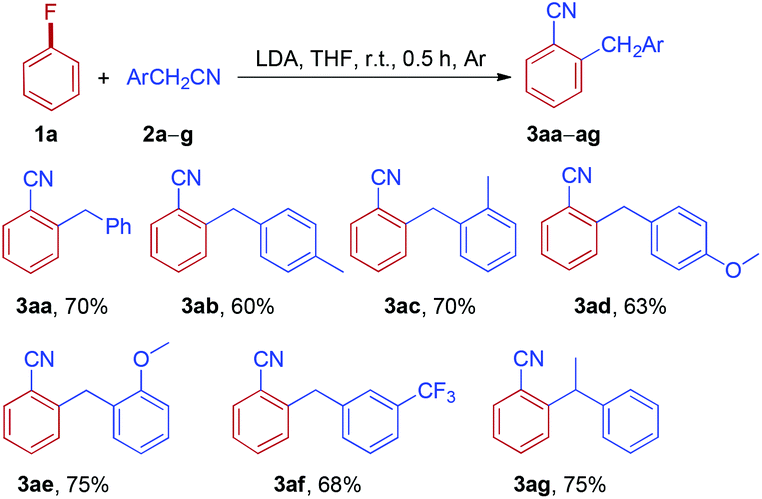
Subsequently, the reactions of phenylacetonitrile 2a with various fluoroarenes were examined (Table 3). Despite the inertness of the C–F bond of non-activated fluoroarenes, most of the reactions proceeded rapidly and was complete at room temperature within 0.5 h, affording ortho-cyanated diarylmethanes in moderate to good yields. Generally, a mixture of regioisomers was generated due to random aryne formation and subsequent random nucleophilic addition to aryne. Unfortunately, regioisomers could not be separated because of their similarity in affinity to chromatographic silica gel. The regioisomer ratio was based on the 1H and 19F NMR spectra, and GC-MS. However, we were unable to distinguish between two isomers owing to the similarity of their 1H NMR spectra (3ba and 3ca). 1-Fluoro-4-methoxybenzene 1d could provide 2-benzyl-4-methoxybenzonitrile 3da as the major product along with a small amount of 2-benzyl-5-methoxybenzonitrile 3da′ (10%, GC). Curiously, the reaction of 2-fluoro-1,4-dimethoxybenzene 1e with phenylacetonitrile 2a afforded two isomers, rearranged product 3ea and simple anion nitrile addition product 3ea′, which were isolated in 40% and 38% yields, respectively. The treatment of 1-fluoro-3-methylbenzene 1f with 2a could generate two arynes, which led to the formation of a mixture of four rearranged products 3fa in 77% yield and no simple anion nitrile addition product was observed. In the case of 1,4-difluorobenzene 1g, two rearranged products (3ga-I and 3ga-II) and two simple anion nitrile addition products (3ga-III and 3ga-IV) were obtained in moderate yields. Disappointingly, 1,3,5-trifluorobenzene (1h) failed to give the desired product. However, when highly electron-deficient polyfluoroarenes (1i and 1j) were used as substrates, the reaction proceeded through nucleophilic aromatic substitution (SNAr) and nucleophilic aromatic substitution products (3ia and 3ja) were isolated in good yields, which is in accordance with the previous report.27 Meanwhile, when two ortho positions of fluorobenzene were blocked by methyl groups (1k), no reaction could take place. This result further indicated that the reaction of electron-rich fluoroarenes with phenylacetonitrile 2a proceeded via an aryne intermediate.
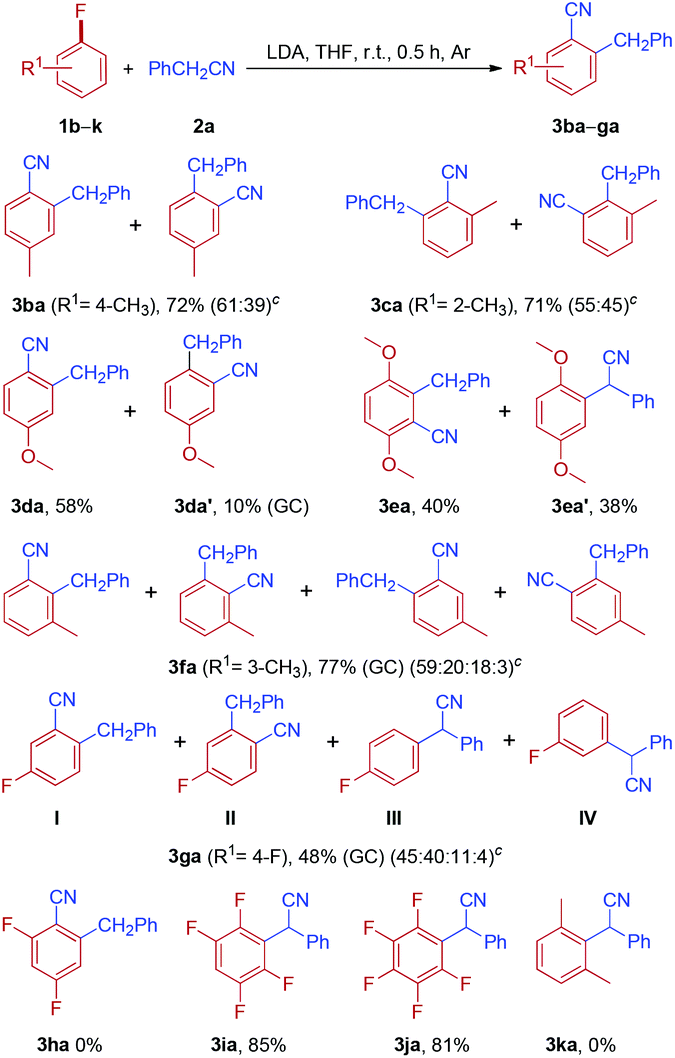
To further probe the generality and limitation of the reaction, various fluoroarenes were applied to the reaction with different arylacetonitriles, and the results are summarized in Table 4. In most cases, the reaction worked smoothly, giving a mixture of two isomers in moderate to good yields. Interestingly, cyanation and arylmethylation of 1-fluoro-4-methoxybenzene 1d with arylacetonitriles (2b, 2c and 2f) produced 2-arylmethyl-4-methoxybenzonitriles (3db, 3dc and 3df) as the major products in moderate isolated yields, along with small amounts of 2-arylmethyl-5-methoxybenzonitriles (3db′, 3dc′ and 3df′). Gratifyingly, 1-fluoro-4-methoxybenzene (1d) was reacted with sterically hindered 2-phenylpropanenitrile 2g to furnish 3dg as the sole product in 71% yield. When perfluorobenzene 1j was treated with 2-(4-methoxyphenyl)acetonitrile 2d, nucleophilic substitution products 3jd were obtained in 82% isolated yield.
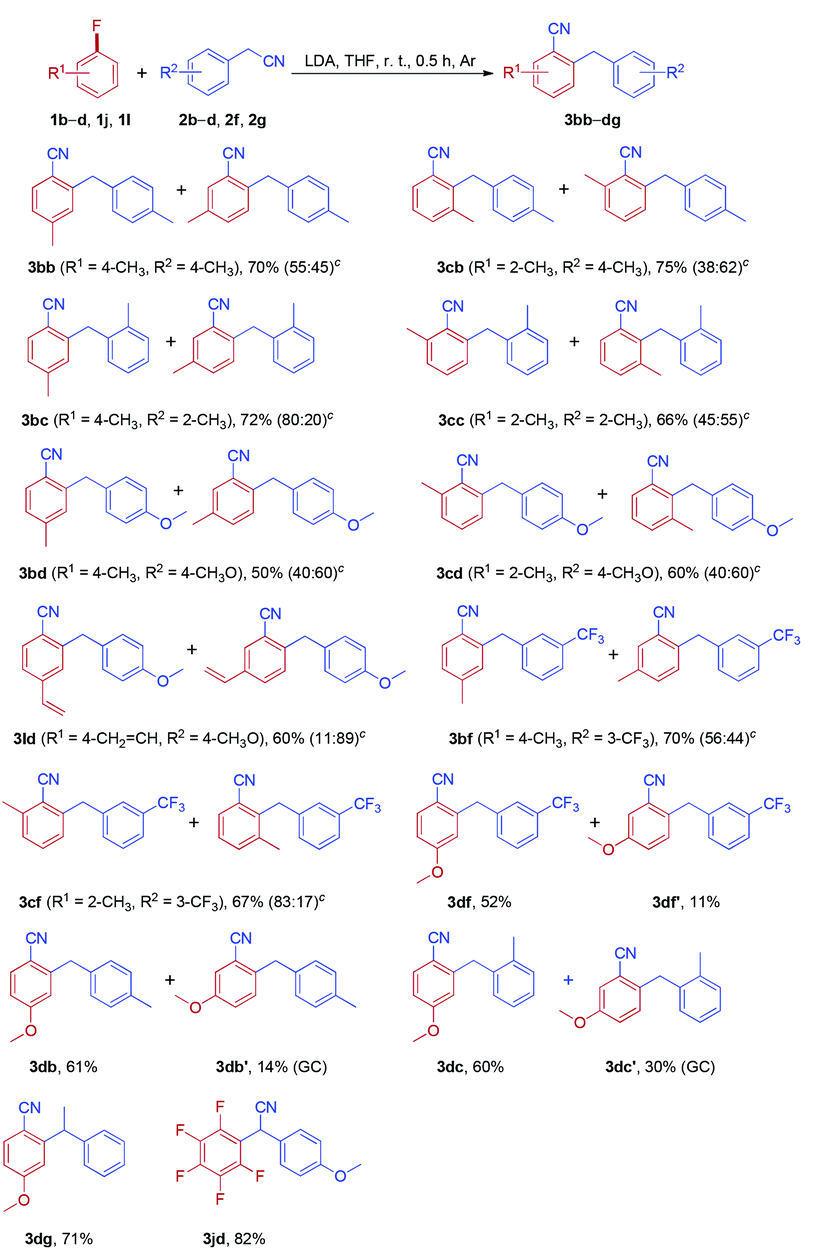
Inspired by the success in the reaction of fluoroarenes with arylacetonitriles, we extended this protocol to a variety of chloroarenes and bromoarenes (Table 5). Both chloroarenes and bromoarenes could react with arylacetonitriles smoothly to provide ortho-cyanated diarylmethanes in good yields.
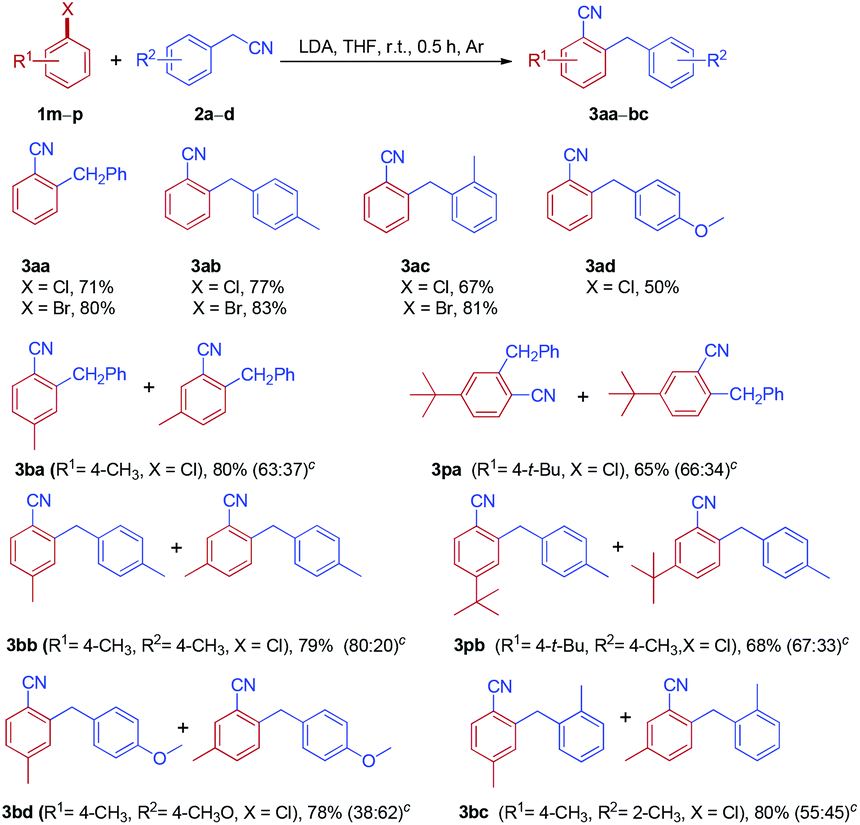
Importantly, no significant differences were observed in the reactivity of fluoroarenes, chloroarenes and bromoarenes under the optimized conditions.
Based on the above experimental results and related literatures,25a–c,28 a plausible mechanism of the reaction of an electron-rich haloarene is depicted in Scheme 2. Firstly, deprotonation of arylacetonitrile with LDA generates α-lithiated arylacetonitriles I. The cleavage of the carbon–halogen bond in the presence of LDA furnishes the key intermediate, aryne II. Addition of α-lithiated arylacetonitriles I to aryne II yields two aryne–nitrile adducts, III and IV. Intermediates III and IV might follow two pathways at the same time. In path A, the simple anion nitrile addition products VI and XIV are obtained via the usual aryne pathway, which involves the formation of intermediates, lithiated 2,2-diarylacetonitriles V and XIII. Alternatively, the reaction proceeds via a tandem addition–rearrangement pathway (path B). Cyclization of intermediates III and IV generates lithiated benzocyclobutanimines VII and X. Ring opening of VII and X provides α-lithiated benzylbenzonitriles VIII and XI which, after neutralization, gives rearranged nitrile products IX and XII. It should be noted that two pathways compete with each other. The reaction pathway, the ratio of isomers and the regiochemistry of the addition are greatly dependent on the electronic nature and position of the substituents on the phenyl ring of both haloarenes and arylacetonitriles.
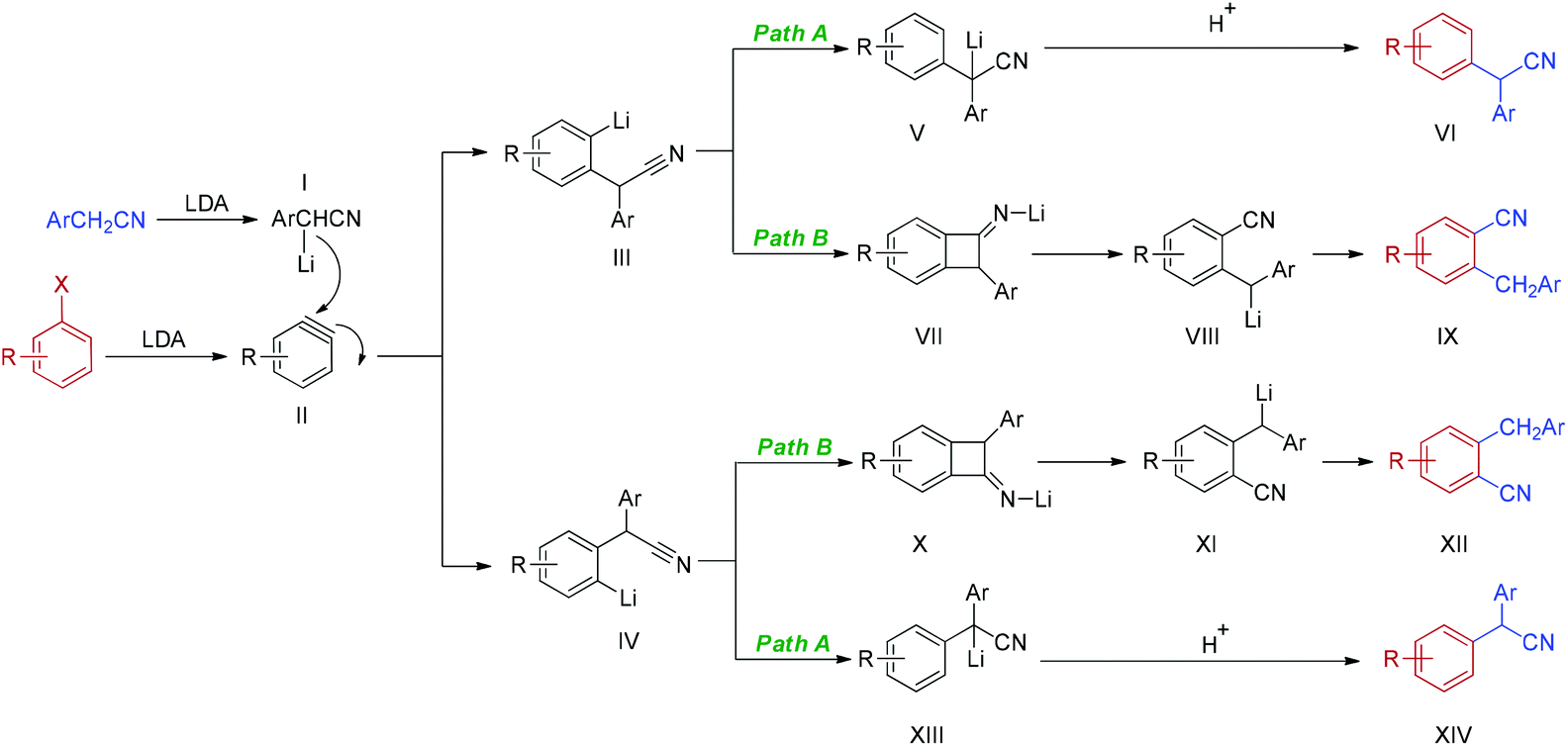 | ||
| Scheme 2 Plausible mechanism of electron-rich haloarene with arylacetonitrile. | ||
In summary, we have developed a rapid, atom-economical and scalable method for the synthesis of ortho-cyanated diarylmethanes from various readily available non-activated fluoroarenes and arylacetonitriles in the presence of LDA at room temperature. The reaction of electron-rich fluoroarenes proceeds via aryne intermediates, whereas highly electron-deficient fluoroarenes proceed via a typical SNAr pathway. Remarkably, the substrate scope could be expanded to a variety of chloroarenes and bromoarenes. The advantage of this method is that the cyano functional group and arylmethyl group are incorporated into the phenyl ring simultaneously, despite the fact that the regioselectivity of this protocol is low. We anticipated that the regioselectivity can be controlled by tuning the position, and the steric and electronic properties of the substituents on the aryl ring. Notably, the industrially important chemical raw materials fluorobenzene, chlorobenzene and bromobenzene could be transformed to useful ortho-cyanated diarylmethanes in good yields by this methodology.
Acknowledgements
Financial support from the National Natural Science Foundation of China (Grant No. 21472043 and 21272070) is gratefully acknowledged.Notes and references
- (a) A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substances: Syntheses, Patents, Applications, Thieme, Stuttgart, New York, 4th edn, 2001 Search PubMed; (b) F. F. Fleming, L.-H. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS PubMed.
- (a) Z. Rappoport, The Chemistry of the Cyano Group, Interscience, New York, 1970 Search PubMed; (b) R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, New York, 2nd edn, 1988 Search PubMed.
- (a) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC; (b) Q.-D. Wen, J.-S. Jin, L.-P. Zhang, Y. Luo, P. Lu and Y.-G. Wang, Tetrahedron Lett., 2014, 55, 1271 CrossRef CAS; (c) Z.-B. Shu, W.-Z. Ji, X. Wang, Y.-J. Zhou, Y. Zhang and J.-B. Wang, Angew. Chem., Int. Ed., 2014, 126, 2218 CrossRef.
- (a) T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 1633 CrossRef; (b) J. Von Braun and G. Manz, Justus Liebigs Ann. Chem., 1931, 488, 111 CrossRef; (c) K. W. Rosenmund and E. Struck, Ber. Dtsch. Chem. Ges. B, 1919, 52, 1749 CrossRef.
- (a) Z.-Q. Jiang, Q. Huang, S. Chen, L.-S. Long and X.-G. Zhou, Adv. Synth. Catal., 2012, 354, 589 CrossRef CAS; (b) Y.-M. Zhu, L.-Y. Li and Z.-M. Shen, Chem. – Eur. J., 2015, 21, 13246 CrossRef CAS PubMed; (c) S.-Y. Zheng, C.-H. Yu and Z.-W. Shen, Org. Lett., 2012, 14, 3644 CrossRef CAS PubMed; (d) S.-T. Ding and N. Jiao, J. Am. Chem. Soc., 2011, 133, 12374 CrossRef CAS PubMed; (e) J.-W. Dong, Z.-J. Wu, Z.-Y. Liu, P. Liu and P.-P. Sun, J. Org. Chem., 2015, 80, 12588 CrossRef CAS PubMed.
- (a) M. Sundermeier, A. Zapf, S. Mutyala, W. Baumann, J. Sans, S. Weiss and M. Beller, Chem. – Eur. J., 2003, 9, 1828 CrossRef CAS PubMed; (b) M. Sundermeier, A. Zapf, M. Beller and J. Sans, Tetrahedron Lett., 2001, 42, 6707 CrossRef CAS; (c) Y.-Z. Zhu and C. Cai, Eur. J. Org. Chem., 2007, 2401 CrossRef CAS; (d) A. V. Ushkov and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 10999 CrossRef CAS PubMed.
- T. Schareina, A. Zapf, A. Cotté, M. Gotta and M. Beller, Adv. Synth. Catal., 2011, 353, 777 CrossRef CAS.
- C.-D. Pan, H.-M. Jin, P. Xu, X. Liu, Y.-X. Cheng and C.-J. Zhu, J. Org. Chem., 2013, 78, 9494 CrossRef CAS PubMed.
- S.-L. Zhang and L. Huang, Org. Biomol. Chem., 2015, 13, 9963 CAS.
- C. Zhu, J.-B. Xia and C. Chen, Org. Lett., 2014, 16, 247 CrossRef CAS PubMed.
- S.-X. Lin, Y. Wei and F.-S. Liang, Chem. Commun., 2012, 48, 9879 RSC.
- J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948 CrossRef CAS PubMed.
- (a) Q.-D. Wen, J.-S. Jin, Y.-C. Mei, P. Lu and Y.-G. Wang, Eur. J. Org. Chem., 2013, 4032 CrossRef CAS; (b) Q.-D. Wen, J.-S. Jin, B.-B. Hu, P. Lu and Y.-G. Wang, RSC Adv., 2012, 2, 6167 RSC; (c) L.-P. Zhang, Q.-D. Wen, J.-S. Jin, C. Wang, P. Lu and Y.-G. Wang, Tetrahedron, 2013, 69, 4236 CrossRef CAS; (d) Y. Luo, Q.-D. Wen, Z.-Y. Wu, J.-S. Jin, P. Lu and Y.-G. Wang, Tetrahedron, 2013, 69, 8400 CrossRef CAS; (e) J.-S. Jin, Q.-D. Wen, P. Lu and Y.-G. Wang, Chem. Commun., 2012, 48, 9933 RSC.
- A. Yada, T. Yukawa, Y. Nakao and T. Hiyama, Chem. Commun., 2009, 3931 RSC.
- (a) I. Iovel, K. Mertins, J. Kischel, A. Zapf and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 3913 CrossRef CAS PubMed; (b) V. Nair, S. Thomas, S. C. Mathew and K. G. Abhilash, Tetrahedron, 2006, 62, 6731 CrossRef CAS; (c) J.-C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS PubMed.
- (a) J.-Y. Yu and R. Kuwano, Org. Lett., 2008, 10, 973 CrossRef CAS PubMed; (b) H. Yi, Q. Liu, J. Liu, Z.-Q. Zeng, Y.-H. Yang and A.-W. Lei, ChemSusChem, 2012, 5, 2143 CrossRef CAS PubMed; (c) F.-Q. Yuan, L.-X. Gao and F.-S. Han, Chem. Commun., 2011, 47, 5289 RSC.
- (a) J. J. Dunsford, E. R. Clark and M. J. Ingleson, Angew. Chem., Int. Ed., 2015, 54, 5688 CrossRef CAS PubMed; (b) R. Kuwano and M. Yokogi, Chem. Commun., 2005, 5899 RSC; (c) M. Amatore and C. Gosmini, Chem. Commun., 2008, 5019 RSC.
- T. Tsuchimoto, K. Tobita, T. Hiyama and S. Fukuzawa, J. Org. Chem., 1997, 62, 6997 CrossRef CAS.
- (a) G. A. Molander and M. D. Elia, J. Org. Chem., 2006, 71, 9198 CrossRef CAS PubMed; (b) C. C. Kofink and P. Knochel, Org. Lett., 2006, 8, 4121 CrossRef CAS PubMed; (c) C. Duplais, A. Krasovskiy, A. Wattenberg and B. H. Lipshutz, Chem. Commun., 2010, 46, 562 RSC.
- Q. Shen, Y.-G. Huang, C. Liu, J.-C. Xiao, Q.-Y. Chen and Y. Guo, J. Fluorine Chem., 2015, 179, 14 CrossRef CAS.
- H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed.
- (a) F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2012, 51, 8012 CrossRef CAS PubMed; (b) X.-G. Meng, Z.-Y. Cai, S. Xiao and W.-C. Zhou, J. Fluorine Chem., 2013, 146, 70 CrossRef CAS.
- A. D. Sun and J. A. Love, Dalton Trans., 2010, 39, 10362 RSC.
- (a) R. D. Clark and J. M. Caroon, J. Org. Chem., 1982, 47, 2804 CrossRef CAS; (b) W. F. Bailey and S. C. Longstaff, J. Org. Chem., 1998, 63, 432 CrossRef CAS PubMed; (c) K. N. Singh, P. Singh, P. Singh and Y. S. Deol, Org. Lett., 2012, 14, 2202 CrossRef CAS PubMed.
- (a) J. H. Waggenspack, L. Tran, S. Taylor, L. K. Yeung, M. Morgan, A. R. Deshmukh, S. P. Khanapure and E. R. Biehl, Synthesis, 1992, 765 CrossRef CAS; (b) W. Han, J. Tran, H.-M. Zhang, S. Jeffrey, D. Swartling, G. P. Ford and E. R. Biehl, Synthesis, 1995, 827 CrossRef CAS; (c) S. P. Khanapure, L. Crenshaw, R. T. Reddy and E. R. Biehl, J. Org. Chem., 1988, 53, 4915 CrossRef CAS; (d) H. Pellissier and M. Santelli, Tetrahedron, 2003, 59, 701 CrossRef CAS.
- (a) X.-F. Ji, T. Huang, W. Wu, F. Liang and S. Cao, Org. Lett., 2015, 17, 5096 CrossRef CAS PubMed; (b) G.-Y. Jin, X.-X. Zhang and S. Cao, Org. Lett., 2013, 15, 3114 CrossRef CAS PubMed; (c) X.-X. Zhang, W.-P. Dai, W. Wu and S. Cao, Org. Lett., 2015, 17, 2708 CrossRef CAS PubMed; (d) J. Zhang, C.-Y. Xu, W. Wu and S. Cao, Chem. – Eur. J., 2016, 22, 9902 CrossRef CAS PubMed.
- G.-H. Yang, M. Liu, N. Li, R. Wu, X. Chen, L.-L. Pan, S. Gao, X. Huang, C. Wang and C.-M. Yu, Eur. J. Org. Chem., 2015, 616 CrossRef CAS.
- (a) S. Tandel, A.-L. Wang, H.-M. Zhang, P. Yousuf and E. R. Biehl, J. Chem. Soc., Perkin Trans. 1, 2000, 3149 RSC; (b) S. Tandel, H. Zhang, N. Qadri, G. P. Ford and E. R. Biehl, J. Chem. Soc., Perkin Trans. 1, 2000, 587 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00363j |
| This journal is © the Partner Organisations 2016 |