
Copper-catalyzed 8-amido chelation-induced regioselective C–H fluoroalkylation of quinolines†
Li-Kun
Jin
a,
Guo-Ping
Lu
a and
Chun
Cai
*ab
aChemical Engineering College, Nanjing University of Science and Technology, 200 Xiao Ling Wei, Nanjing 210094, People's Republic of China. E-mail: c.cai@njust.edu.cn
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 20032, People's Republic of China
First published on 12th August 2016
Abstract
An efficient and highly regioselective protocol for copper(II)-catalyzed remote C–H fluoroalkylation of 8-aminoquinoline amides with RfSO2Na (Rf = CF3, CF2H, C4F9, C5F11, and C6F13) is described. This reaction provides an effective strategy for the direct construction of fluorine-containing six-membered heteroaromatic core structures.
To date, the incorporation of fluorine-containing groups into biologically active molecules has emerged as a widely employed strategy in medicinal chemistry, in order to perturb the chemical, physical, and biological properties of parent compounds.1 Among them, the trifluoromethyl group is of extensive interest owing to its strong electron-withdrawing power and high lipophilicity.2 Therefore, substantial efforts have been devoted to the development of new synthetic methods for the direct assembly of CF3-containing small molecules.3
Transition-metal-catalyzed or -mediated trifluoromethylations of aryl halides,4a–c alkynes4d–f and the corresponding boronic acids4g,h are all early examples of this transformation. In recent years, transition-metal-catalyzed C–H functionalization has become a facile and robust approach to the construction of C–C and C-heteroatom bonds.5 Its power originates in the ability to accomplish site-selective derivatization of inert C–H bonds without the need for prefunctionalized starting materials. As such, new approaches of direct transformation from C–H to C–CF3 have been realized, including palladium-catalyzed ortho C–H trifluoromethylation by PdII/PdIV redox catalysis,6a–c copper-catalyzed or -mediated trifluoromethylation of heterocyclic compounds,6d,e as well as methods which take advantage of CF3 radical formation.6f–h
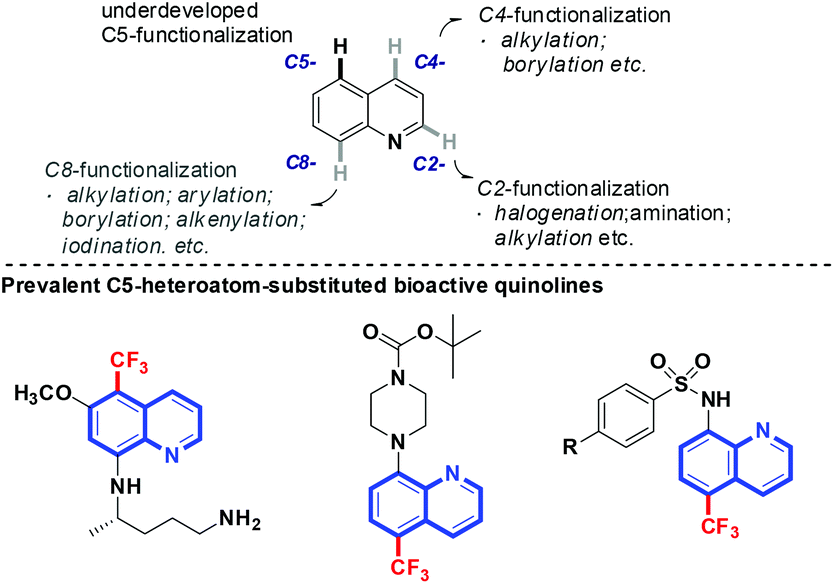
Derivatives of quinolines are ubiquitously found in naturally occurring and synthetic bioactive compounds (Scheme 1).7 Straightforward access for their functionalization has aroused great interest based on the C–H activation strategy.8 However, the implementation of such an atom-economical technique is challenging due to the uncontrolled regioselectivity problem. Although the direct functionalization of C–H bonds at the C2-,8d,eC4-,8f,g and C8-positions8h,i of quinolines has been well-developed, the selective functionalization at the C5-position remains an intriguing problem. Pioneered by the work of Stahl and co-workers demonstrated in 2013,9 some progress has been made focused on the functionalization of this distal and geometrically inaccessible C–H bond in quinolines. Zeng and coworkers demonstrated a C5-allylation of quinolines in the presence of an iron catalyst.10a Shortly thereafter, the copper-catalyzed direct C–H bond sulfonylation of aminoquinolines with arylsulfonyl chlorides was reported by the Wei and Wu group, which has a broad substrate scope for both aromatic and aliphatic sulfonyl chlorides.10b–d Very recently, Baidya and coworkers developed a direct C(sp2)–H amination of quinolines by the use of azodicarboxylates.10e However, not a single approach for the direct selective remote C–H fluoroalkylation of quinolines has been reported (Scheme 2).
 | ||
| Scheme 1 Direct C–H functionalization of quinolines. | ||
 | ||
| Scheme 2 Chelation-induced remote C–H functionalization of quinolines. | ||
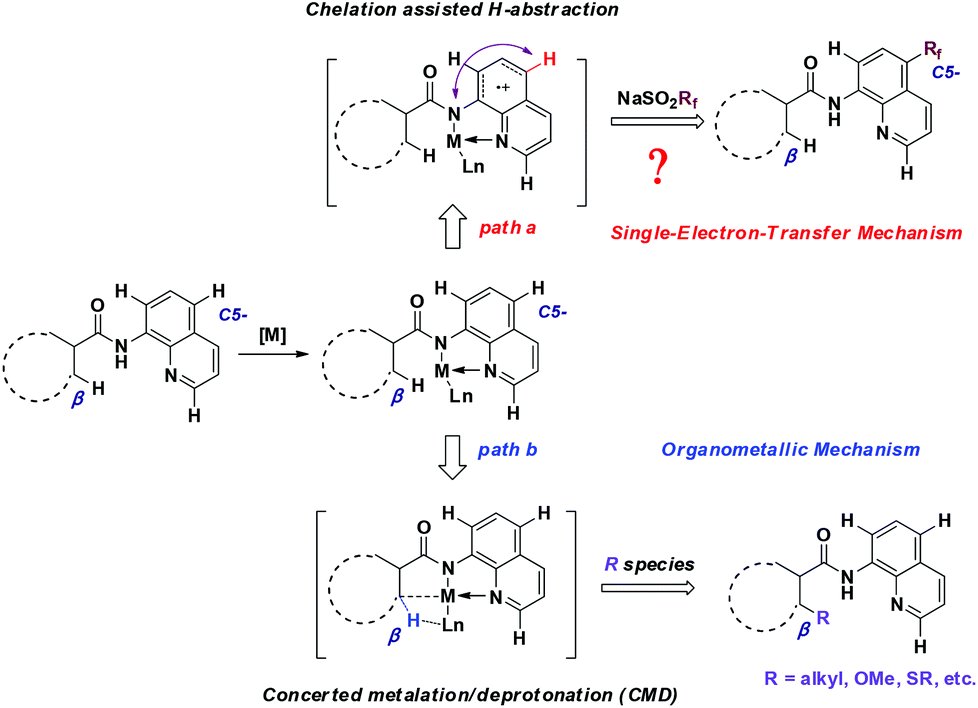
Our previous studies on the selective introduction of fluorine-containing groups into inert C–H bonds11 led us to wonder if the CF3 group could be inserted into quinoline derivatives and thus offer a direct entry towards the C5-functionalized aminoquinoline-derived antimalarial and anti-cancer drug candidates. It was anticipated that the formation of a metal-chelated complex would affect the electron contribution in quinoline, which ends up with the regioselective assembly of the CF3 group at the C5-position. Herein, we detailed a highly regioselective C5–H trifluoromethylation of 8-aminoquinolines by using NaSO2CF3 as a trifluoromethylating reagent in the presence of AIBN as an oxidant. This protocol tolerates a wide range of functional groups and is also compatible with RfSO2Na to offer perfluoroalkylated quinolines.
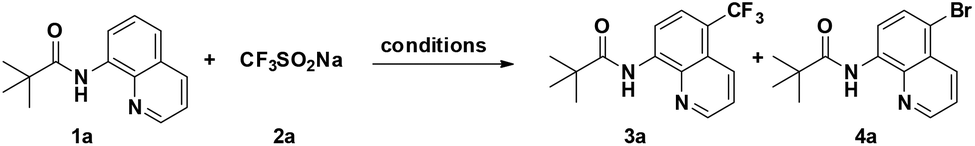
We commenced our investigation by examining the reaction of 8-aminoquinoline derivative 1a with NaSO2CF3 (2a) (Table 1). After a careful evaluation of various reaction parameters, the desired trifluoromethylated quinoline 3a was isolated in 73% yield in the presence of CuBr2 as a catalyst and AIBN as an oxidant under air for 12 h (Table 1, entry 4). However, compared to other copper salts such as Cu(OAc)2, CuCl2, CuO, and Cu(OTf)2, CuCl showed lower reactivity (entries 1–3, 5–6). As expected, copper played a pivotal role in this reaction, the desired product could not be obtained under copper-catalyst-free conditions and reducing the catalyst loading resulted in a decreased yield of the C5-trifluoromethylation product (entries 7 and 8), while using 50 mol% CuBr2 resulted in a mixture of trifluoromethylation and bromination products 3a and 4a (entry 9). Control experiments suggest that 3a formed under these conditions arises from direct C–H trifluoromethylation, not C–H bromination followed by CF3 substitution of bromide in 4a. Among the oxidants examined, including TBHP, K2S2O8, AIBN, and H2O2, AIBN was the best choice (entry 4 vs. 10–12). Besides, the addition of a base (e.g., K2CO3, Na2CO3, and Cs2CO3) would disfavor the generation of the desired product but promote the bromination at the C5 position (entries 13–15). The reaction yield showed a decrease in DCE (entry 16), while screening of other solvents resulted in recovery of the starting material 1a (entries 17–19). Finally, air can be well-tolerated in the process, and the yield of 3a dropped to 51% when the reaction temperature was altered from 120 °C to 100 °C (entry 20).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Oxidant | Solvent | Yieldb (%) | |
| 3a | 4a | |||||
| a Reaction conditions: 1a (0.20 mmol), 2a (0.40 mmol), copper catalyst (10 mol%), oxidant (0.24 mmol), base (0.20 mmol), solvent (1 mL) at 120 °C under air for 12 h. b Isolated yield. c Copper catalyst (5 mol%). d Copper catalyst (50 mol%). e At 100 °C. | ||||||
| 1 | Cu(OAc)2 | AIBN | MeCN | 31 | ||
| 2 | Cu(OTf)2 | AIBN | MeCN | 53 | ||
| 3 | CuCl2 | AIBN | MeCN | 15 | ||
| 4 | CuBr2 | AIBN | MeCN | 73 | ||
| 5 | CuO | AIBN | MeCN | 12 | ||
| 6 | CuCl | AIBN | MeCN | 36 | ||
| 7 | AIBN | MeCN | ||||
| 8 | CuBr2 | AIBN | MeCN | 58c | ||
| 9 | CuBr2 | AIBN | MeCN | 68d | 21 | |
| 10 | CuBr2 | TBHP | MeCN | <5 | 17 | |
| 11 | CuBr2 | K2S2O8 | MeCN | 12 | 15 | |
| 12 | CuBr2 | H2O2 | MeCN | Trace | ||
| 13 | CuBr2 | K2CO3 | AIBN | MeCN | 24 | 35 |
| 14 | CuBr2 | Na2CO3 | AIBN | MeCN | 22 | 40 |
| 15 | CuBr2 | Cs2CO3 | AIBN | MeCN | 13 | 51 |
| 16 | CuBr2 | AIBN | DCE | 56 | ||
| 17 | CuBr2 | AIBN | PhCl | Trace | ||
| 18 | CuBr2 | AIBN | DME | |||
| 19 | CuBr2 | AIBN | Dioxane | Trace | ||
| 20 | CuBr2 | AIBN | MeCN | 51e | ||
With the optimized reaction conditions in hand, we next sought to investigate the substrate scope. As shown in Table 2, the reaction was quite general for a wide variety of 8-amino-quinolines. Aliphatic amides (1a–1i), aromatic amides (1j–1l, 1n), and carboxamides with heterocyclic substitutions (1m) are compatible with the reaction, though aliphatic amides would generate the corresponding product in relatively lower yields. Aliphatic amides bearing either linear chains or cyclic chains with different ring-sizes were trifluoromethylated in satisfactory yields. Note that the amide with an electrophile-sensitive functional group, 4-(1H-indol-3-yl)-butanamide (1i), remained intact in the reaction. Besides, the electronic effect was not obvious on the benzene ring of benzamides, such desired products were afforded in moderate to good yields (3j–3l). The functional groups such as Me and MeO on the quinoline ring would change the electron contribution which showed increases of product yields (3o–3r). In addition, compound 3k was crystallized and X-ray analysis unambiguously confirmed the regioselective C-5 trifluoromethylation of the quinoline (Scheme 3).
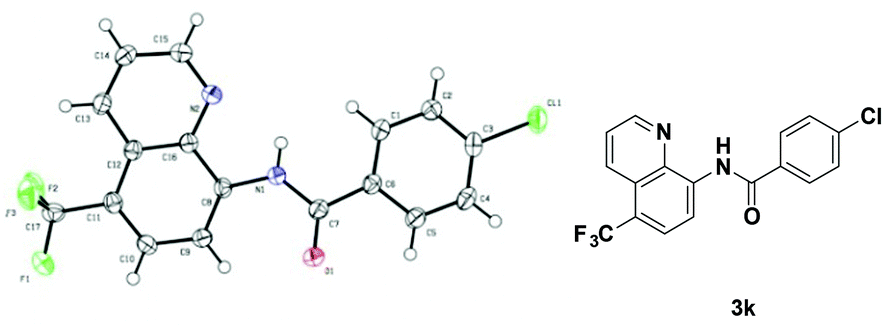 | ||
| Scheme 3 The crystal structure of 3k. | ||
|
Moreover, the reaction is not restricted only to NaSO2CF3 (2a), we further extended the substrate scope to investigate the direct introduction of CH2F, CHF2, and Rf groups into the C5-position of quinolines by using CH2FSO2Na, CHF2SO2Na and RfSO2Na as fluorinating reagents (Table 2). Sodium perfluoroalkanesulfinates (RfSO2Na) with different Rf groups were synthesized according to the procedure reported by Hu and DesMarteau.12 Remarkably, it turns out that this single-electron transfer activation mode enables the formation of fluoroalkyl radicals and further facilitates bond formation regioselectively at the C5-position of quinolines. In each case, perfluoroalkylation products were forged with moderate levels of efficiency (3u–3w), while the reactions proceeded less effectively with CH2FSO2Na. In the case of CHF2SO2Na, only a trace amount of 3s was produced, probably because CH2FSO2Na is less reactive in generating fluoroalkyl radicals.13
To gain some insight into the mechanism of the copper-catalyzed 8-amido chelation-induced remote C–H trifluoromethylation of quinolines, control experiments were carried out (Scheme 4). It should be mentioned that coordination of 1a to CuII as an amidate is prior to trifluoromethylation, which can be supported by the lack of reaction of the N-methyl substrate 5a under standard conditions. Meanwhile, if the C5–H position was blocked by a bromo group (4a), the target product would not occur, which demonstrates that the trifluoromethylation product is formed not by CF3 substitution of 4a. The addition of the radical inhibitor TEMPO resulted in an obvious inhibition of the reaction, indicating that a radical pathway might be involved in the process. Besides, in the case of non-protecting 8-aminoquinoline, the reaction gave a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of 7-CF3 and 5-CF3 products, respectively, with 16% yield of the mixture.
:2 mixture of 7-CF3 and 5-CF3 products, respectively, with 16% yield of the mixture.
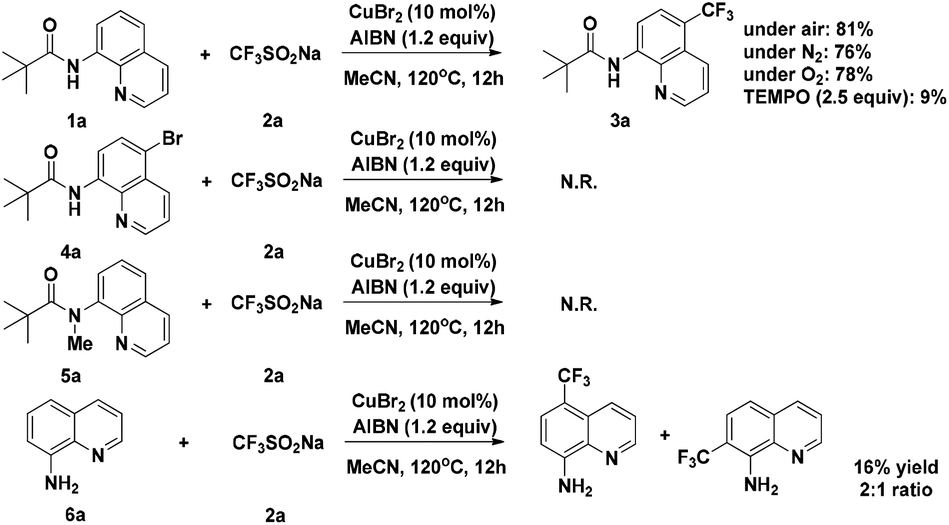 | ||
| Scheme 4 Control experiment. | ||
When the quinoline trifluoromethylation reaction was performed in the presence of D2O, no deuterium incorporation was observed in the recovered starting material or the product (Scheme 5). This result indicates that cleavage of the C–H bond is irreversible. Besides, an intermolecular competition experiment was carried out to obtain the kinetic isotope effect (KIE). Analysis of the H/D ratio of quinoline revealed a nearly equal mixture of isotopes, corresponding to a KH/KD = 1.17.
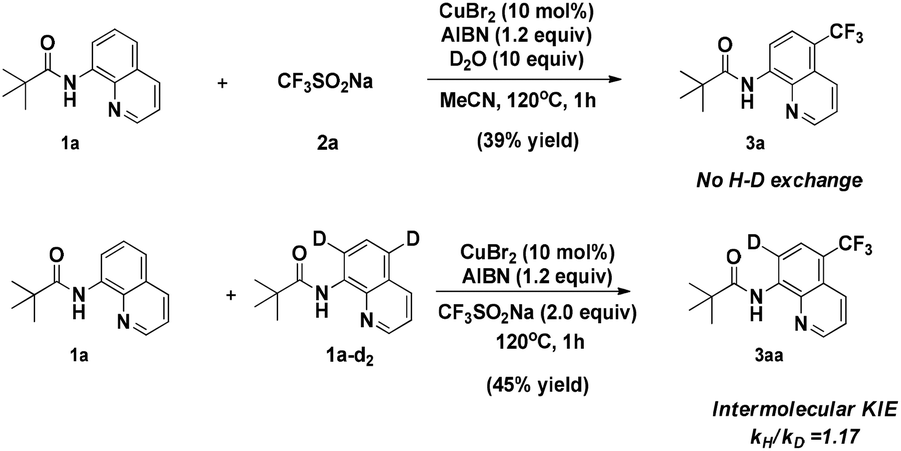 | ||
| Scheme 5 Deuteration and isotope effect experiments. | ||
This observation implies that the cleavage of C–H is not the rate-limiting step in the nonchelate-directed trifluoromethylation process.
On the basis of the above-mentioned results and related precedents,8a,9,10a–e a tentative mechanism is proposed as shown in Scheme 6. We believe that the quinoline trifluoromethylation in our study is governed by a single-electron transfer (SET) mechanism. The process is initiated from the coordination of 8-aminoquinoline derived 1 with CuBr2 and subsequently deprotonation to afford the chelated intermediate B. Meanwhile, the homolytic cleavage of AIBN releases 2-cyano-2-propyl radicals which are employed to initiate the trifluoromethyl radical via a single electron transfer process. Then, the addition of the trifluoromethyl radical to the intermediate B occurs at the C5 position to form the complex C. Thereafter, the CuI complex C is oxidized followed by the generation of complex E through a proton transfer process. Finally, the decomposition of E provides the desired product 3, along with the regeneration of intermediate A to continue the catalytic cycle.
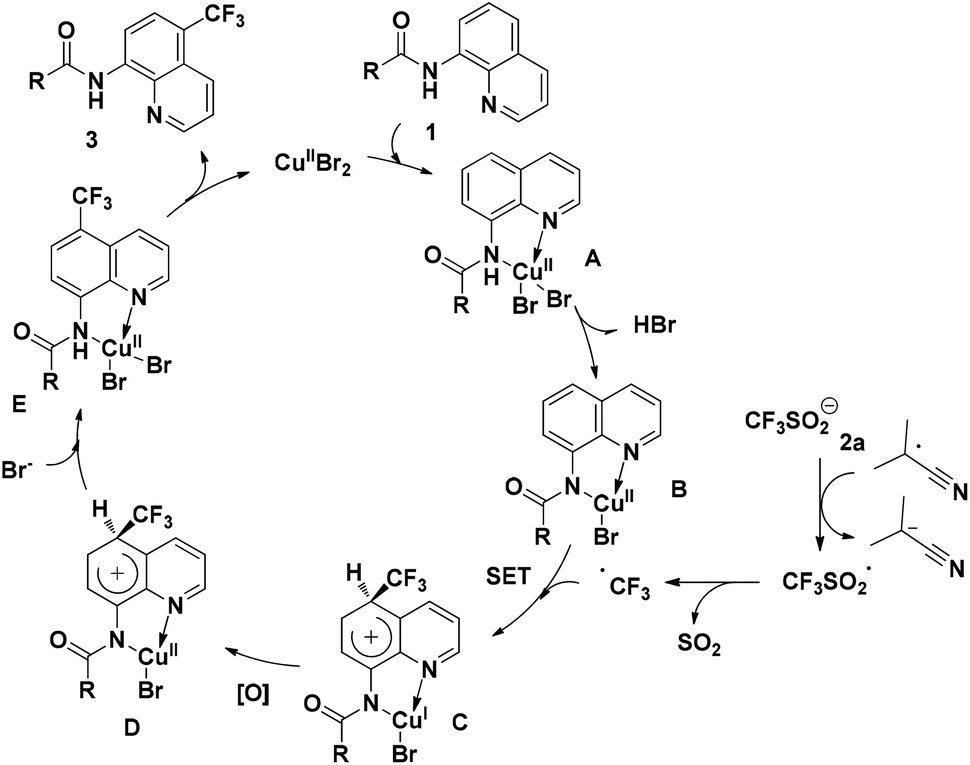 | ||
| Scheme 6 Plausible mechanism for the remote C–H trifluoromethylation of quinolines. | ||
Conclusions
In conclusion, an efficient copper-catalyzed 8-amido chelation induced strategy is described to realize the C-5 direct C–H functionalization of quinolines with fluorinated sulfinate salts RfSO2Na. The reaction features a broad substrate scope, excellent functional group tolerance and high regioselectivity. Considering the critical role of fluoro-containing moieties in pharmaceutical and agrochemical research, this approach could potentially offer new strategies to the synthesis of fluoroalkylated quinolines. Further efforts in our laboratory are devoted to the exploitation of regio- and stereocontrol in fluoroalkylation reactions, particularly regarding the direct functionalization of more challenging inactive sp3 C–H bonds.Acknowledgements
We gratefully acknowledge the Natural Science Foundation of Jiangsu Province (BK 20131346, BK 20140776) for financial support. This work was also supported by the National Natural Science Foundation of China (21476116, 21402093) and the Chinese Postdoctoral Science Foundation (2015M571761, 2016T90465).Notes and references
- For selected reviews or books on organofluorine chemistry, see: (a) K. Uneyama, Organofluorine Chemistry, Blackwell, Oxford, 2006 Search PubMed; (b) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Blackwell, Oxford, 2009 Search PubMed; (c) M. Egli, Acc. Chem. Res., 2012, 45, 1237 CrossRef CAS PubMed; (d) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed; (e) R. Littich and P. J. H. Scott, Angew. Chem., Int. Ed., 2012, 51, 1106 CrossRef CAS PubMed; (f) M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 31 RSC; (g) B. Koksch, Chem. Soc. Rev., 2012, 41, 2135 RSC; (h) T. Nakajima, J. Fluorine Chem., 2013, 149, 104 CrossRef CAS.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (b) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455 CrossRef CAS PubMed; (c) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (d) T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048 CrossRef CAS PubMed.
- For selected reviews and references on new synthetic methods for the construction of CF3-containing small molecules, see: (a) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (b) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (c) Y. Cheng, X. Yuan, H. Jiang, R. Wang, J. Ma, Y. Zhang and S. Yu, Adv. Synth. Catal., 2014, 356, 2859 CrossRef CAS; (d) J. Zheng, L. Wang, J.-H. Lin, J.-C. Xiao and S. H. Liang, Angew. Chem., Int. Ed., 2015, 54, 13236 CrossRef CAS PubMed; (e) Z. Huang, C. Wang, E. Tokunaga, Y. Sumii and N. Shibata, Org. Lett., 2015, 17, 5610 CrossRef CAS PubMed; (f) H. Luo, G. Wu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 14503 CrossRef CAS PubMed; (g) H. Zhang, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2014, 53, 10174 CrossRef CAS PubMed; (h) J.-B. Liu, X.-H. Xu and F.-L. Qing, Org. Lett., 2015, 17, 5048 CrossRef CAS PubMed.
- (a) M. Oishi, H. Kondo and H. Amii, Chem. Commun., 2009, 1909 RSC; (b) A. Zanardi, M. A. Novikov, E. Martin, J. Benet-Buchholz and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 20901 CrossRef CAS PubMed; (c) M. Chen and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 11628 CrossRef CAS PubMed; (d) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2010, 132, 7262 CrossRef CAS PubMed; (e) N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539 CrossRef CAS PubMed; (f) L. He and G. C. Tsui, Org. Lett., 2016, 18, 2800 CrossRef CAS PubMed; (g) T. Liu and Q. Shen, Org. Lett., 2011, 13, 2342 CrossRef CAS PubMed; (h) J. Xu, D.-F. Luo, B. Xiao, Z.-J. Liu, T.-J. Gong, Y. Fu and L. Liu, Chem. Commun., 2011, 47, 4300 RSC.
- For selected reviews, books and references on C–H bond functionalizations, see: (a) J.-Q. Yu and Z.-J. Shi, C–H activation, Springer, Berlin, Germany, 2010 Search PubMed; (b) D. Seidel, Acc. Chem. Res., 2015, 48, 317 CrossRef CAS PubMed; (c) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2010, 111, 1293 CrossRef PubMed; (d) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (e) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (f) W. Zhu, D. Zhang, N. Yang and H. Liu, Chem. Commun., 2014, 50, 10634 RSC; (g) L.-K. Jin, L. Wan, J. Feng and C. Cai, Org. Lett., 2015, 17, 4726 CrossRef CAS PubMed.
- (a) X. Wang, L. Truesdale and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3648 CrossRef CAS PubMed; (b) Y. Ye, N. D. Ball, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 14682 CrossRef CAS PubMed; (c) X. Mu, S. Chen, X. Zhen and G. Liu, Chem. – Eur. J., 2011, 17, 6039 CrossRef CAS PubMed; (d) M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590 CrossRef CAS PubMed; (e) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2011, 134, 1298 CrossRef PubMed; (f) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed; (g) J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6 Search PubMed; (h) J. C. Fennewald and B. H. Lipshutz, Green Chem., 2014, 16, 1097 RSC.
- (a) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, Wiley-VCH, Weinheim, Germany, 2003 CrossRef; (b) V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488 CrossRef CAS PubMed; (c) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC.
- (a) T. Iwai and M. Sawamura, ACS Catal., 2015, 5, 5031 CrossRef CAS; (b) J. Jayakumar, K. Parthasarathy, Y.-H. Chen, T.-H. Lee, S.-C. Chuang and C.-H. Cheng, Angew. Chem., Int. Ed., 2014, 53, 9889 CrossRef CAS PubMed; (c) L. Kong, S. Yu, X. Zhou and X. Li, Org. Lett., 2016, 18, 588 CrossRef CAS PubMed; (d) Z. Li, S.-S. Wu, Z.-G. Luo, W.-K. Liu, C.-T. Feng and S.-T. Ma, J. Org. Chem., 2016, 81, 4386 CrossRef CAS PubMed; (e) Y. Siddaraju, M. Lamani and K. R. Prabhu, J. Org. Chem., 2014, 79, 3856 CrossRef CAS PubMed; (f) J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565 CrossRef CAS PubMed; (g) M. Nagase, Y. Kuninobu and M. Kanai, J. Am. Chem. Soc., 2016, 138, 6103 CrossRef CAS PubMed; (h) J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598 CrossRef CAS PubMed; (i) X. Shao, C. Xu, L. Lu and Q. Shen, J. Org. Chem., 2015, 80, 3012 CrossRef CAS PubMed.
- A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797 CrossRef CAS PubMed.
- (a) X. Cong and X. Zeng, Org. Lett., 2014, 16, 3716 CrossRef CAS PubMed; (b) H.-W. Liang, K. Jiang, W. Ding, Y. Yuan, L. Shuai, Y.-C. Chen and Y. Wei, Chem. Commun., 2015, 51, 16928 RSC; (c) J. Xu, C. Shen, X. Zhu, P. Zhang, M. J. Ajitha, K. W. Huang, Z. An and X. Liu, Chem. – Asian J., 2016, 11, 882 CrossRef CAS PubMed; (d) H. Qiao, S. Sun, F. Yang, Y. Zhu, W. Zhu, Y. Dong, Y. Wu, X. Kong, L. Jiang and Y. Wu, Org. Lett., 2015, 17, 6086 CrossRef CAS PubMed; (e) H. Sahoo, M. K. Reddy, I. Ramakrishna and M. Baidya, Chem. – Eur. J., 2016, 22, 1592 CrossRef CAS PubMed.
- (a) L. Jiang, J. Qian, W. Yi, G. Lu, C. Cai and W. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14965 CrossRef CAS PubMed; (b) Y.-B. Wu, G.-P. Lu, B.-J. Zhou, M.-J. Bu, L. Wan and C. Cai, Chem. Commun., 2016, 52, 5965 RSC.
- (a) L. Q. Hu and D. D. DesMarteau, Inorg. Chem., 1993, 32, 5007 CrossRef CAS; (b) Z. He, P. Tan, C. Ni and J. Hu, Org. Lett., 2015, 17, 1838 CrossRef CAS PubMed.
- (a) J. Hu, B. Gao, L. Li, C. Ni and J. Hu, Org. Lett., 2015, 17, 3086 CrossRef CAS PubMed; (b) Y. Zeng, C. Ni and J. Hu, Chem. – Eur. J., 2016, 22, 3210 CrossRef CAS PubMed; (c) C. S. Thomoson, X.-J. Tang and W. R. Dolbier, J. Org. Chem., 2015, 80, 1264 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all products. CCDC 1478785. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00369a |
| This journal is © the Partner Organisations 2016 |