
Bovine serum albumin catalyzed one-pot, three-component synthesis of dihydropyrano[2,3-c]pyrazole derivatives in aqueous ethanol†
Kiran S. Dalala,
Yogesh A. Tayadeb,
Yogesh B. Waghb,
Darshak R. Trivedic,
Dipak S. Dalalb and
Bhushan L. Chaudhari *a
*a
aSchool of Life Sciences, North Maharashtra University, Jalgaon 425 001, MS, India. E-mail: blchaudhari@nmu.ac.in; blchaudhari@hotmail.com; Fax: +91-2572258403; Tel: +91 2572257424
bSchool of Chemical Sciences, North Maharashtra University, Jalgaon 425 001, MS, India
cSupramolecular Chemistry Laboratory, Department of Chemistry, National Institute of Technology Karnataka (NITK), Srinivasnagar, Surathkal, Mangalore 575025, Karnataka, India
First published on 26th January 2016
Abstract
Bovine serum albumin (BSA) catalyzed synthesis of dihydropyrano[2,3-c]pyrazole derivatives via a one pot, three component reaction of an aldehyde/ketone/isatin, malononitrile and 3-methyl-1H-pyrazol-5-(4H)-one in H2O–EtOH (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) at ambient temperature was developed in this work. The catalyst was found to work efficiently for aldehydes, ketones and isatins to give the corresponding dihydropyrano[2,3-c]pyrazole and spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives in high yields. BSA showed a broad range of catalytic promiscuity towards various aldehydes, aromatic/aliphatic ketones and substituted isatins. The use of an environmentally benign protocol, reusability of the catalyst, avoidance of hazardous solvents, excellent yields, easy work up and no byproduct formation make BSA an attractive candidate for further applications as a biocatalyst.
:3) at ambient temperature was developed in this work. The catalyst was found to work efficiently for aldehydes, ketones and isatins to give the corresponding dihydropyrano[2,3-c]pyrazole and spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives in high yields. BSA showed a broad range of catalytic promiscuity towards various aldehydes, aromatic/aliphatic ketones and substituted isatins. The use of an environmentally benign protocol, reusability of the catalyst, avoidance of hazardous solvents, excellent yields, easy work up and no byproduct formation make BSA an attractive candidate for further applications as a biocatalyst.
Introduction
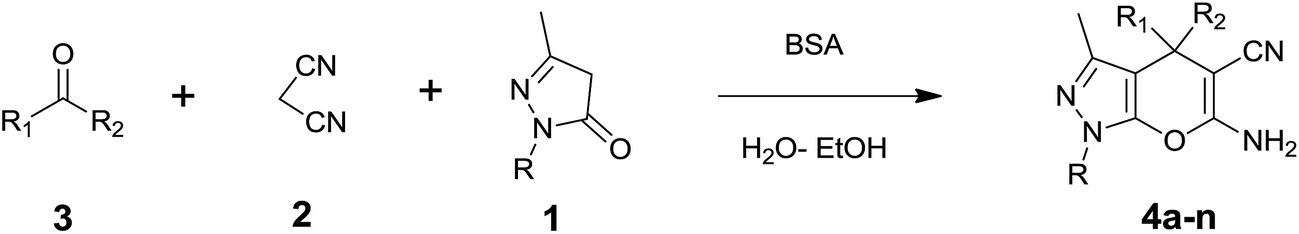
Dihydropyrano[2,3-c]pyrazoles are a promising class of heterocyclic compounds and a structural unit in a variety of therapeutic agents.1 The synthesis of dihydropyrano[2,3-c]pyrazole derivatives receives great attention among synthetic chemists due to their diverse bioactivity profiles with various medicinal properties, including as a potential inhibitor of human Chk1 kinase,2a and their anti-inflammatory,2b anticancer,3a analgesic,3b molluscicidal4 and antimicrobial activities.5 Because of the above-mentioned important properties of pyranopyrazoles, the development of new methods for the synthesis of this heterocyclic moiety has gained huge significance and is a demanding task in organic synthesis. Generally, they are synthesized by a multicomponent reaction of aldehydes/ketones, malononitrile, ethyl acetoacetate, and hydrazine hydrate in the presence of environmentally non-compatible base catalysts.6–9 Recently, some environmentally friendly catalysts such as L-proline,10 γ-alumina,11 per-6-amino-cyclodextrin,12 Amberlyst A21,13 piperidine,14a meglumine,14b cocamidopropyl betaine (CAPB),15a cetyltrimethylammonium chloride (CTACl),15b molecular sieves (MS 4 Å)16a and saccharose16b have also been used to achieve this transformation. Their diverse biological and pharmacological properties and high cost necessitate the search for economically efficient and environmentally benign synthetic methodology for the preparation of these heterocyclic compounds.Biocatalysis is a green chemistry approach to chemical transformations because of its high selectivity, mild reaction conditions, low energy requirements and few by-products.17–20 Biocatalytic promiscuity provides a new tool for extending the applications of enzymes in organic synthesis.21 Hence, the search for possible new biocatalysts to achieve environmentally friendly reactions continues, that avoid the use of hazardous organic solvents, toxic metals, and other harsh reaction conditions and produce different types of chemical and biological substance with higher yields.22 In the past few decades, several examples of exploiting biocatalysts like lipase,23a DNA,23b whole cell23c and laccase23d for catalyzing organic synthesis, have been demonstrated that offer high catalytic activity and simple operation. It is important to increase enzyme promiscuity and develop cost effective as well as green protocols for the synthesis of dihydropyrano[2,3-c]pyrazole compounds. The literature states that Bovine Serum Albumin (BSA) is an efficient biocatalyst in many organic reactions, which include the aerobic oxidative coupling of thiols into disulphides,24 nitro-aldol addition,25 Gewald reaction,26 stereoselective thio-Michael addition to chalcones,27 Knoevenagel condensation between diethylmalonate with aldehydes,28 and Biginelli reaction29 for the synthesis of valuable compounds. BSA or “Fraction V” is the most abundant, low cost serum albumin protein derived from cows. It binds several organic molecules, due to the presence of hydrophobic binding sites and it’s basic character, which are attractive features.30,31 Recently Bora et al.32 reported a biocatalytic route for the synthesis of dihydropyrano[2,3-c]pyrazoles from aldehydes and ketones using lipase from Aspergillus niger. However, there is scope left to explore BSA as a better biocatalyst. Therefore, the need to search for new biocatalysts with ecofriendly protocols, atom economy and broad substrate range is obvious. In general, ketones are less reactive than aldehydes and less explored, hence ketone based dihydropyrano[2,3-c]pyrazole synthesis was investigated.
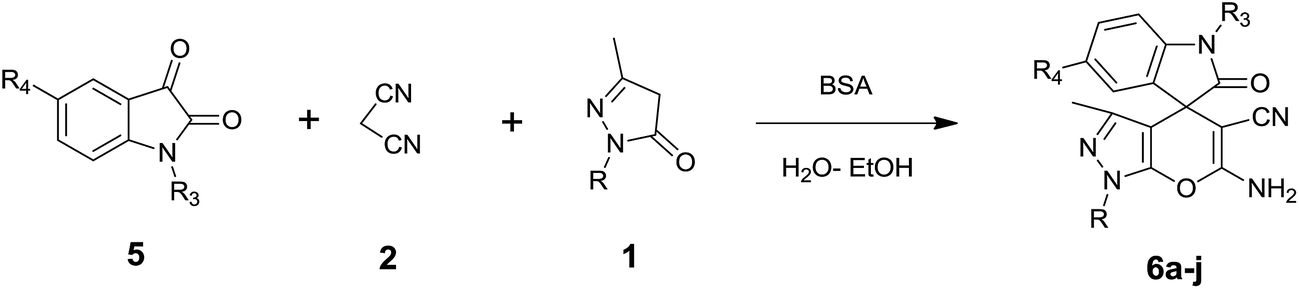
In continuation of our work on protein isolation33 and biomimetic heterocycle synthesis,34 recently we developed an efficient method for the synthesis of dihydropyrano[2,3-c]pyrazoles and spirooxindoles using β-cyclodextrin in an ethanol–water mixture.35 Herein, we report an highly efficient and environmentally benign protocol for the synthesis of dihydropyrano[2,3-c]pyrazoles and spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derived from aldehydes, ketones and isatins by using BSA at ambient temperature as a biocatalyst (Scheme 1).
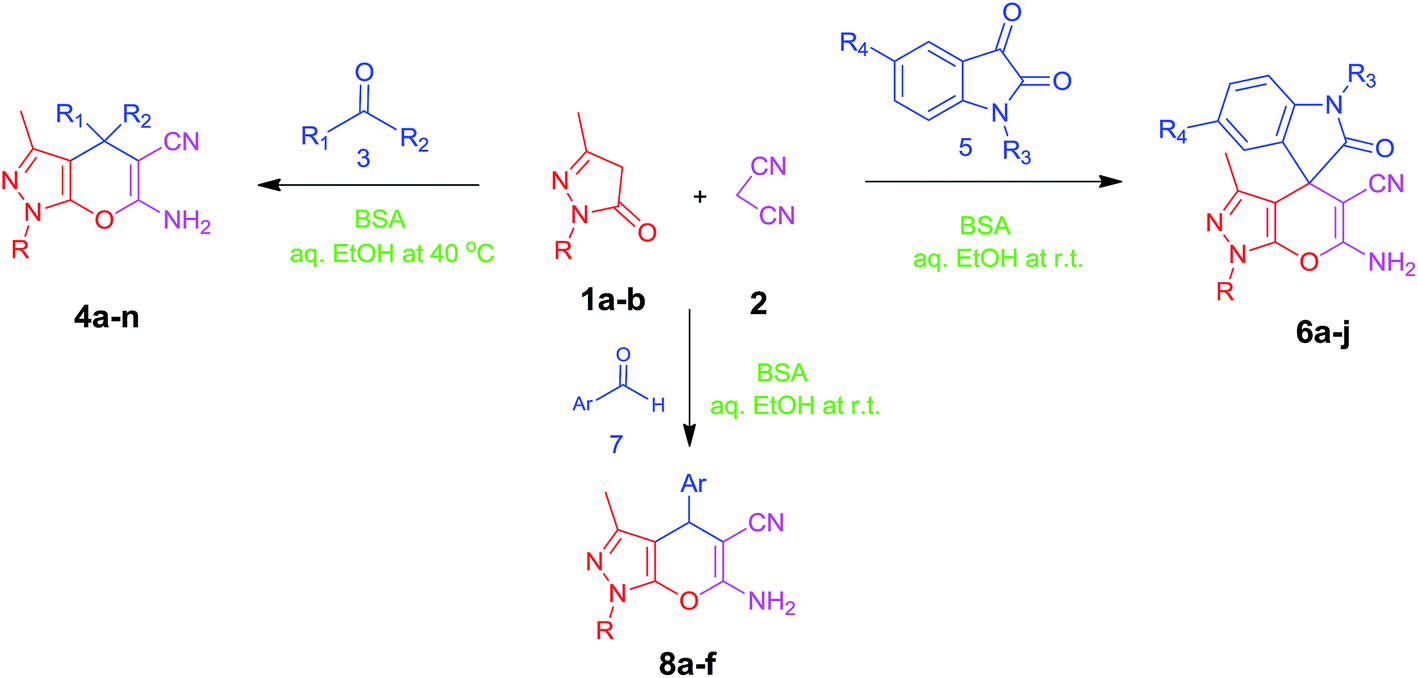 | ||
| Scheme 1 Synthesis of dihydropyrano[2,3-c]pyrazoles and spiro[indoline-3,4′-pyrano[2,3-c]pyrazoles in aqueous ethanol. | ||
Results and discussion
Screening of enzymes
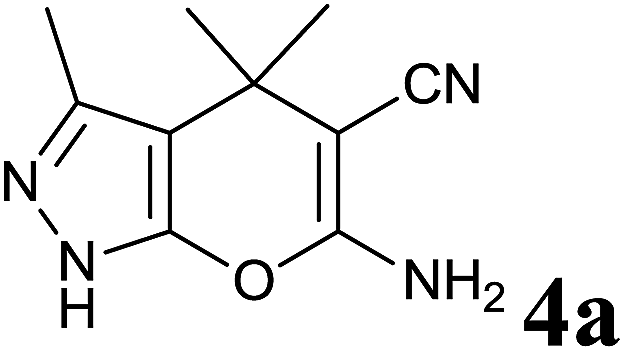
Different commercially available enzymes (purchased from Sigma-Aldrich and HiMedia) were selected to screen for their role as a catalyst in the synthesis of dihydropyrano[2,3-c]pyrazole compounds. We choose an equimolar mixture of acetone, malononitrile and pyrazolone. Acetone (2 mmol) and malononitrile (2 mmol) were mixed together in a round bottom flask containing a mixture of water and ethanol (7:3) and stirred vigorously for 10 min to make a homogeneous solution. Then pyrazolone (2 mmol) and BSA (60 mg) were added. The reaction was carried out at a temperature of 40 °C with continuous stirring for 1 h. The formation of product was monitored by thin layer chromatography (TLC). The product obtained was solid and insoluble in water. So, the reaction mixture was diluted with 10 mL of water and separated by filtration. Different enzymes were used to catalyze the synthesis of 4a and the results are summarized in Table 1.
|
||
|---|---|---|
| Entry | Enzyme | Yieldb (%) |
| a Reaction conditions: acetone (2 mmol), malononitrile (2 mmol), pyrazolone (2 mmol), BSA (60 mg) and H2O–EtOH (7:3, 10 mL) at 40 °C for 1 h.b Isolated yield. |
||
| 1 | No catalyst | 20 |
| 2 | Lipase from Candida rugosa | 13 |
| 3 | Lipase from porcine pancreas | 35 |
| 4 | Trypsin from bovine | 57 |
| 5 | Trypsin from porcine pancreas | 30 |
| 6 | Papain | 40 |
| 7 | Diastase α-amylase | 25 |
| 8 | Bovine serum albumin | 94 |
When lipase from Candida rugosa was used as a catalyst, only 13% yield was obtained in 1 h while lipase from porcine pancreas gave 35% yield. We also used some digestive enzymes like trypsin from bovine, for which surprisingly 57% yield was obtained but in the case of trypsin from porcine pancreas, the yield was decreased (30%). When papain was used 40% product formation was observed while diastase α-amylase gave a lower yield (25%). If no catalyst was added, only 20% yield was detected. When BSA was used, the yield increased dramatically to 94%, probably bought about by catalysis due to the rich diversity of amino acids present on the surface of BSA.36
Optimization of solvent
The reaction medium plays an important role in enzyme catalyzed reactions owing to its effects on enzyme activity as well as substrate solubility.37a The effects of different solvents with different polarities on a model reaction were investigated. The results in Table 2 exhibit the diverse effects of different solvents on the activity of BSA in the reaction. Independently, water gave ∼39% yield after 1 h whereas the rest of the solvents such as ethanol, methanol, acetonitrile, THF, and DMF gave trace yields. However, it was observed that mixing water and ethanol at different ratios enhances the yield of the reaction. The best result was obtained by using a mixture of water and ethanol (7:3) with a highest yield of 94%. Therefore, BSA in a water and ethanol mixture (7:3) was used to carry out further reactions.
|
||
|---|---|---|
| Entry | Solvent | Yieldb (%) |
| a Reaction conditions: acetone (2 mmol), malononitrile (2 mmol), pyrazolone (2 mmol), BSA (60 mg) and H2O–EtOH (7:3, 10 mL), reaction time: 1 h at 40 °C.b Isolated yield. |
||
| 1 | Water | 39 |
| 2 | Ethanol | Trace |
| 3 | Methanol | Trace |
| 4 | Acetonitrile | Trace |
| 5 | THF | Trace |
| 6 | DMF | Trace |
| 7 | H2O:EtOH (1:1) |
Trace |
| 8 | H2O:EtOH (9:1) |
43 |
| 9 | H2O:EtOH (8:2) |
50 |
| 10 | H2O:EtOH (7:3) |
94 |
Effect of catalyst loading
In order to optimize the catalyst loading of BSA, a model reaction was performed with variation of the quantity of BSA between 20 to 70 mg per mmol of substrate (Fig. 1) while the rest of the reaction conditions were kept the same. It was observed that an increase in enzyme concentration up to 60 mg increased the yield of reaction, however, no significant improvement in the yield of the reaction was observed further. The best yield of 94% was obtained with 60 mg of BSA per 2 mmol of substrate in a duration of 1 h.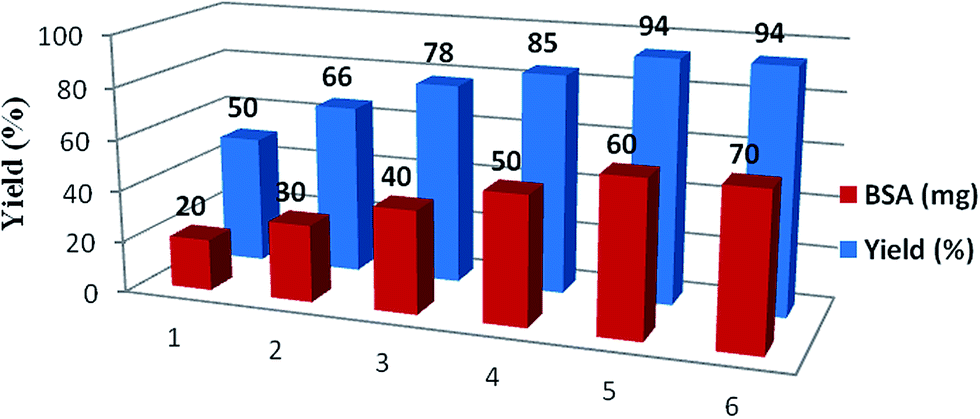 | ||
| Fig. 1 Study of the amount of catalyst on reaction yields. | ||
Effect of temperature
Temperature is an important factor in every biocatalytic reaction. When the reaction temperature is increased, the collisions between enzyme and substrate molecules may be accelerated to form enzyme–substrate complexes resulting in an improved reaction rate.38 In the present study, the reaction temperature was changed from 20 to 50 °C to investigate its effect. The results shown in Fig. 2 indicate that the yield was greatly improved by raising the temperature from 20 to 30 °C, and reached a maximum of 94% at 40 °C in a duration of one hour; further increasing the temperature to 50 °C resulted in the same yield.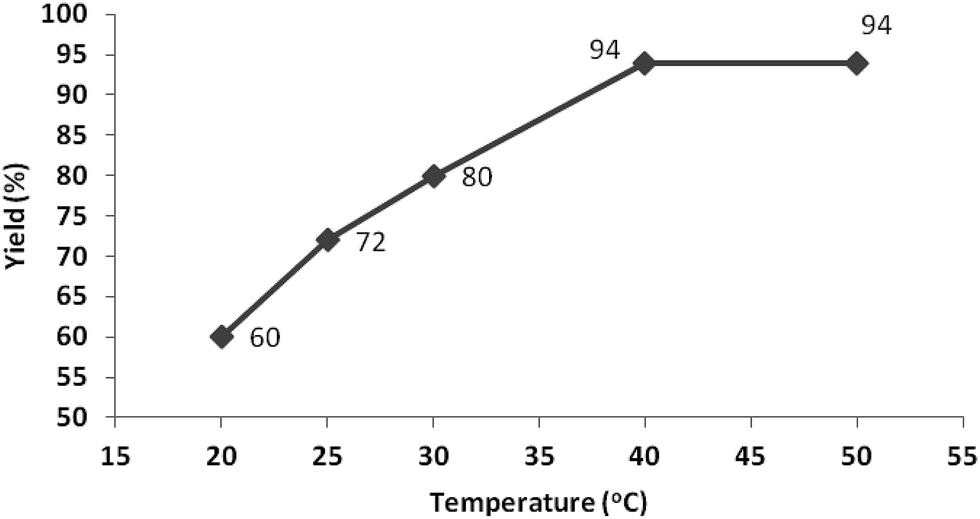 | ||
| Fig. 2 Study of the effect of temperature on reaction yields. | ||
Substrate scope
Having set optimized parameters, the substrate scope for BSA-catalyzed dihydropyrano[2,3-c]pyrazole synthesis was investigated by varying the nature of ketones. As shown in Table 3, the BSA-catalyzed reaction proceeded smoothly with aliphatic, cyclic and aromatic ketones. The chain length of ketone showed a great influence on the reaction time. In the case of acetone (entry 1, Table 3), compound 4a was obtained in excellent yield of 94% in one hour, while when 2-butanone was used as a ketone under the same conditions, a slightly decreased yield of 90% was observed (entry 2, Table 3) in 15 h. When ethyl acetoacetate and methyl acetoacetate (entries 3–4, Table 3) were used as a ketone, 94% and 95% yields in 12 h and 15 h respectively were obtained for compounds 4c and 4d. This indicates that BSA chemoselectively reacts with the ketone carbonyl in the presence of an ester carbonyl group. Furthermore, the ring size of cyclic ketones might be exerting an obvious influence. For example, the yield of the reaction between cyclohexanone, malononitrile and pyrazolone after 21 h was 80% (entry 5, Table 3). But, when cyclopentanone was used as a substrate, the yield dramatically increased to 86% (entry 6, Table 3) in only 10 h. This phenomenon might be ascribed to higher internal ring strain. When the methodology was tested on aromatic ketones (entries 7–12, Table 3) with electron donating and electron withdrawing substituents, reaction times for complete conversion to products were found to be higher, with good yields, than the previously reported method.32
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ketone | Product | Time (h) | Yieldb (%) | MP (°C)[Lit.] | |
| Found | Reported | |||||
| a Reaction conditions: ketone (2 mmol), malononitrile (2 mmol), pyrazolone (2 mmol), BSA (60 mg) and H2O–EtOH (7:3, 10 mL) at 40 °C.b Isolated yield. |
||||||
| 1 | 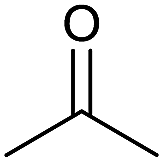 |
 |
1 | 94 | 174–176 | 176–177 (ref. 14b) |
| 2 | 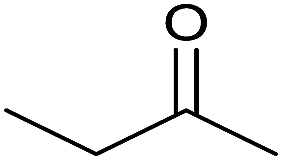 |
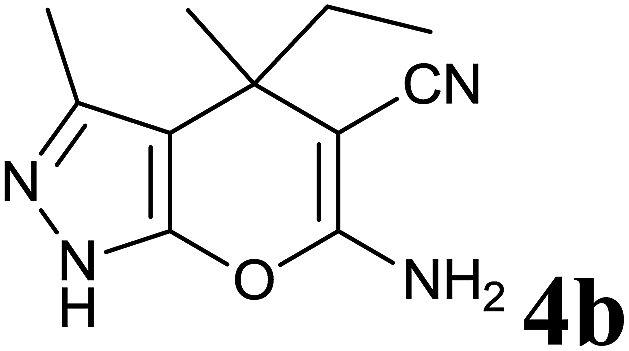 |
15 | 90 | 164–166 | — |
| 3 | 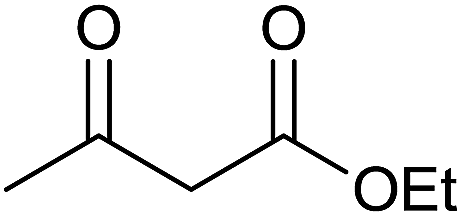 |
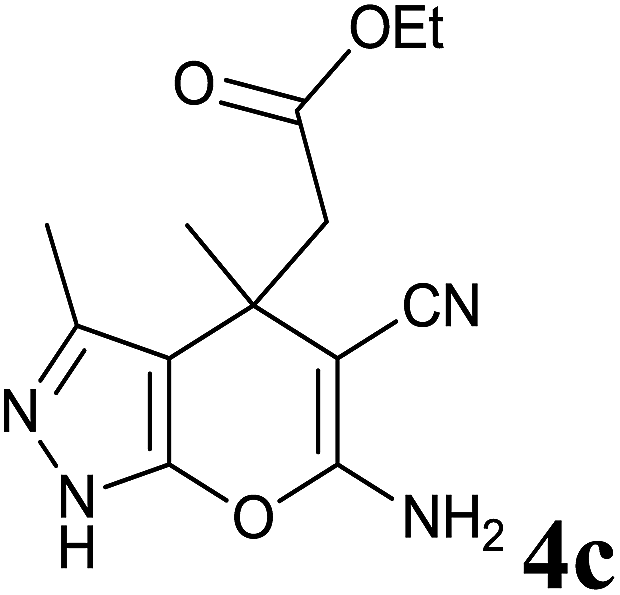 |
12 | 94 | 204–206 | — |
| 4 | 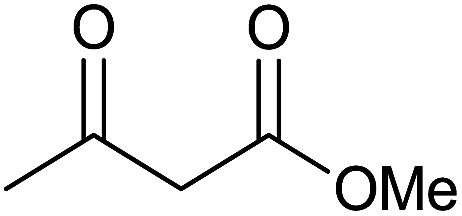 |
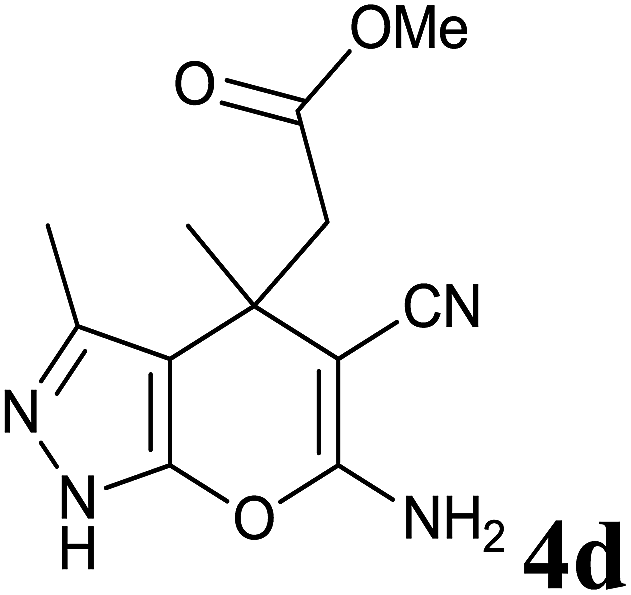 |
15 | 95 | 200–202 | — |
| 5 | 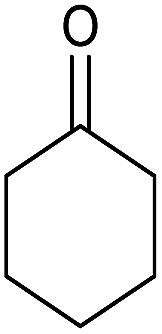 |
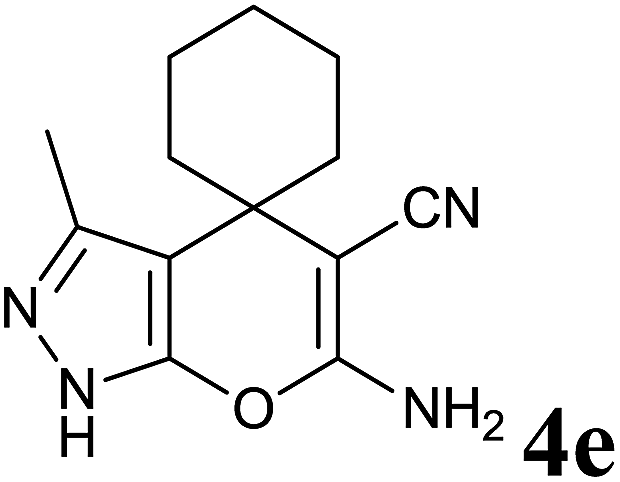 |
21 | 80 | 146–148 | 147–148 (ref. 14b) |
| 6 | 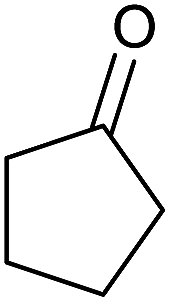 |
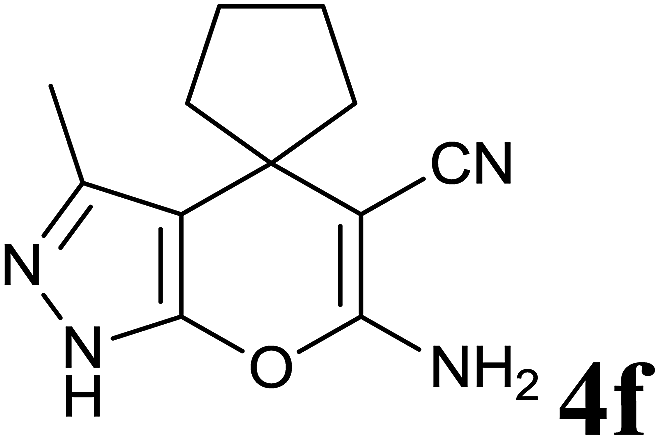 |
10 | 86 | 164–166 | 164–165 (ref. 14b) |
| 7 | 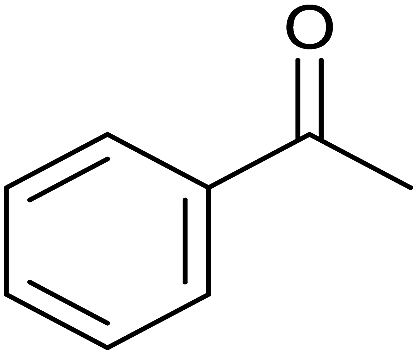 |
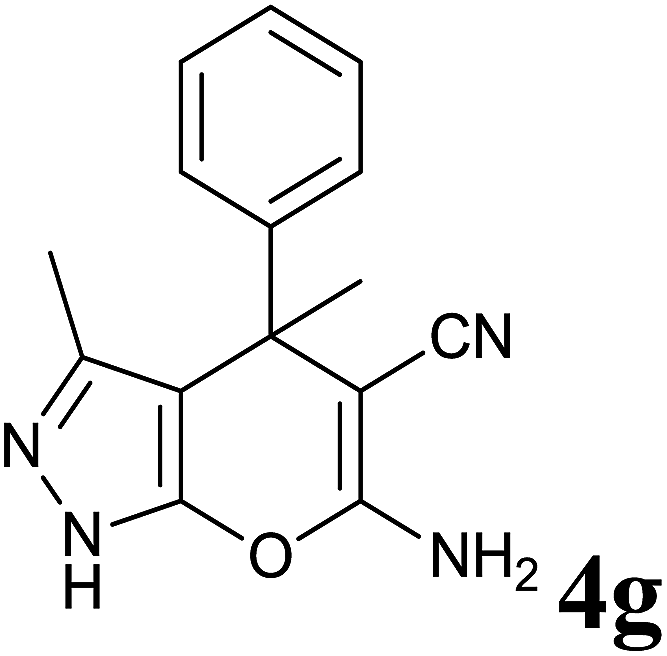 |
30 | 94 | 175–176 | 174–175 (ref. 32) |
| 8 | 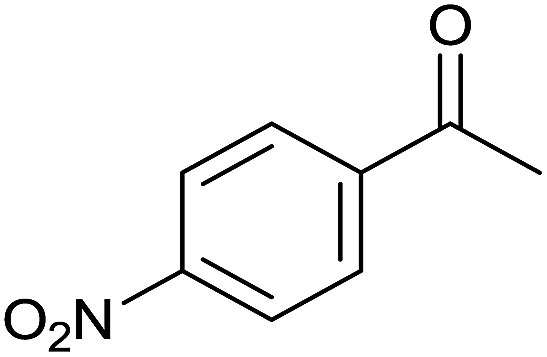 |
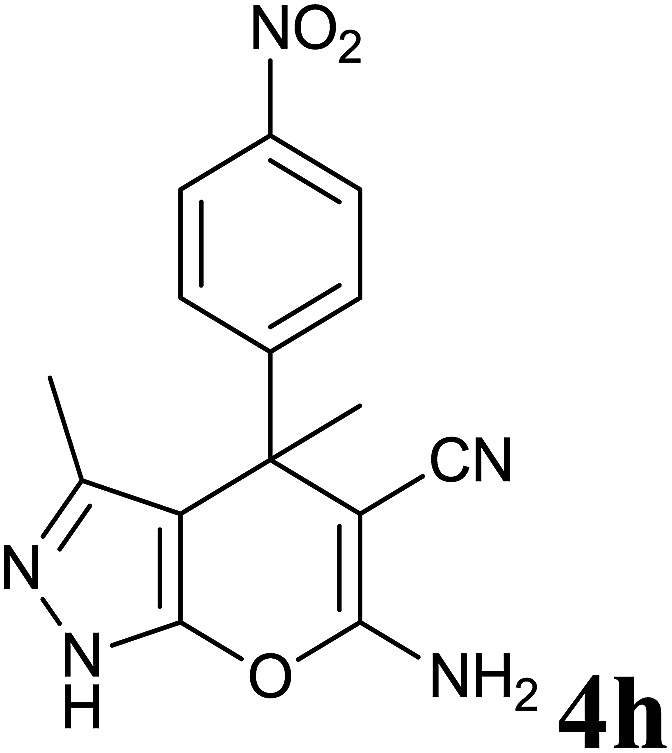 |
36 | 86 | 210–212 | 220–221 (ref. 14b) |
| 9 | 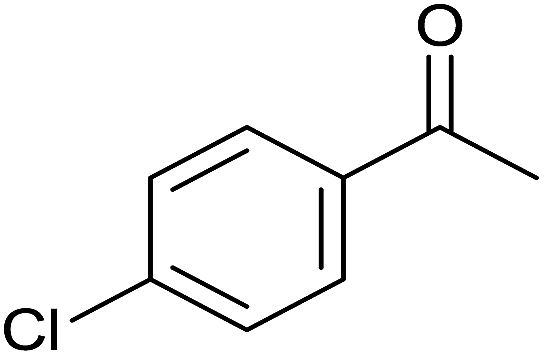 |
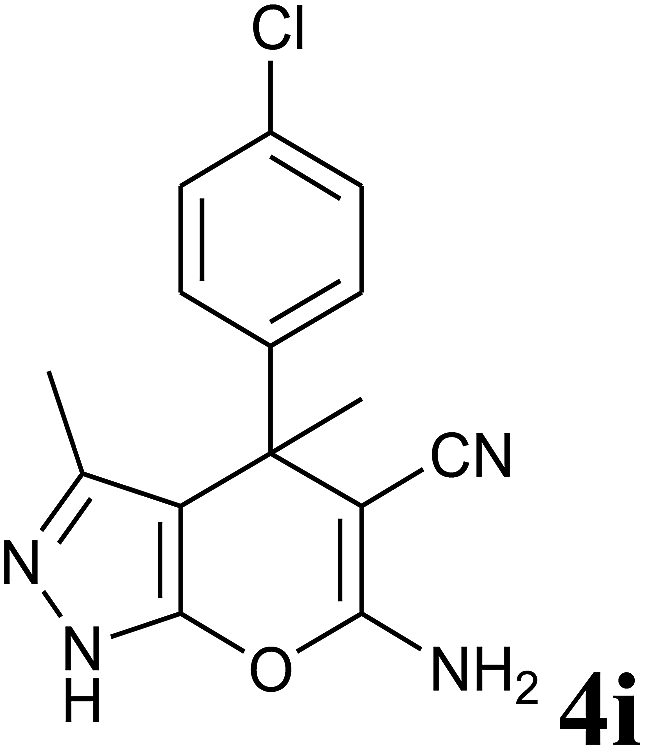 |
24 | 95 | 182–184 | 181–182 (ref. 13) |
| 10 | 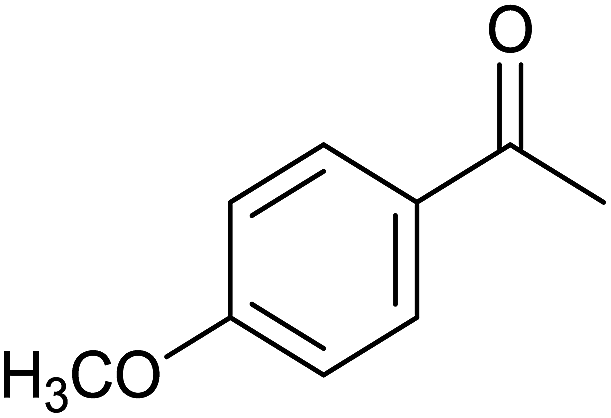 |
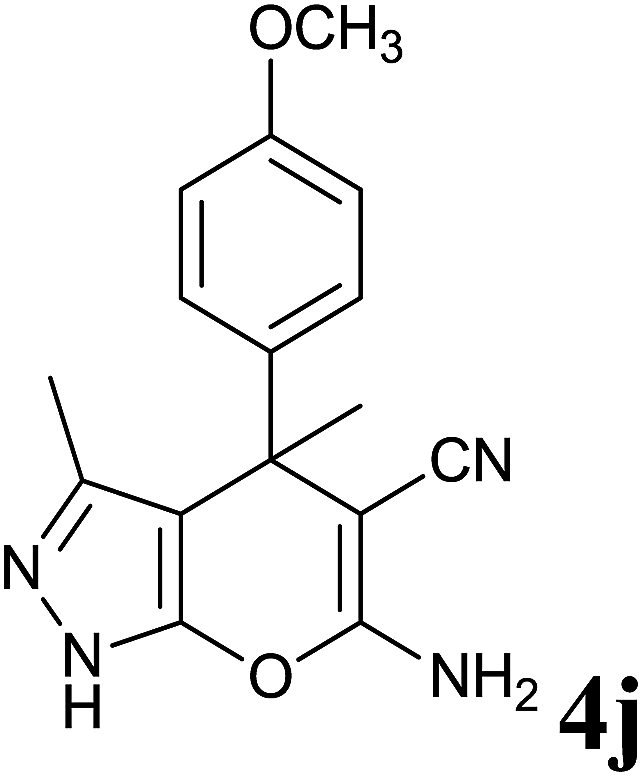 |
48 | 76 | 193–194 | 194–195 (ref. 13) |
| 11 | 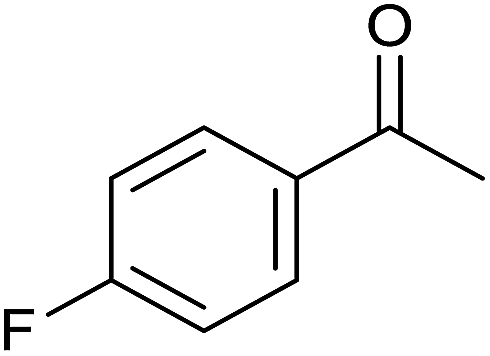 |
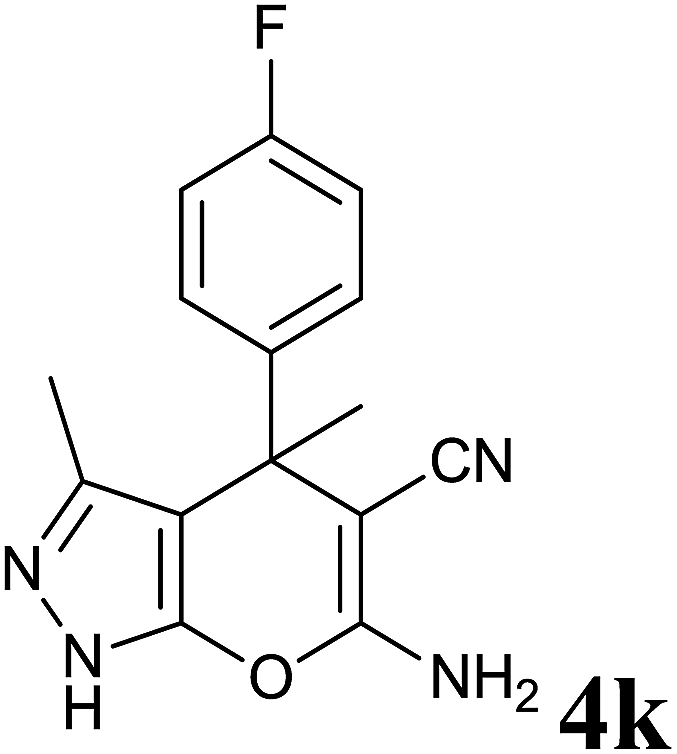 |
36 | 83 | 208–210 | — |
| 12 | 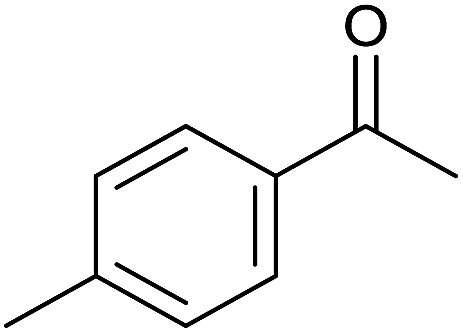 |
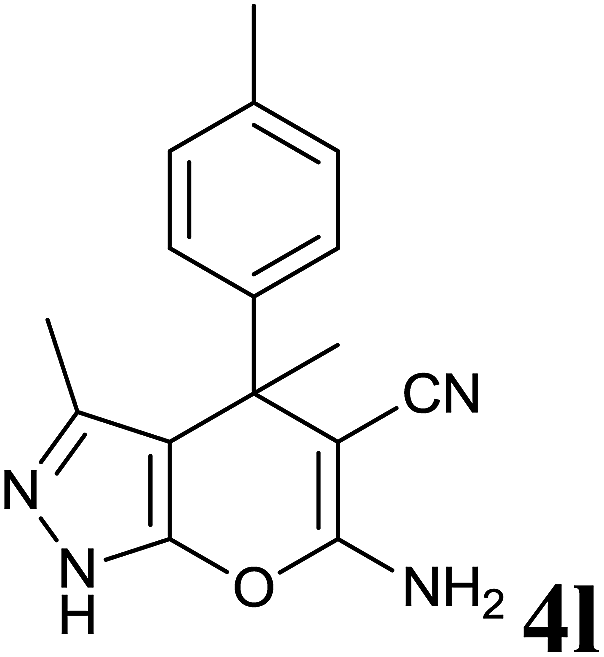 |
48 | 86 | 182–183 | 183–184 (ref. 13) |
| 13 | 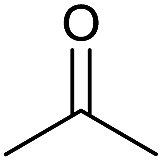 |
 |
24 | 72 | 140–142 | — |
| 14 | 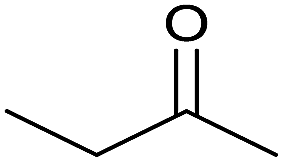 |
 |
24 | 85 | 158–160 | — |
The efficiency of the protocol on a multigram scale was established by using equimolar (100 mmol) amounts of acetone, malononitrile and pyrazolone. Acetone (100 mmol, 5.8 g) and malononitrile (100 mmol, 6.6 g) were mixed together in a round bottom flask containing a 200 mL mixture of water and ethanol (7:3) and stirred vigorously for 10 min to make a homogeneous solution. Then pyrazolone (100 mmol, 9.8 g) and BSA (3 g) were added and stirred at 40 °C for 1 h. The reaction promptly proceeded to afford 4a in 95% yield (19.4 g).
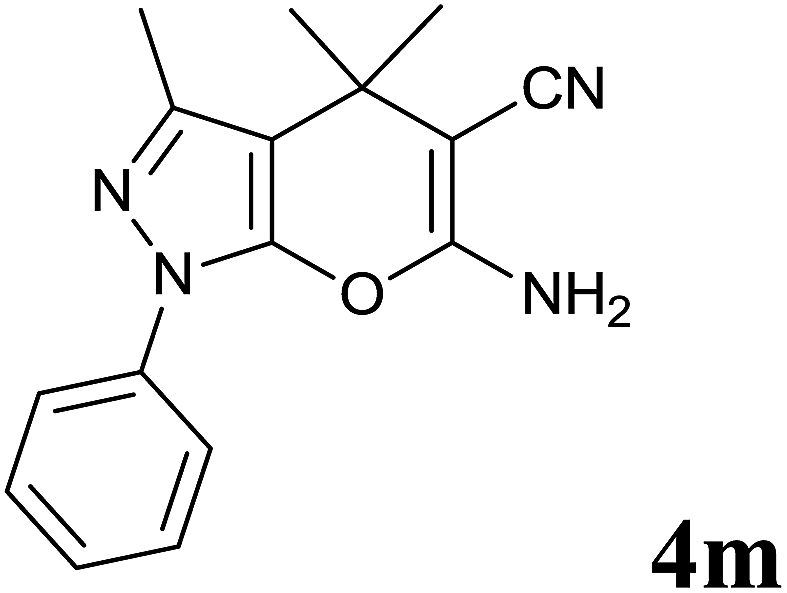
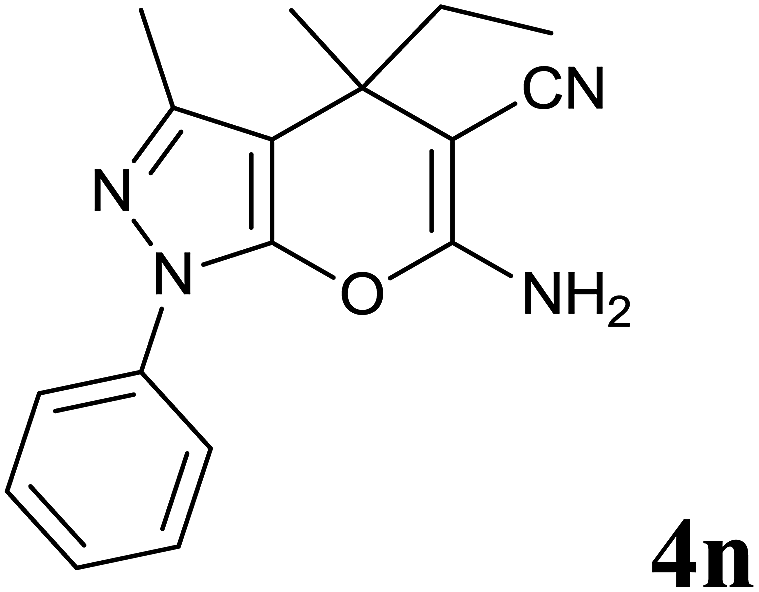
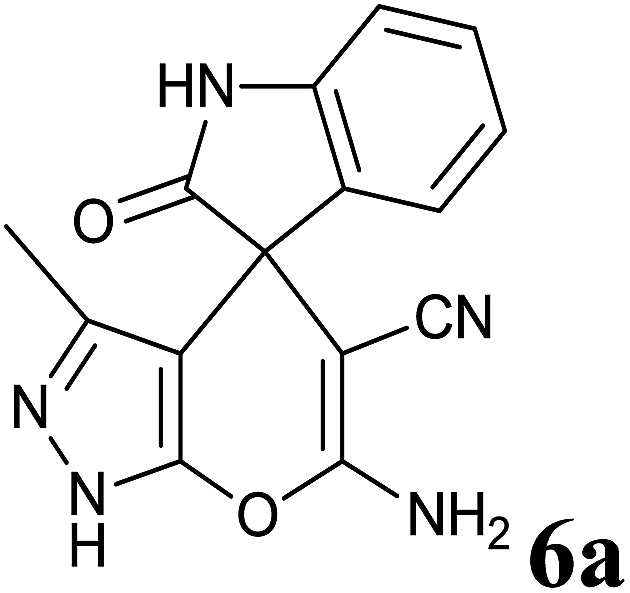
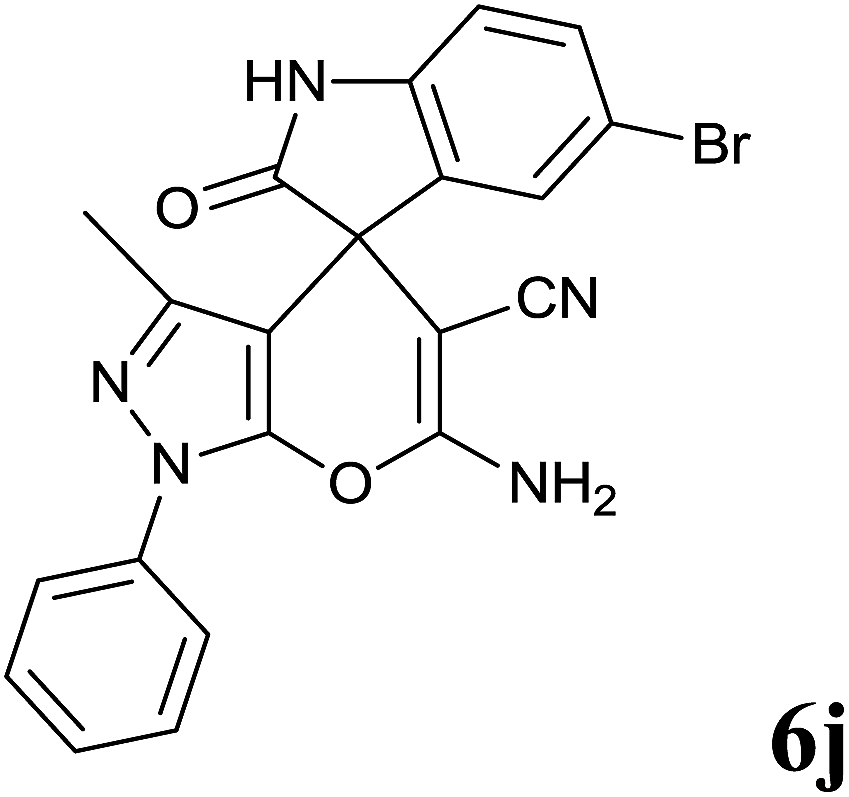
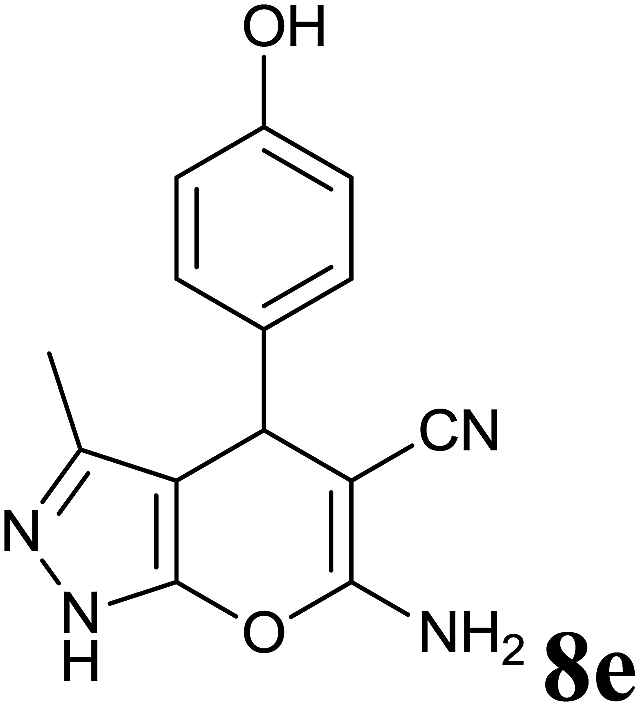
Isatin is an important molecule for designing potential pharmacological agents, and its derivatives have shown a broad range of bioactivities and have been involved in the synthesis of various heterocyclic compounds.39 Spiro compounds are naturally occurring compounds present in many pharmacological agents and natural alkaloids.40 The indole nucleus shows antibacterial and antifungal activities and it is an important heterocyclic agent present in diverse types of medicinal agents and natural products.41 These interesting properties prompted us to widen the applicability of this procedure with isatins. To explore the scope, we used isatin as a ketone substrate to react with malononitrile, and 3-methyl-1H-pyrazol-5(4H)-one (1a) under the previously optimized conditions. Interestingly, reactions with isatin proceed speedily at room temperature to spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] with excellent yields (Table 4). The reactions for isatin, 5-chloroisatin and 5-bromoisatin (entries 1–3, Table 4) were completed in 15 minutes where as N-alkyl isatins (entries 4–7, Table 4) required slightly longer reaction times. Overall, reactions with isatin resulted in excellent yields within 35 minutes, except for 5-bromoisatin which resulted in a somewhat lower yield of 84%. However, when a four component, one pot reaction was attempted using acetone, malononitrile, hydrazine and ethyl acetoacetate, the reaction was completed in a longer time with lower yields. In order to explore substrate scope, the carbonyl compounds malononitrile and 3-methyl-1-phenyl-1H-pyrazol-5(4H)-one (1b) were reacted in an aqueous ethanol medium to give the products 4m–n and 6h–j in good yields. X-ray studies and the molecular structure of pyranopyrazole (4c and 6e) revealed that it exists in the crystal phase as a 2H, instead of a 1H tautomer as shown in Fig. S1 and S2 (see ESI†), which is in congruence with the literature reports on pyranopyrazoles.43
|
||||||
|---|---|---|---|---|---|---|
| Sr. No. | Isatin | Product | Time (min) | Yieldb (%) | MP | |
| Found | Ref. | |||||
| a Reaction conditions: isatin (2 mmol), malononitrile (2 mmol), pyrazolone (2 mmol), BSA (60 mg) and H2O–EtOH (7:3, 10 mL) at rt.b Isolated yield. |
||||||
| 1 |  |
 |
15 | 94 | 284–286 | 286–287 (ref. 14b) |
| 2 | 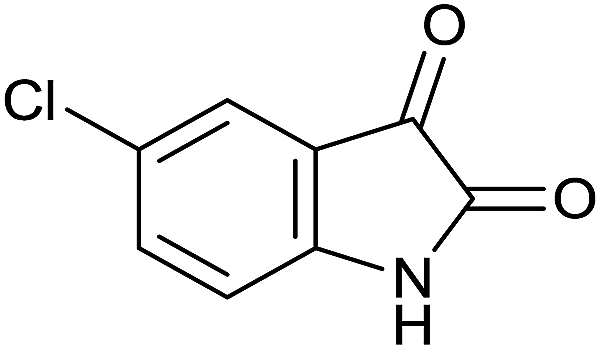 |
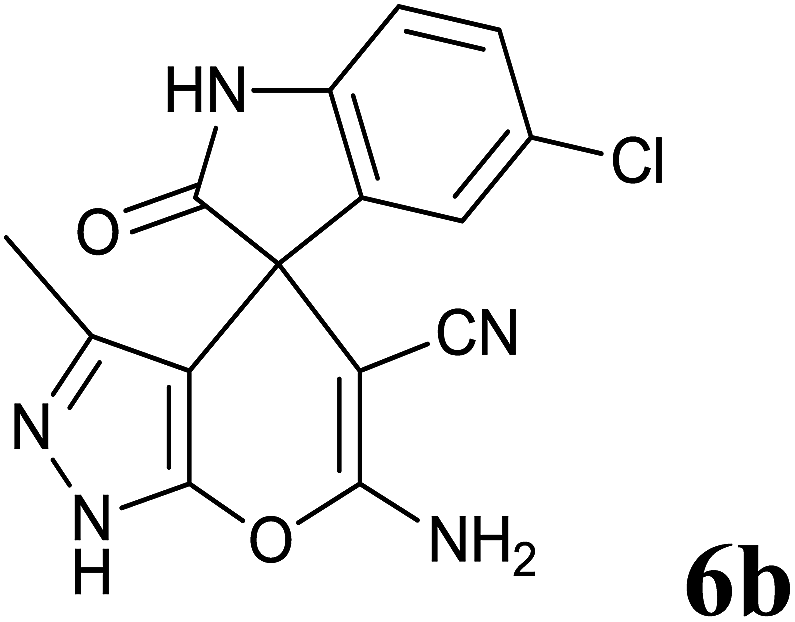 |
15 | 98 | 296–298 | 298–299 (ref. 14b) |
| 3 | 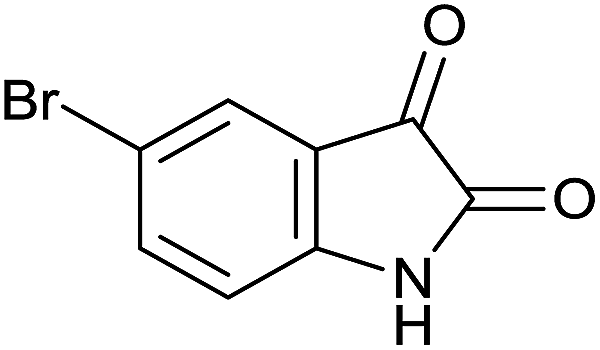 |
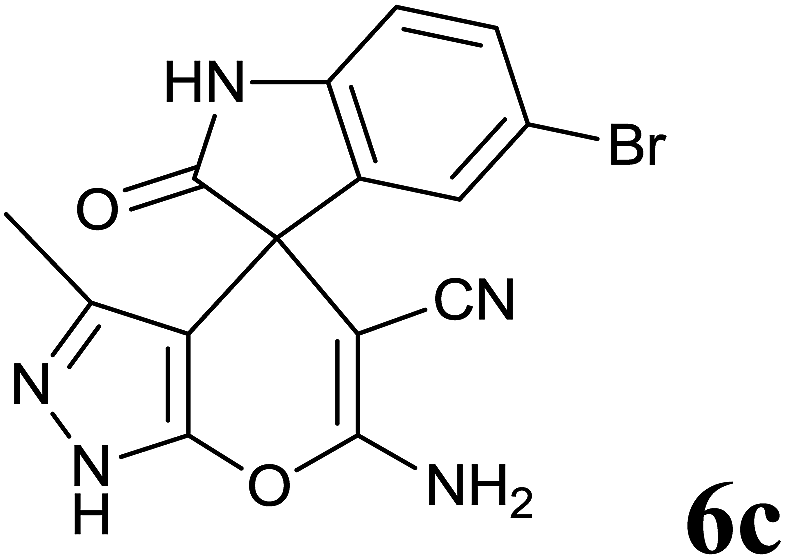 |
15 | 84 | 278–280 | 283–284 (ref. 14b) |
| 4 | 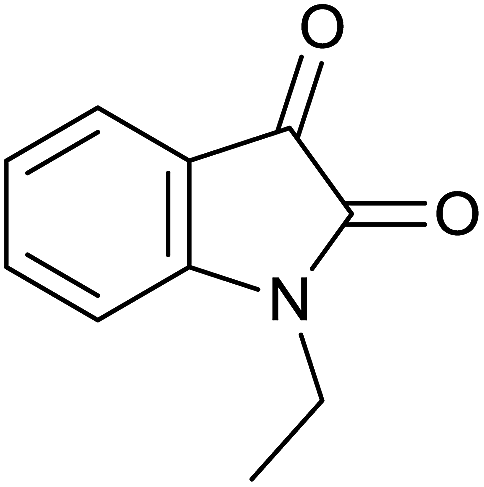 |
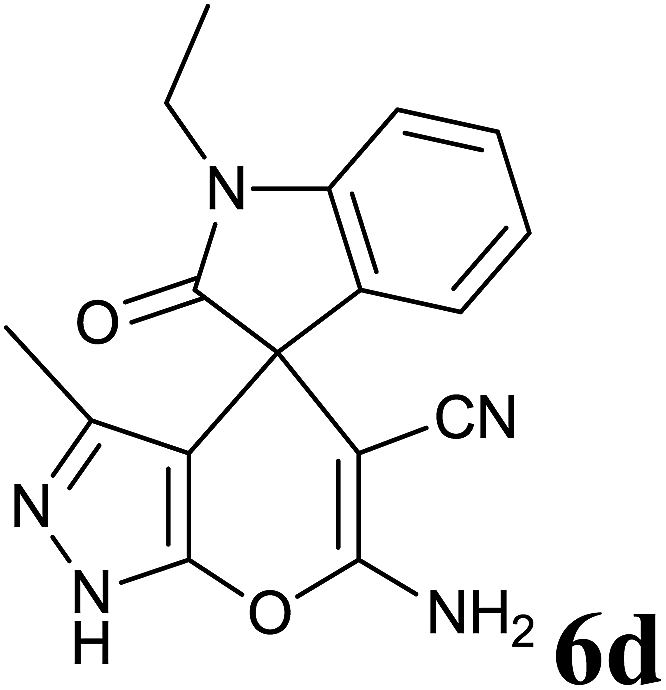 |
30 | 90 | 284–286 | 284–286 (ref. 35) |
| 5 | 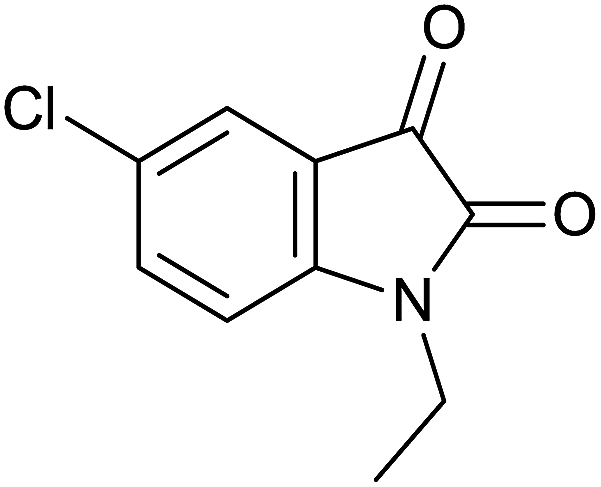 |
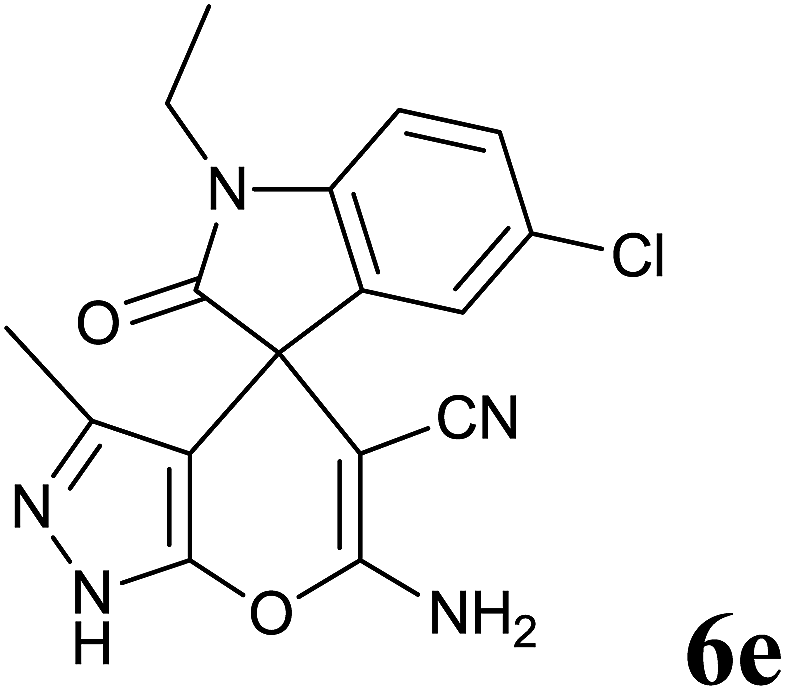 |
35 | 93 | 267–268 | — |
| 6 | 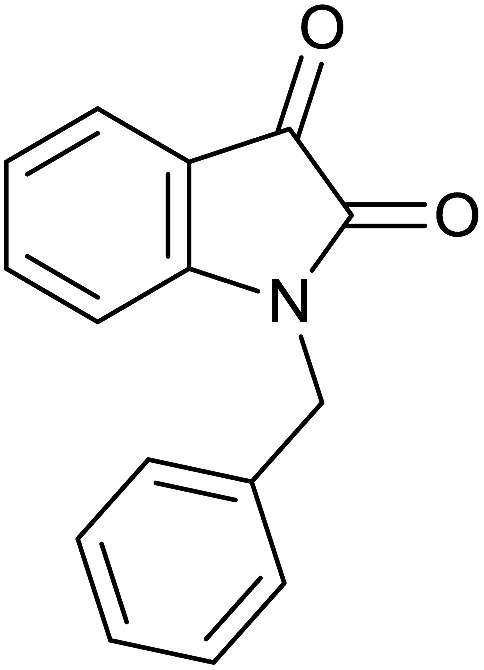 |
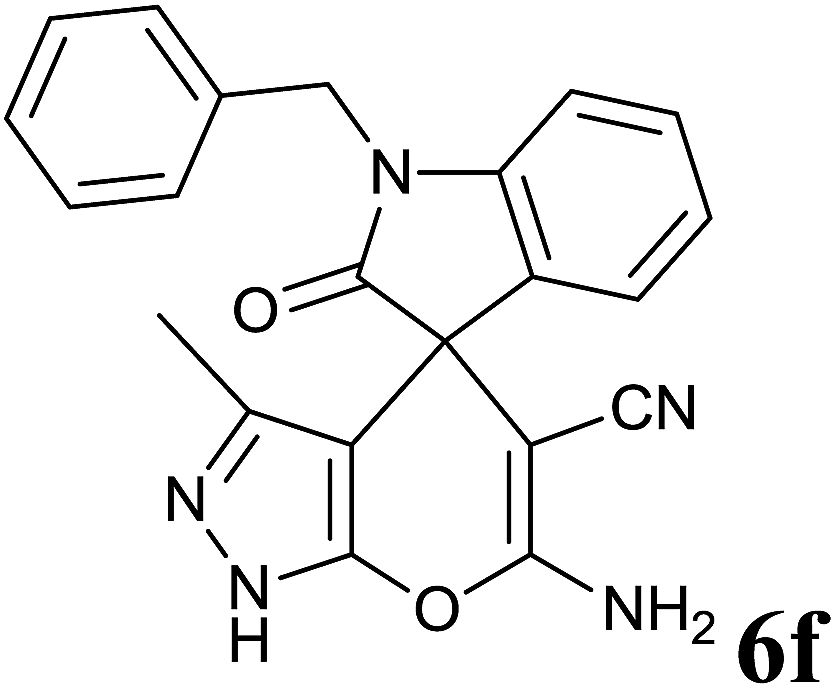 |
20 | 94 | 274–276 | 276–278 (ref. 35) |
| 7 | 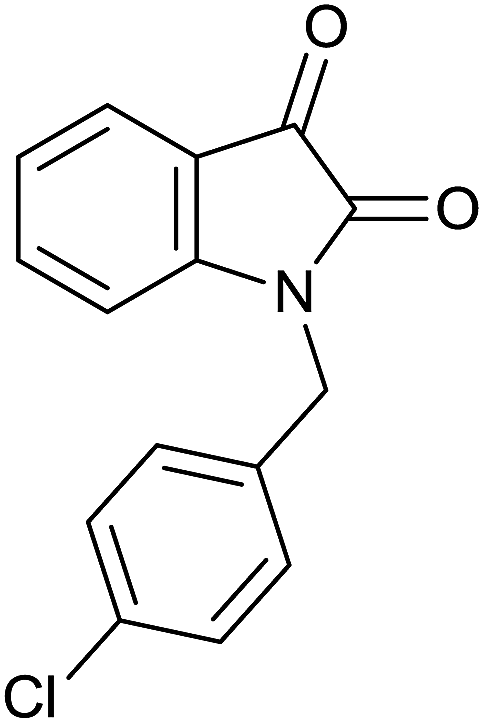 |
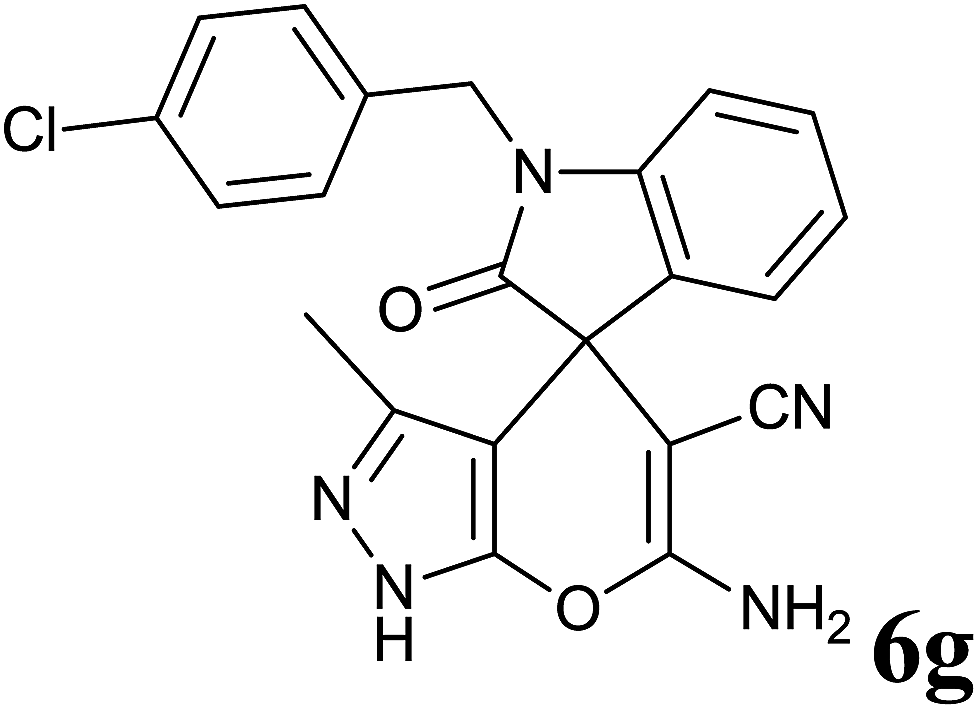 |
25 | 97 | 274–276 | — |
| 8 | 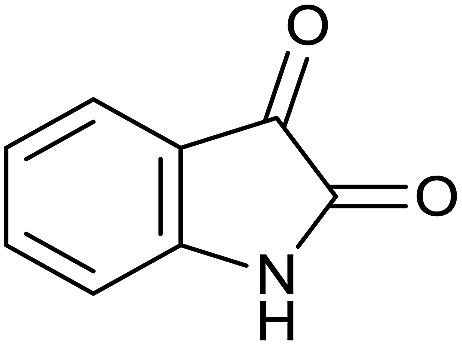 |
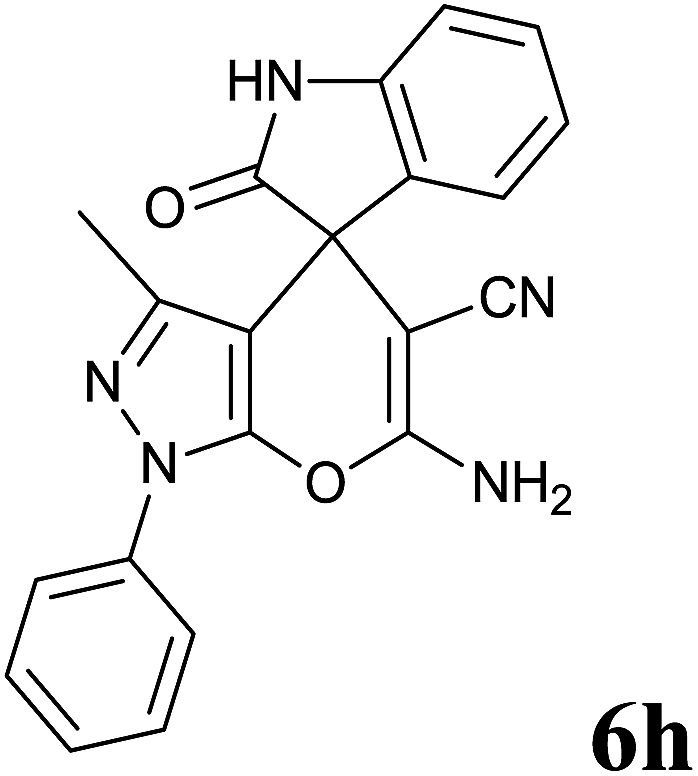 |
30 | 98 | 226–228 | 228–230 (ref. 37b) |
| 9 | 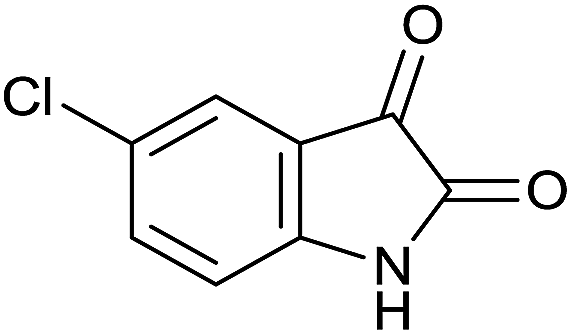 |
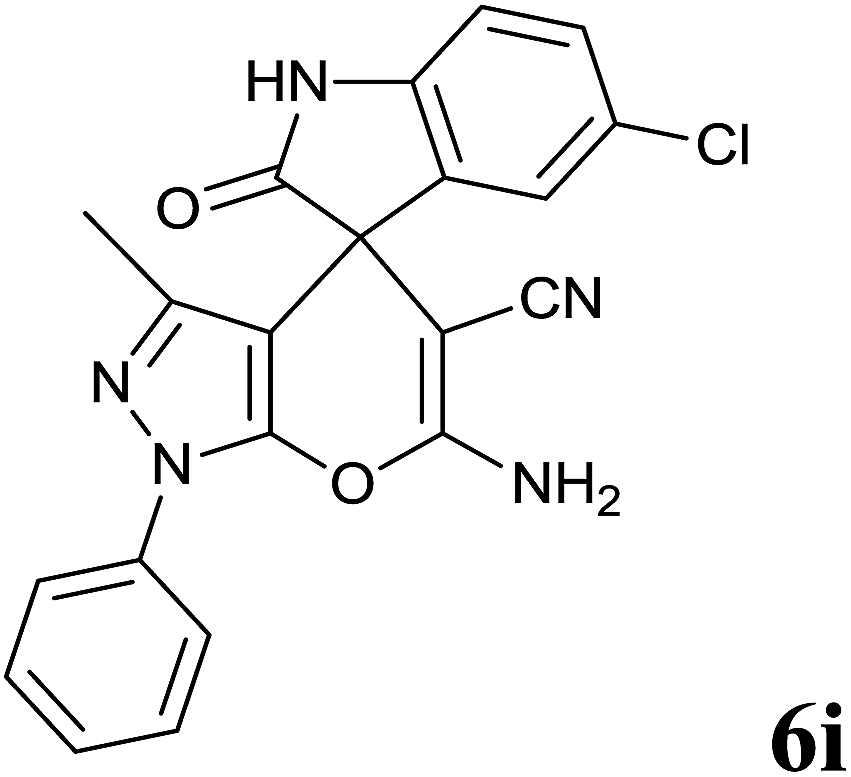 |
30 | 93 | 230–232 | 232–234 (ref. 37c) |
| 10 | 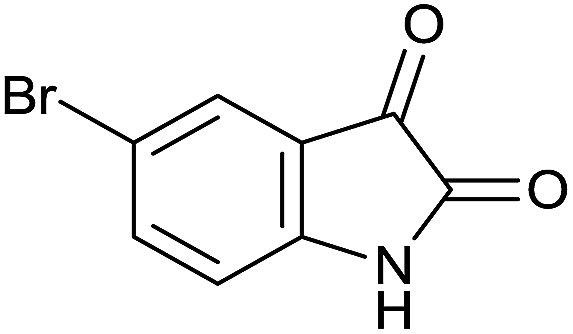 |
 |
35 | 89 | 230–232 | 232–234 (ref. 37c) |
Inspired by the initial success above, we generalized the applicability of this method for the synthesis of dihydropyrano[2,3-c]pyrazoles from various aromatic aldehydes. Interestingly, a variety of aromatic aldehydes including those with electron withdrawing and donating abilities participated well in this reaction and gave good to excellent yields (Table 5).
| Sr. No. | Aldehyde | Product | Time (h) | Yieldb (%) | MP | |
|---|---|---|---|---|---|---|
| Found | Reference | |||||
| a Reaction conditions: aldehyde (2 mmol), malononitrile (2 mmol), pyrazolone (2 mmol), BSA (60 mg) and H2O–EtOH (7:3, 10 mL) at rt.b Isolated yield. |
||||||
| 1 | 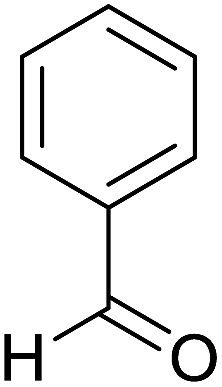 |
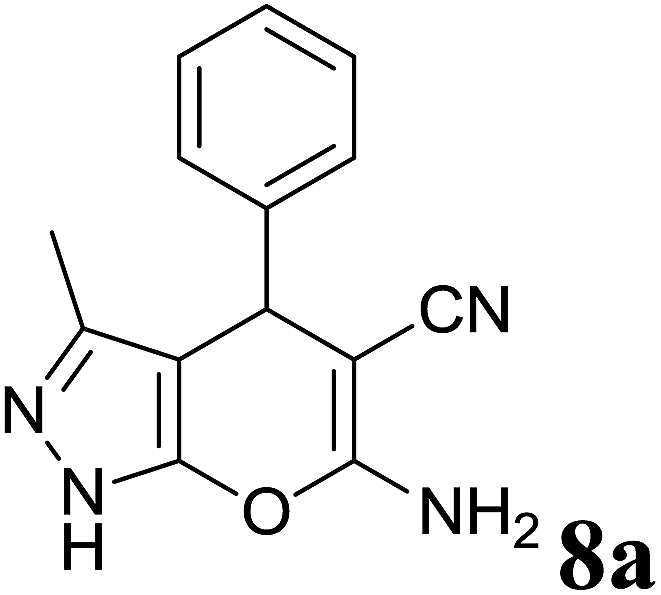 |
1.5 | 94 | 244–246 | 244–246 (ref. 35) |
| 2 | 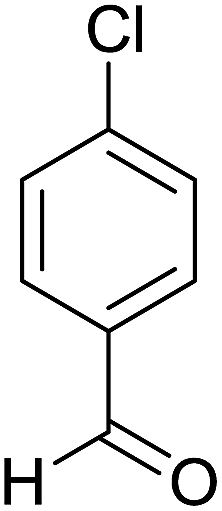 |
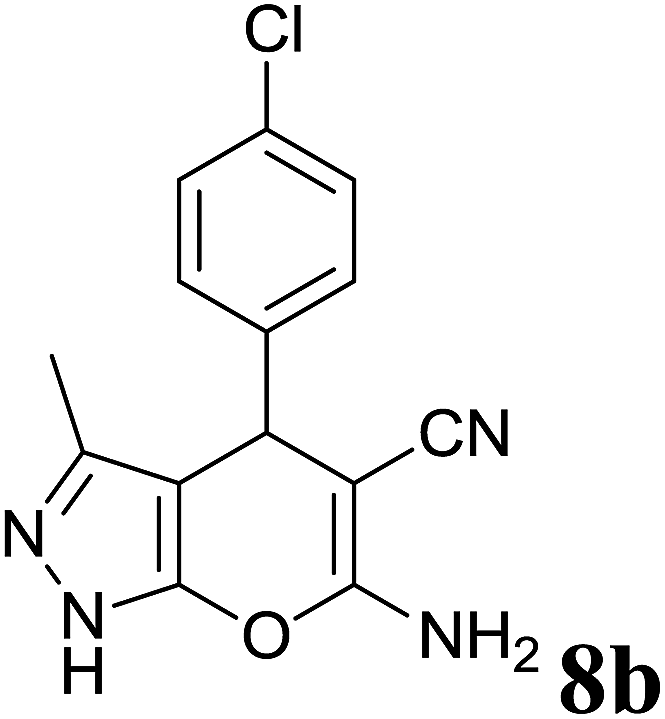 |
2 | 88 | 236–238 | 234–236 (ref. 35) |
| 3 |  |
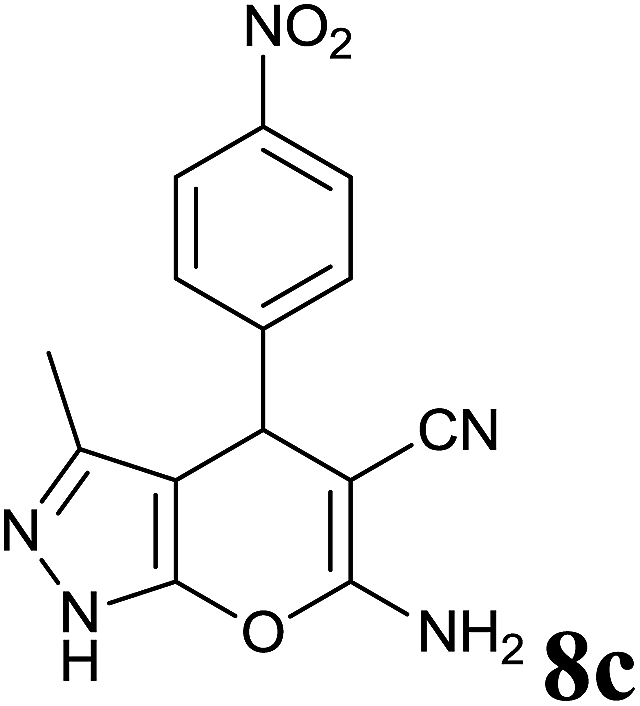 |
2.5 | 82 | 246–248 | 248–250 (ref. 35) |
| 4 | 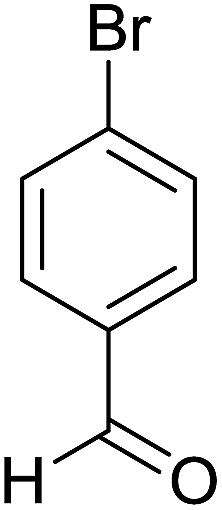 |
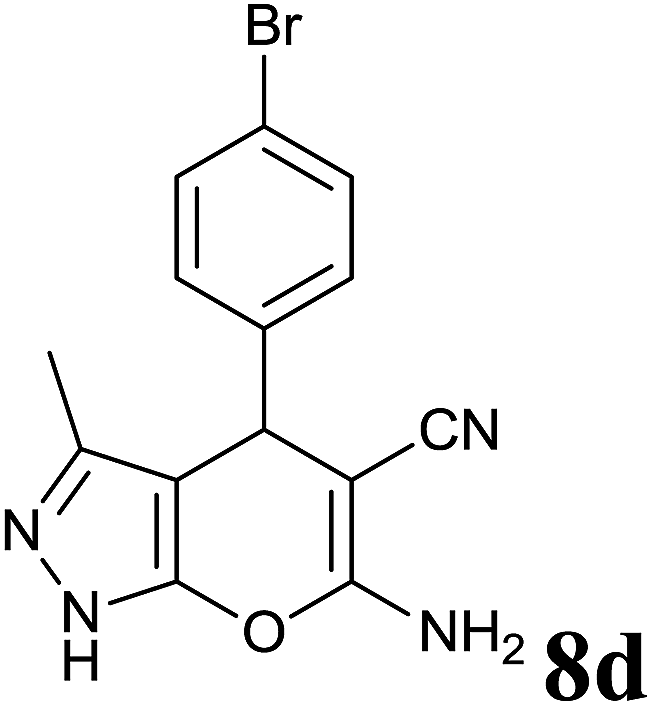 |
2 | 95 | 178–180 | 178–180 (ref. 35) |
| 5 | 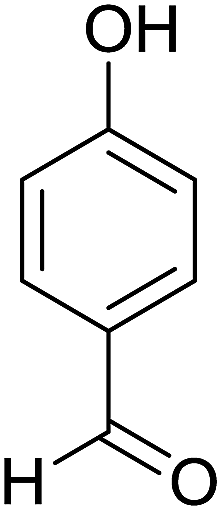 |
 |
2 | 97 | 218–220 | 223–224 (ref. 35) |
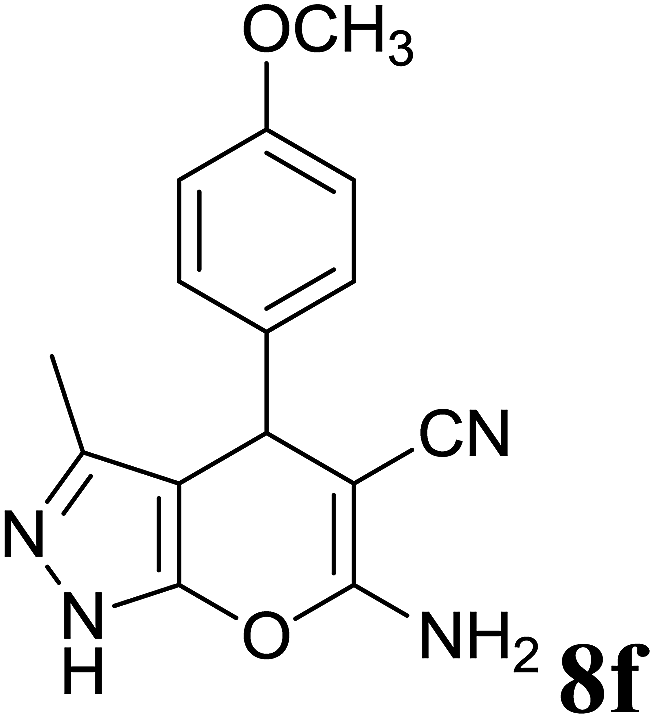
| 6 | 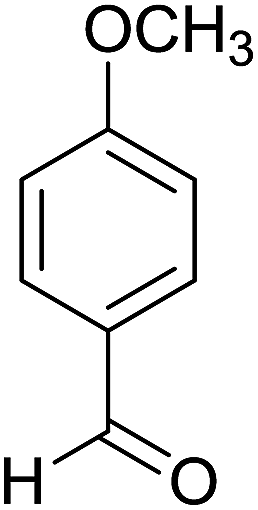 |
 |
3 | 94 | 210–212 | 210–212 (ref. 35) |
Test of recyclability
The enzyme BSA remained in the aqueous medium after the reaction. This aqueous medium was reused to recycle BSA for three runs for which the product isolated was good enough, with the potential to make it cost effective, as shown in Fig. 3. A slight decrease in the yield of product was observed in the first, second and third reaction runs (92%, 88% and 82% respectively).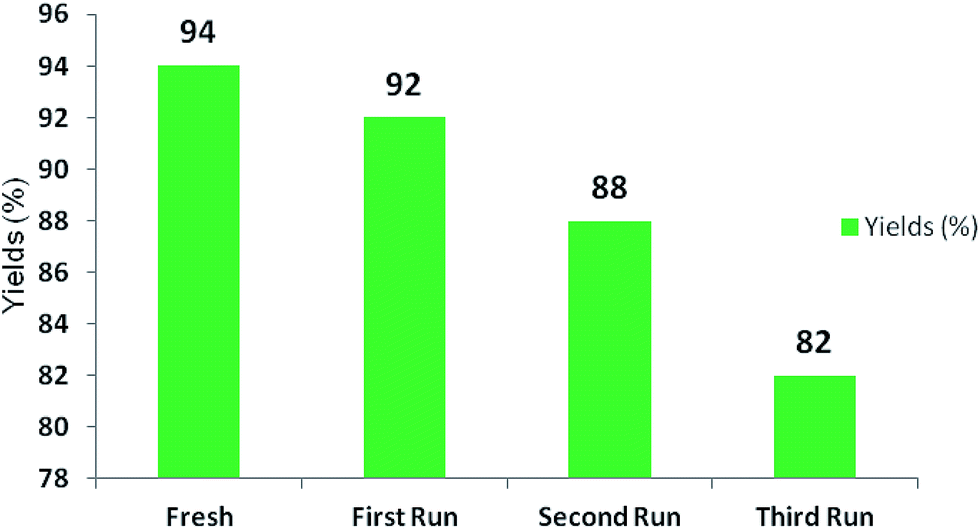 | ||
| Fig. 3 Recycling studies of BSA and the effect on yields. | ||
Plausible mechanism
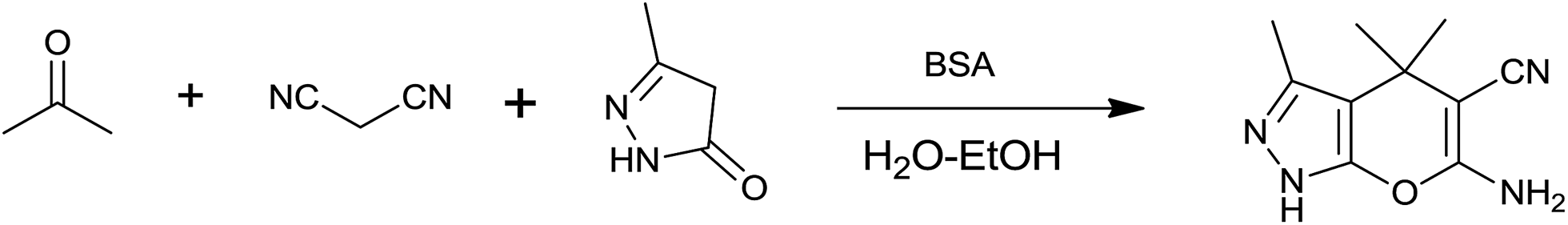
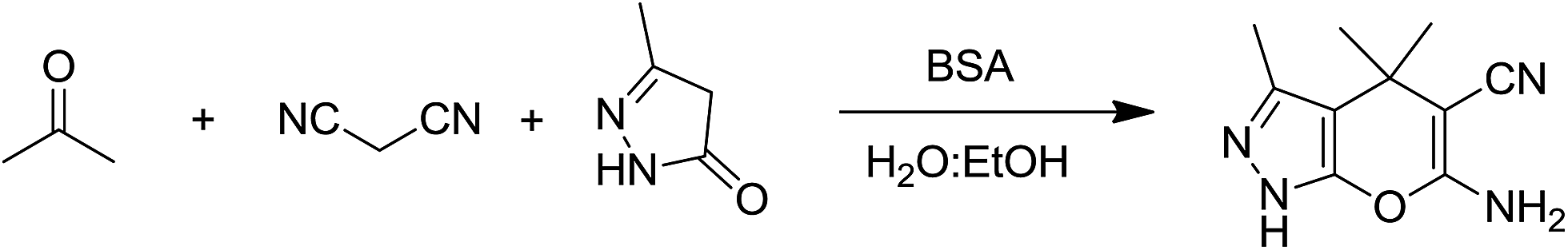
On the basis of the above results and previous studies, a plausible mechanism is proposed for the synthesis of dihydropyrano[2,3-c]pyrazole from ketones. According to the literature reports,42 the catalytic activity of BSA is based on the basic character of the amino groups present in the side chains of some amino acid residues, especially the lysine residue. It is proposed that firstly, the lysine’s amine attacks the carbonyl group of a ketone leading to the formation of iminium ion 3. At same time, the removal of a proton from malononitrile by the free basic amino group of BSA forms a carbanion which is subsequently condensed with the iminium ion to give α,β-unsaturated nitrile 5. This step is known as the Knoevenagel condensation. Then, activated 3-methyl-1H-pyrazol-5(4H)-one 7 and α,β-unsaturated nitrile 5 react with each other through Michael addition to form 8 which undergoes cyclization to the desired product 9 as shown in Scheme 2.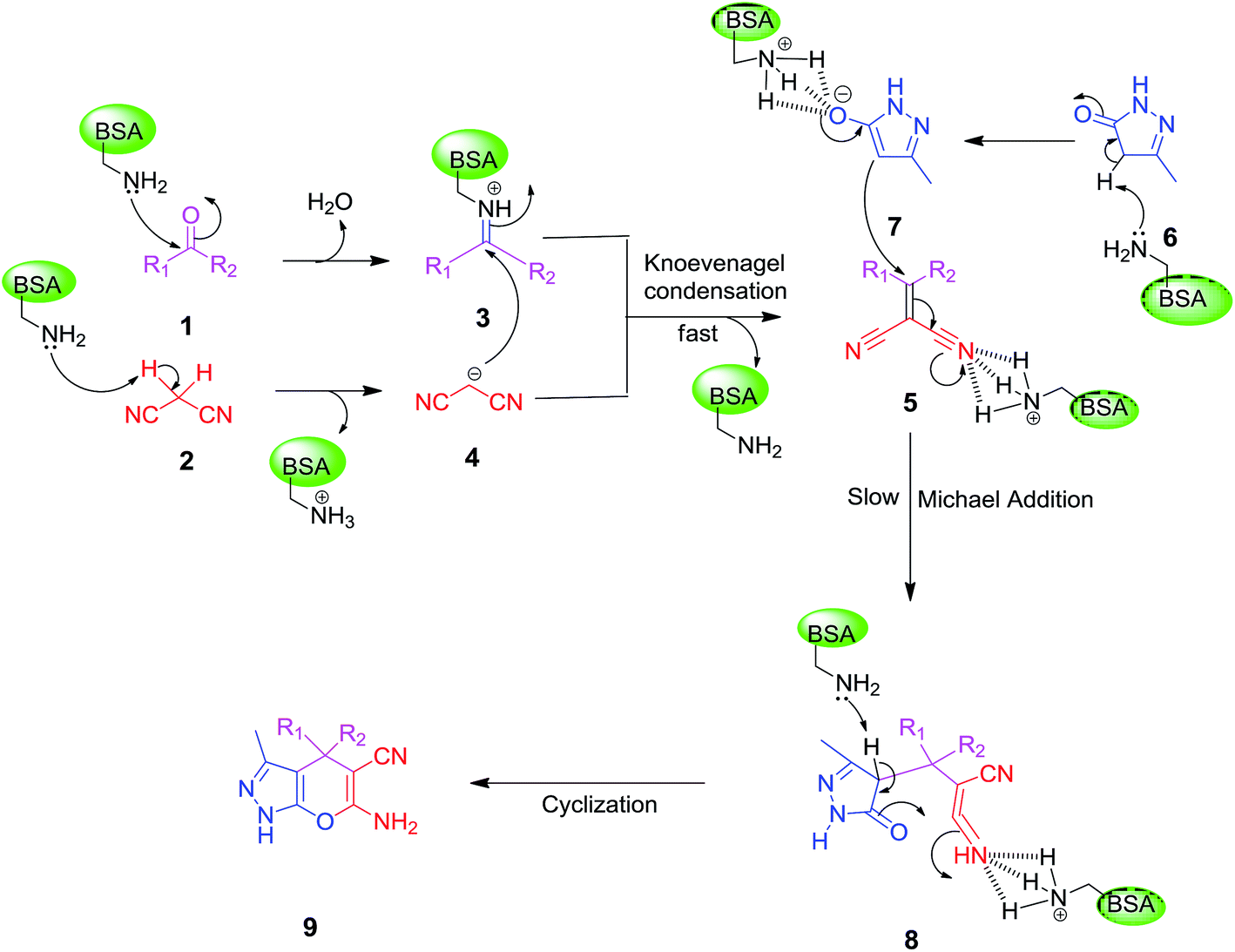 | ||
| Scheme 2 Plausible mechanism for the synthesis of dihydropyrano[2,3-c]pyrazole derivatives in the presence of BSA in an aqueous ethanol medium. | ||
Conclusion
In conclusion, we describe a simple, efficient and biocatalytic route for the synthesis of various dihydropyrano[2,3-c] pyrazoles and spiro[indoline-3,4′-pyrano[2,3-c]pyrazoles using inexpensive BSA. The advantages of this protocol include good to high yields, operational simplicity, and simple filtration. No metal catalyst or activation, and no extraction or separation by column chromatography is necessary. The overall green chemistry approach and the use of BSA as a biocatalyst in the reaction is of practical significance in expanding its applications in biocatalysis.Acknowledgements
The authors are thankful to the University Grants Commission (UGC) New Delhi, India for financial assistance under the Special Assistance Programme (SAP) and to the Department of Science and Technology (DST), Government of India for infrastructural facilities under the FIST programme to NMU. We also thank the Department of Science and Technology (DST), Government of India for providing the SCXRD facility under the FIST program to Department of Chemistry, NITK Surathkal, Mangaluru and SAIF-Punjab University, Chandigarh for characterization support for this work.References
-
(a) M. A El Aleem and A. A. El-Remaily, Tetrahedron, 2014, 70, 2971–2975 CrossRef
; (b) A. M. Soliman, S. K. Mohamed, M. A. El Remaily and H. Abdel-Ghany, Eur. J. Med. Chem., 2012, 47, 138–142 CrossRef CAS PubMed
-
(a) N. Foloppe, L. M. Fisher, R. Howes, A. Potter, A. G. Robertson and A. E. Surgenor, Bioorg. Med. Chem., 2006, 14, 4792–4802 CrossRef CAS PubMed
-
(a) J. L. Wang, D. Liu, Z. J. Zheng, S. Shan, X. Han, S. M. Srinivasula, C. M. Croce, E. S. Alnemri and Z. Huang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7124–7129 CrossRef CAS PubMed
- F. M. Abdelrazek, P. Metz, O. Kataeva, A. Jaeger and S. F. El-Mahrouky, Arch. Pharm., 2007, 340, 543–548 CrossRef CAS PubMed
- E. S. El-Tamany, F. A. El-Shahed and B. H. Mohamed, J. Serb. Chem. Soc., 1999, 64, 9–19 CAS
-
(a) Y. M. Litvinov, A. A. Shestopalov, L. A. Rodinovskaya and A. M. Shestopalov, J. Comb. Chem., 2009, 11, 914–919 CrossRef CAS PubMed
-
(a) F. Lehman, M. Holm and S. Laufer, J. Comb. Chem., 2008, 10, 364–367 CrossRef PubMed
- Y. Peng, G. Song and R. Dou, Green Chem., 2006, 8, 573–575 RSC
- G. Vasuki and K. Kumaravel, Tetrahedron Lett., 2008, 49, 5636–5638 CrossRef CAS
- H. Mecadon, M. R. Rohman, I. Kharbangar, B. M. Laloo, I. Kharkongor, M. Rajbangshi and B. Myrboh, Tetrahedron Lett., 2011, 52, 3228–3231 CrossRef CAS
- H. Mecadon, M. R. Rohman, M. Rajbangshi and B. Myrboh, Tetrahedron Lett., 2011, 52, 2523–2525 CrossRef CAS
- K. Kanagaraj and K. Pitchumani, Tetrahedron Lett., 2010, 51, 3312–3316 CrossRef CAS
- M. Bihani, P. P. Bora, G. Bez and H. Askari, ACS Sustainable Chem. Eng., 2013, 1, 440–447 CrossRef CAS
-
(a) Y. Zou, Y. Hu, H. Liu and D. Shi, ACS Comb. Sci., 2012, 14, 38–43 CrossRef CAS PubMed
-
(a) F. Tamaddon and M. Alizadeh, Tetrahedron Lett., 2014, 55, 3588–3591 CrossRef CAS
-
(a) J. B. Gujar, M. A. Chaudhari, D. S. Kawade and M. S. Shingare, Tetrahedron Lett., 2014, 55, 6030–6033 CrossRef CAS
- Z. B. Xie, N. Wang, G. F. Jiang and X. Q. Yu, Tetrahedron Lett., 2013, 54, 945–948 CrossRef CAS
- C. Li, X. W. Feng, N. Wang, Y. J. Zhou and X. Q. Yu, Green Chem., 2008, 10, 616–618 RSC
- Z. Xiang, Z. Liu, X. Chen, Q. Wu and X. Lin, Amino Acids, 2013, 45, 937–945 CrossRef CAS PubMed
- Y. Xue, L. Li, Y. He and Z. Guan, Sci. Rep., 2012, 2, 761 Search PubMed
-
(a) Z. Xiang, Y. Liang, X. Chen, Q. Wu and X. Lin, Amino Acids, 2014, 46, 1929–1937 CrossRef CAS PubMed
-
(a) P. V. Rizzo, L. A. Boarin, I. O. Freitas, R. S. Gomes, A. Beatriz, A. W. Rinaldi, A. W. Rinaldi and N. L. C. Domingues, Tetrahedron Lett., 2014, 55, 430–434 CrossRef CAS
-
(a) Y. Zhang, Y. Dai, M. Hou, T. Li, J. Ge and Z. Liu, RSC Adv., 2013, 3, 22963–22966 RSC
- A. G. Lavekar, R. Kumar and A. K. Sinha, J. Mol. Catal. B: Enzym., 2015, 116, 113–123 CrossRef
- E. Busto, V. Gotor-Fernandez and V. Gotor, Org. Process Res. Dev., 2011, 15, 236–240 CrossRef CAS
- D. D. Zhao, L. Li, F. Xu, Q. Wu and X. F. Lin, J. Mol. Catal. B: Enzym., 2013, 95, 29–35 CrossRef CAS
- N. Gaggero, D. C. M. Albanese, G. Celentano, S. Banfi and A. Aresi, Tetrahedron: Asymmetry, 2011, 22, 1231–1233 CrossRef CAS
- P. Ramesh, B. Shalini and N. W. Fadnavis, RSC Adv., 2014, 4, 7368–7373 RSC
- U. K. Sharma, N. Sharma, R. Kumar and A. K. Sinha, Amino Acids, 2013, 44, 1031–1037 CrossRef CAS PubMed
- F. Benedetti, F. Berti, I. Donati and M. Fregonese, Chem. Commun., 2002, 8, 828–829 RSC
-
(a) D. C. M. Albanese and N. Gaggero, RSC Adv., 2015, 5, 10588–10598 RSC
- P. P. Bora, M. Bihani and G. Bez, J. Mol. Catal. B: Enzym., 2013, 92, 24–33 CrossRef CAS
-
(a) P. N. Chaudhari, B. L. Chaudhari and S. B. Chincholkar, Process Biochem., 2013, 48, 144–151 CrossRef CAS
-
(a) Y. B. Wagh, A. Kuwar, S. K. Sahoo, J. Gallucci and D. S. Dalal, RSC Adv., 2015, 5, 45528–45534 RSC
- Y. A. Tayade, S. A. Padvi, Y. B. Wagh and D. S. Dalal, Tetrahedron Lett., 2015, 56, 2441–2447 CrossRef CAS
-
(a) S. S. Noinville, M. Revault, H. Quiquampoix and M. H. Baron, J. Colloid Interface Sci., 2000, 221, 273–283 CrossRef PubMed
-
(a) Y. K. Huang and S. W. Tsai, Appl. Microbiol. Biotechnol., 2014, 98, 621–628 CrossRef CAS PubMed
-
(a) R. Tian, C. H. Yang, X. F. Wei, E. N. Xun, R. Wang, S. G. Cao, Z. Wang and L. Wang, Biotechnol. Bioprocess Eng., 2011, 16, 337–342 CrossRef CAS
-
(a) L. R. Chena, Y. C. Wanga, Y. W. Lina, S. Y. Choua, S. F. Chena, L. T. Liua, Y. T. Wub, C. J. Kuob, T. S. S. Chena and S. H. Juanga, Bioorg. Med. Chem. Lett., 2005, 15, 3058–3062 CrossRef PubMed
-
(a) T. H. Kang, K. Matsumoto, M. Tohda, Y. Murakami, H. Takayama, M. Kitajima, N. Aimi and H. Watanabe, Eur. J. Pharmacol., 2002, 444, 39–45 CrossRef CAS PubMed
-
(a) Y. Zou, Y. Hu, H. Liu and D. Shi, ACS Comb. Sci., 2012, 14, 38–43 CrossRef CAS PubMed
-
(a) G. Boucher, R. Sylvain, V. Fargeas, T. Dintinger, M. Allainmat, J. Lebreton and C. Tellier, ChemBioChem, 2005, 6, 807–810 CrossRef CAS PubMed
- S. R. Mandha, S. Siliveri, M. Alla, V. R. Bommena, M. R. Bommineni and S. Balasubramanian, Bioorg. Med. Chem. Lett., 2012, 22, 5272–5278 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1432094 and 1432095. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra13014j |
| This journal is © The Royal Society of Chemistry 2016 |