
Synthesis and biological evaluation of new 2,5-dimethylthiophene/furan based N-acetyl pyrazolines as selective topoisomerase II inhibitors†
Darpanab,
Gaurav Joshia,
Suyog M. Amrutkar c,
Ashish T. Baviskarc,
Harveen Klera,
Sandeep Singhd,
Uttam C. Banerjeec and
Raj Kumar*ab
c,
Ashish T. Baviskarc,
Harveen Klera,
Sandeep Singhd,
Uttam C. Banerjeec and
Raj Kumar*ab
aLaboratory for Drug Design and Synthesis, Centre for Pharmaceutical Sciences and Natural Products, Central University of Punjab, 151 001, Bathinda, India. E-mail: raj.khunger@gmail.com; rajcps@cup.ac.in; Fax: +91-1636-236564; Tel: +91-0164-2864215
bIndo-Soviet Friendship College of Pharmacy, Moga, 141001, Punjab, India
cDepartment of Pharmaceutical Technology (Biotechnology), National Institute of Pharmaceutical Education and Research (NIPER), Mohali, S. A. S. Nagar, Sec 67, 160 062, Punjab, India
dCentre for Human Genetics and Molecular Medicine, Central University of Punjab, 151 001, Bathinda, India
First published on 15th January 2016
Abstract
Based on reported pharmacophores as topoisomerase inhibitors, 2,5-dimethylthiophene/furan based N-acetyl pyrazolines were designed and envisaged as topoisomerase inhibitors. The target compounds were synthesized and tested in vitro against human topoisomerases in decatenation, relaxation, cleavage complex and DNA intercalation assays. Out of 29 compounds, three (10, 11 and 29) showed potent and selective topoisomerase II inhibitory activity with no intercalation with DNA. Further, molecular docking studies also endorsed them as ATP dependent topoisomerase II catalytic inhibitors. These compounds exerted potential anticancer effects on breast, colon, lung and prostate cancer cell lines at a low micromolar level, as compared to etoposide, and low toxicity to normal cells. Apart from the topoisomerase II inhibition, these compounds also induced a reactive oxygen species (ROS) level in cancer cells. The cell cycle analyses showed their apoptotic effect at the G1 phase.
Introduction
Human DNA topoisomerases I and II (hTopoI and hTopoII) are validated drug targets in oncology due to their involvement in central DNA processing steps which include replication, transcription, translation and recombination.1 hTopoI catalyses the temporary breakage of DNA, one strand at a time, and the succeeding re-joining of the strands.2 Whereas, TopoII is a dimeric and ATP-dependant enzyme belonging to the GHKL family.3,4 It is involved in many functional roles related to the management of higher-order DNA structure, modulation of topological state, chromosome segregation, and chromatin condensation.5 It catalyses the transient cleavage of a DNA duplex (G-segment; a transient DNA–enzyme complex is formed) to yield a double-stranded gap through which another duplex (T-segment) is passed. The catalytic process involves the opening and closing of molecular ‘gates’ in the hTopoII structure, which is regulated by ATP binding. The binding of a ligand at the ATPase domain of hTopoII leads to the inhibition of ATP regulated opening and closing of the molecular gates and hence suppresses the activity of DNA hTopoII. An ATP makes three point attachments with the Mg2+ cation in an octahedral fashion. Further, most of the catalytic hTopoII inhibitors occupy the highly conserved Walker A motif composed of residues Arg162, Asn163, Gly164, Tyr165, Gly166 and Ala167 of the ATPase domain.6–9 It has been evident that rapidly proliferating tumour cells express the enzyme TopoI/TopoII at 25–300 times higher levels than those of quiescent cells, recommending them as druggable targets for new anticancer agents.10 Topoisomerase I inhibitors comprise of derivatives from plant extract camptothecin. Irinotecan (CPT-11), which is a semi-synthetic derivative of camptothecin, is approved in the United States for the treatment of colorectal cancer. Other TopoI inhibitors include 9-aminocamptothecin (9-AC), topotecan, etc.2 Various compounds such as doxorubicin (anthracycline),11 etoposide (epipodophyllotoxin),12 (−)-epigallocatechin-3-gallate (catechin),13–15 and genistein (isoflavone)16,17 are called TopoII poisons as they exert a potent cytotoxic effect by stabilizing the DNA–enzyme complex.18 Some synthetic purines and their analogues,19–21 and naturally occurring compounds such as flavones (myricetin),22 and isoflavone (daidzein) block the enzyme23–25 before DNA cleavage or in the last steps of the catalytic cycle after the resealing of the DNA break and are called catalytic TopoII inhibitors.26 Based on our previous experiences in the discovery and development of anticancer agents,27–32 we rationally designed target molecules as topoisomerase inhibitors keeping in view the shape and structural features of chalcones,33,34 flavones,22 isoflavones25 and compound 1 (ref. 35) (Fig. 1) with the following considerations: (i) possess two aromatic or heteroaromatic rings (A and B) joined by a central linker; which could be made rigid by introducing an N-acetyl pyrazoline moiety, (ii) based on the fact that a thienyl/furyl group (at ring A or B) with methyl substitution increases the topoisomerase I and II inhibitory mediated anticancer activity, and (iii) are easy to synthesize.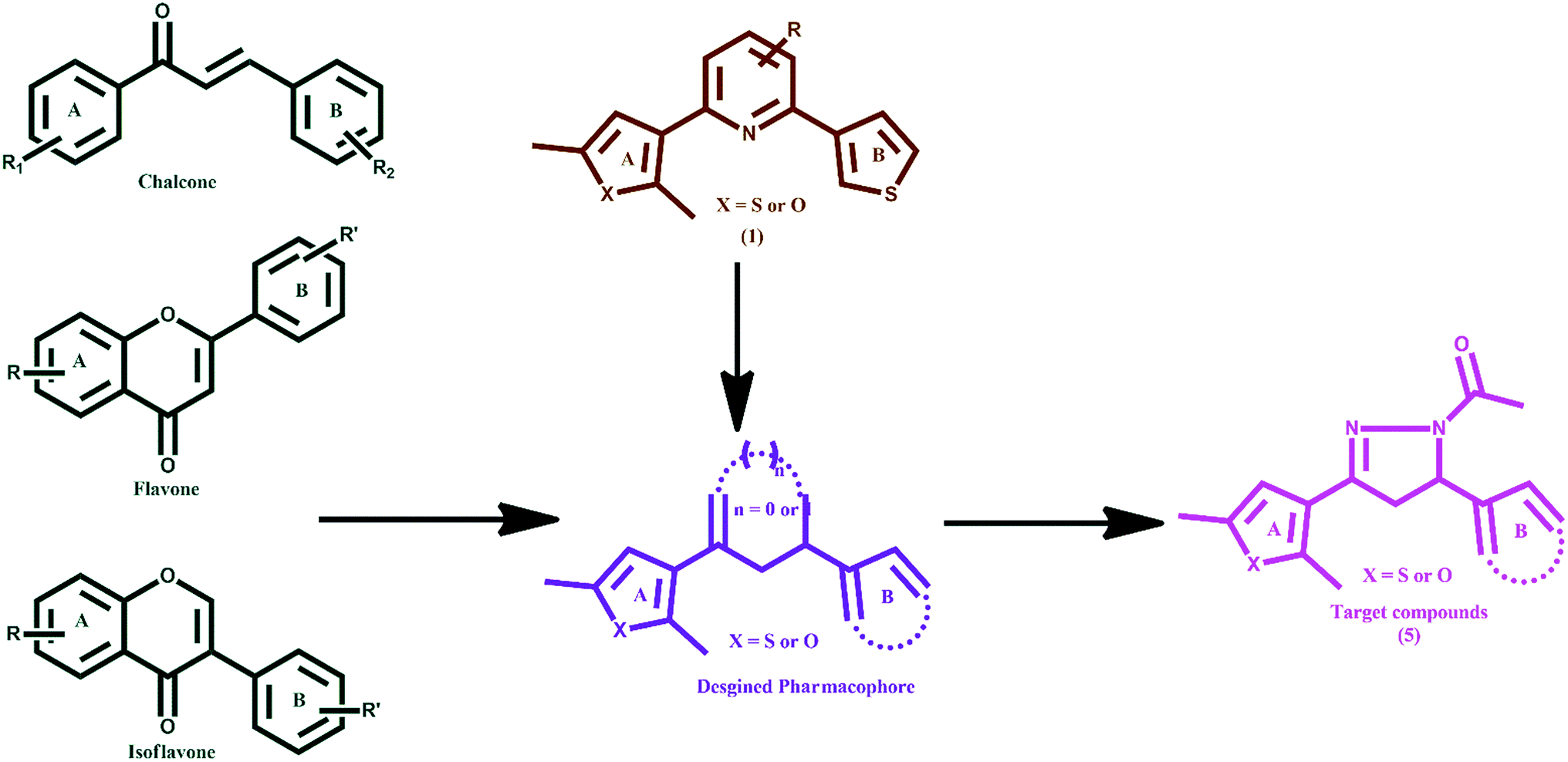 | ||
| Fig. 1 Design of new topoisomerase inhibitors (5–32). | ||
Further, the limited bioavailability of anticancer chalcones and flavonoids/isoflavones, due to glucuronidation and sulfation regardless of their greater absorption36–39 and their Michael acceptor property, has impelled researchers to design new anticancer agents. The present work involves the synthesis of new 2,5-dimethylthiophene/furan based N-acetyl pyrazolines. Their anticancer effects and mechanistic interventions as selective topoisomerase II inhibitors are reported and discussed.
Results and discussion
Synthesis
Synthesis of the target compounds (5–32) (Fig. 2) was accomplished by reacting 1,3-diaryl propenone with hydrazine hydrate and acetic acid. 1,3-Diaryl propenones (4) (E-isomer) were in turn synthesised via a Claisen–Schmidt condensation of aryl aldehydes (3) with aryl ketones (2), in the presence of ethanol under basic conditions (Scheme 1).28 Aryl aldehydes containing a phenolic hydroxyl group were condensed with aryl ketone in the presence of excess base (NaOH), which yielded the sodium salt of 1,3-diarylpropenones, and were neutralised with hydrochloric acid. All of the final compounds were unreported and were fully characterized by melting point, IR, NMR and elemental analyses.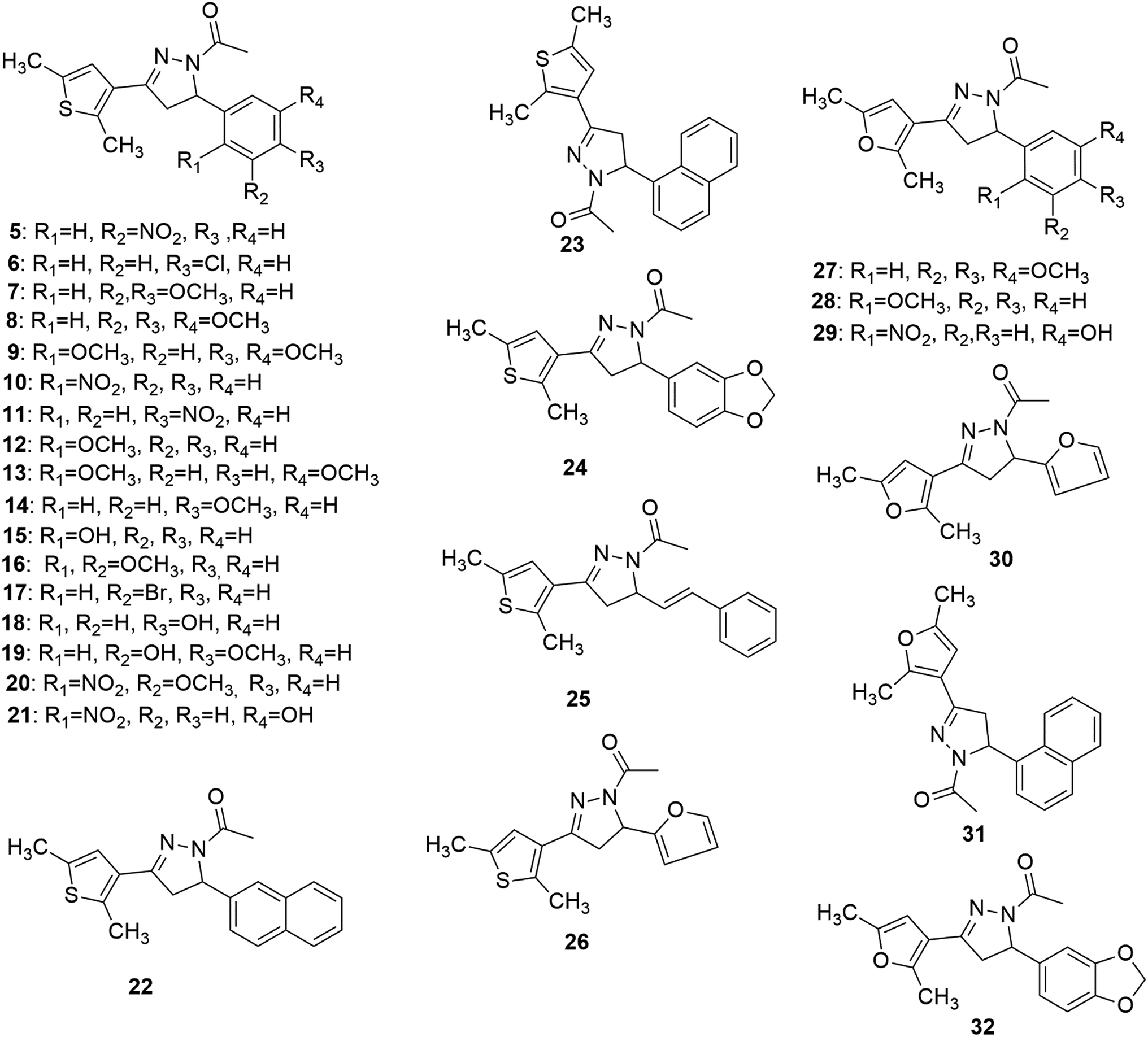 | ||
| Fig. 2 Chemical structures of the synthesized target compounds. | ||
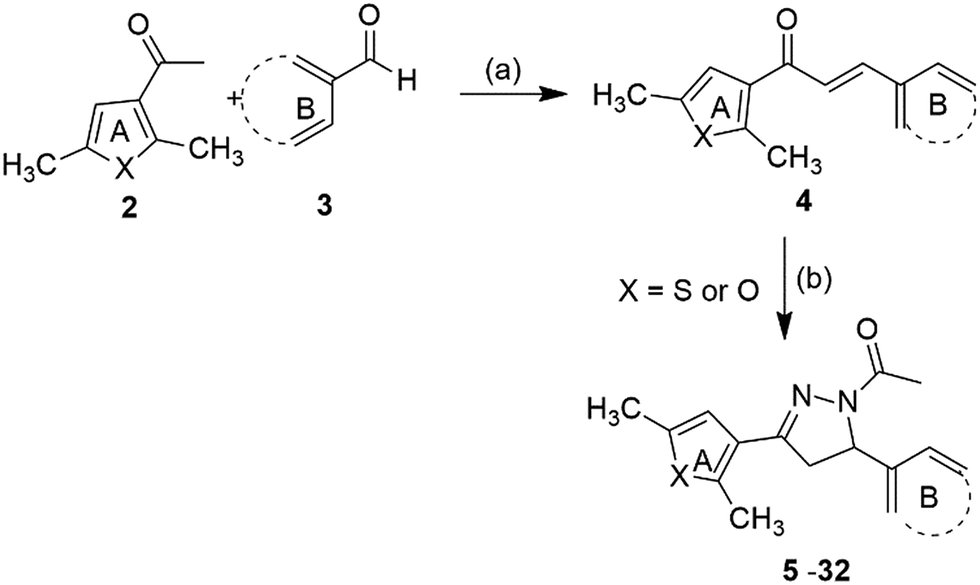 | ||
| Scheme 1 Reagents and conditions: (a) 5% NaOH, ethanol, rt, 4–5 h, 65–82% (b) NH2NH2·H2O, CH3COOH, reflux, 4–6 h, 75–90%. | ||
Interestingly, the reaction of 3-acetyl-2,5-dimethylthiophene either with vanillin or 2,3-dihydroxybenzaldehyde or 2,3,4-trihydroxybenzaldehyde yielded a retro product (33).
Biological studies
Compounds were selective and catalytic TopoIIα inhibitors
To study the topoisomerase inhibitory potential, the investigational compounds (5–33) were screened for inhibition of hTopoIIα mediated kDNA decatenation.29,31 Kinetoplast DNA (kDNA) was used as the substrate with etoposide (TopoII inhibitor) used as the standard (Fig. 3). Incubation of kDNA with hTopoIIα formed two decatenated products. In agarose gel, catenated kDNA appeared at the top as it cannot enter into the gel due to its overall larger size, while other decatenated products moved into the gel. The samples containing etoposide show moderate decatenation, as reported in the literature, because of its known reversible inhibition of topoisomerase IIα. Although compounds 5, 6, 7, 10, 11, 13, 15, 20, 26 and 29 were found to be relatively more active than etoposide against hTopoIIα, 10, 11 and 29 being the most potent, were selected for further mechanistic studies. Relatively less decatenation was observed in the presence of active compounds.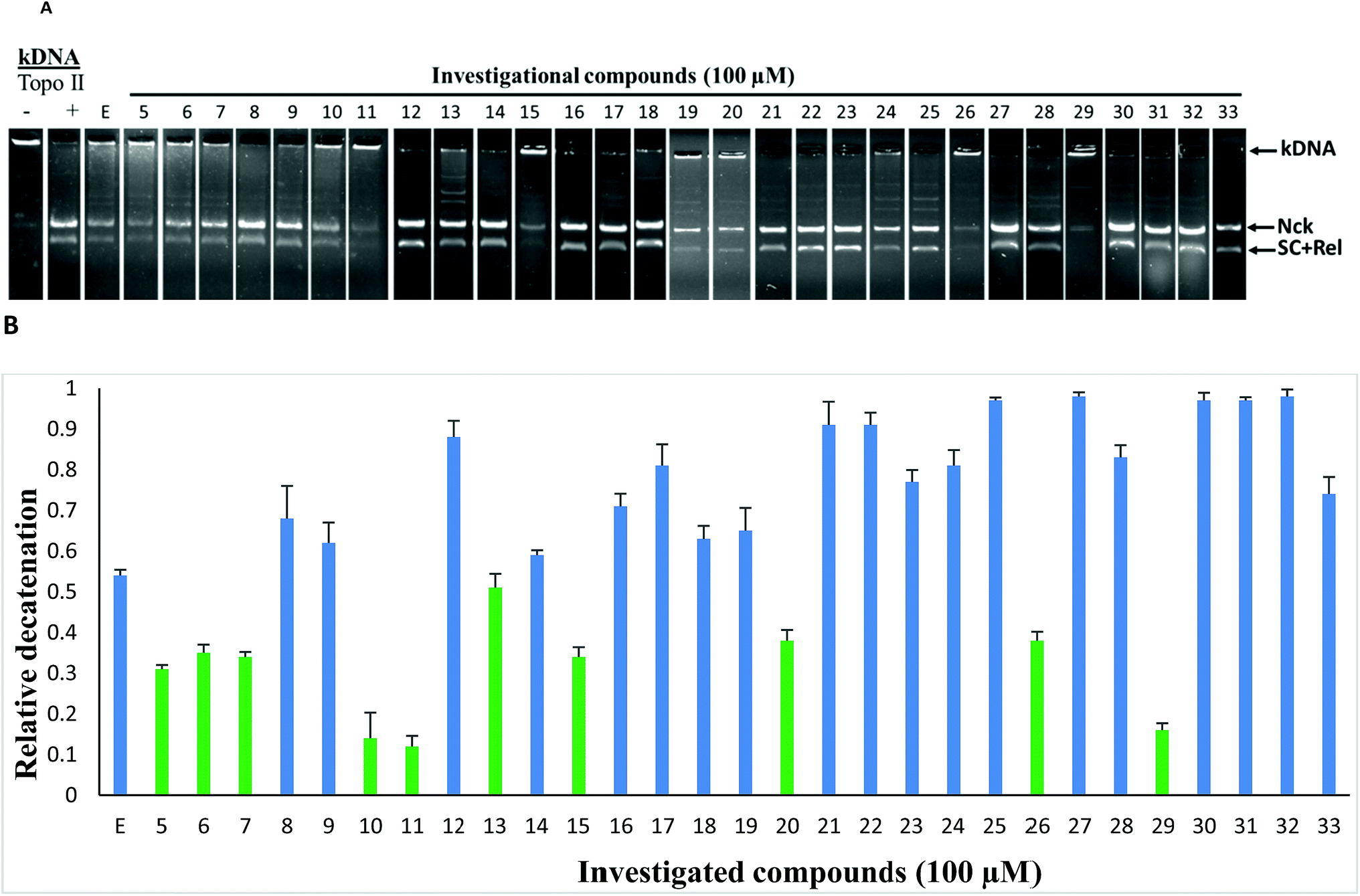 | ||
| Fig. 3 (A) Decatenation assay: kDNA (intact kinetoplast DNA) was used as the substrate, the decatenation products formed were Nck (nicked), Rel (relaxed), and SC (supercoiled) DNA. kDNA was treated with hTopoIIα in the presence of either 100 μM etoposide or investigational compound (5–33). (B) Quantification of products formed in the kDNA decatenation assay. | ||
The representative compound 11 was screened for inhibition of hTopoI mediated relaxation of supercoiled DNA (SC DNA). In this study negatively supercoiled DNA was used as the substrate and camptothecin (a known TopoI inhibitor) was used as the reference standard. Incubation of SC DNA with human topoisomerase-I resulted in the formation of Nck (nicked), Rel (relaxed), and SC (supercoiled) DNA isoforms. In the presence of compound 11, no inhibition of hTopoI mediated relaxation of supercoiled DNA was observed as in the case of camptothecin (Fig. 4). This indicates that the compounds were selective hTopoIIα inhibitors.
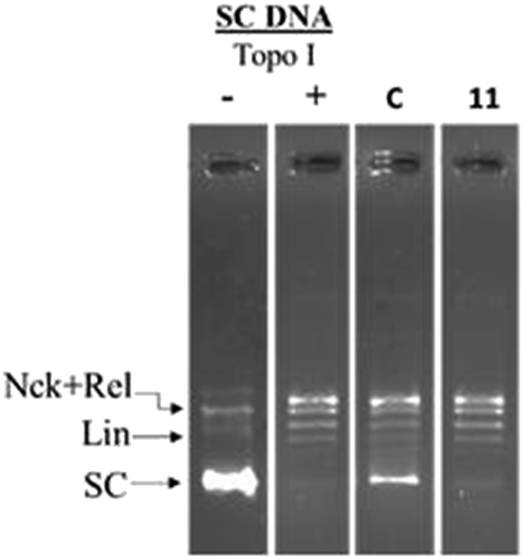 | ||
| Fig. 4 Topoisomerase-I relaxation assay: negatively supercoiled DNA was treated with hTopo-I in the presence of either 100 μM camptothecin (C) or compound 11. | ||
Further, it is known from the literature that hTopoIIα poisons such as etoposide, stabilize a TopoII-cleaved DNA complex (cleavage complex) which generally appears as a linear band (Lin).40 In the presence of 11 no such linear band was observed (Fig. 5) indicating that the investigated compounds were catalytic hTopoIIα inhibitors.
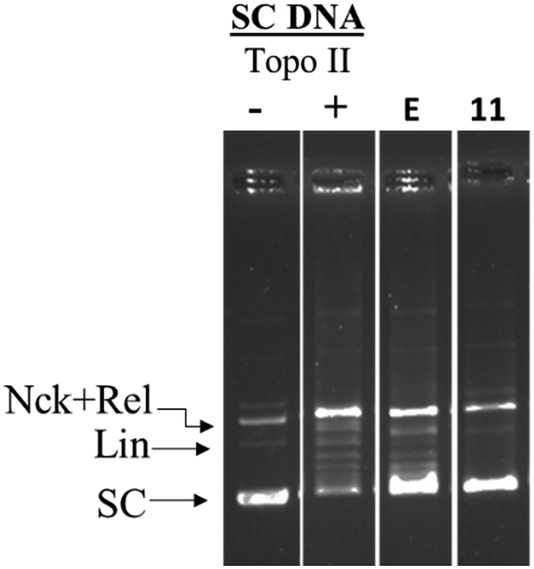 | ||
| Fig. 5 hTopo-IIα DNA cleavage complex assay: negatively supercoiled DNA (pUC19) was treated with hTopo-IIα then immediately incubated with either 100 μM etoposide or compound 11. | ||
In order to find out the possibility of DNA intercalation by the investigational compounds, a DNA intercalation assay was performed. DNA intercalators such as ethidium bromide retard the movement of SC DNA in the gel electrophoresis. In the presence of representative compounds 10, 11 and 29 no such retardation of DNA movement was observed (Fig. 6).
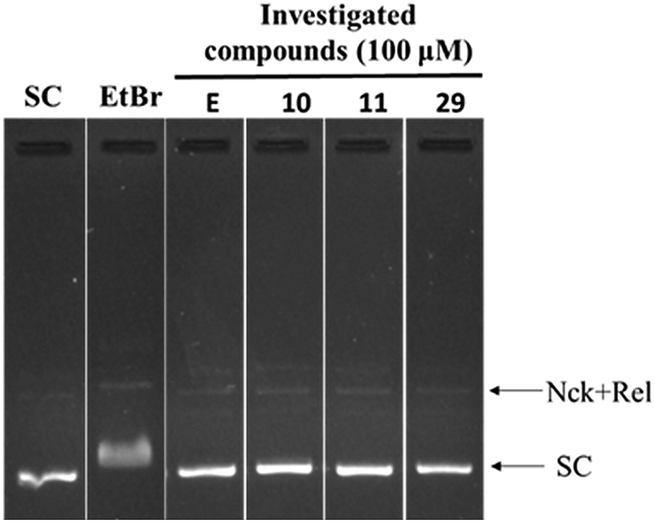 | ||
| Fig. 6 DNA intercalation assay: SC DNA (pUC19) was incubated with 1 μg mL−1 of ethidium bromide, and 100 μM of investigated compounds 10, 11 and 29. | ||
Thus the investigational compounds were found to be non DNA intercalating agents.
Structure–activity relationships (SAR)
Some of the general trends observed with respect to structure–activity relationships which emerged from the TopoII decatenation assay (Fig. 3) with the compounds are as follows: (a) compounds having 2,5-dimethylfuran as ring A were found to be more active against TopoII than corresponding 2,5-dimethylthiophene analogues (compare 8, 23, 24 and 26 with 27, 31, 32 and 30, respectively); (b) a ring size greater than phenyl on ring B (compounds 22–25, 31 and 32) was not found to be tolerable for TopoII inhibitory activity; (c) nitro substituted compounds (5, 10, 11, 20 and 29) at the phenyl on ring B exhibited greater TopoII inhibitory activity (except 21) than methoxy substituted compounds (7–9, 12–14 and 16–19) irrespective of their position(s).Molecular docking study
As the compounds 10, 11 and 29 emerged to be effective inhibitors of TopoII, the compounds were docked into the active site to identify their interactions with the ATPase domain of hTopoII (PDB entry: 1ZXM).6 The favorable interactions between the ligand molecules and receptor were scored using Glide (GLIDE 6.1 module of the Schrödinger Suite). Validation of the docking protocol involved re-docking a phosphoraminophosphonic acid–adenylate ester (ANP), an ATP analog, into the crystal structure of the ATPase domain of hTopoII. The validation results indicated a good agreement between the positioning of the ligand on docking and the already bound ligand. The docked ANP displayed a binding pose similar to the co-crystallized ligand with a root mean square deviation of 0.024 Å. The binding model of ANP with the ATPase domain showed that the amino group of the adenine moiety was involved in the hydrogen bonding with the side chain carbonyl oxygen of Asn120. Also, the hydroxyl group of the ribose unit exhibited a hydrogen bond interaction with the side of Ser149. Remarkably, the oxygen atoms in the three phosphate groups showed hydrogen bonding with the backbone residues; Asn163, Tyr165, Gly166 and Ala167, and with the side chain residues; Glu87, Asn91, Ser148, Lys168 and Gln376. Furthermore, the superimposition of the co-crystallized ligand with docked ANP revealed that the two occupy the ATP binding site in a similar manner. ANP made three point attachments with the Mg2+ cation, hence forming an octahedral geometry. The highly conserved Walker A motif composed of residues Arg162, Asn163, Gly164, Tyr165, Gly166 and Ala167 is essential for binding of catalytic inhibitors that act on the ATPase domain.31 Further, the docking studies revealed that the R configuration of compounds 10, 11 and 29 showed a better glide score than the ones with S configurations. Compound 29R showed the best docking score of −6.033. The superimposition of the co-crystallized ANP with 29R revealed that the nitrophenol ring overlapped with the phosphate groups and the dimethyl furan ring of ANP overlapped with the adenine moiety (Fig. 7A). The interaction model of the top scoring compound, 29R, at the ATPase binding domain revealed that the oxygen atom of the nitro group was involved in hydrogen bonding with the backbone amino group of Ala167 (d = 1.8 Å) and the side chain ammonium ion of Lys168 (d = 2.3 Å). Moreover, the hydroxyl group of the nitrophenol ring showed a hydrogen bond interaction with the side chain amino acid residue Ser149 (d = 2.3 Å). A salt bridge was formed between the magnesium ion (d = 2.0 Å) and the oxygen. The compound could show hydrophobic interactions with Tyr34, Ile141, Ile217, Phe142 and Ile125, and polar interactions with Thr147, Asn150, Asn163, Ser148, Asn91, Asn120 and Asn95 (Fig. 7B). The docking results for compound 11R also showed favorable interactions with the key residues and a docking score of −4.931. A detailed study of its binding model revealed that the oxygen atom of the nitro group formed a hydrogen bond with the backbone amino group of the Ala167 (d = 2.7 Å) and the nitrogen atom of the pyrazole ring formed a hydrogen bond with HOH933 (d = 2.3 Å). Additionally, the compound was also involved in salt bridge formation with the magnesium ion (d = 2.0 Å; Fig. 7C). The binding model of compound 10R indicated that the compound showed interactions with Lys168 and HOH933 (d = 2.3 Å). The docking score of compound 10R was computed to be −4.252 (Fig. 7D). The interactions of compounds 10, 11 and 29 are shown in Table 1.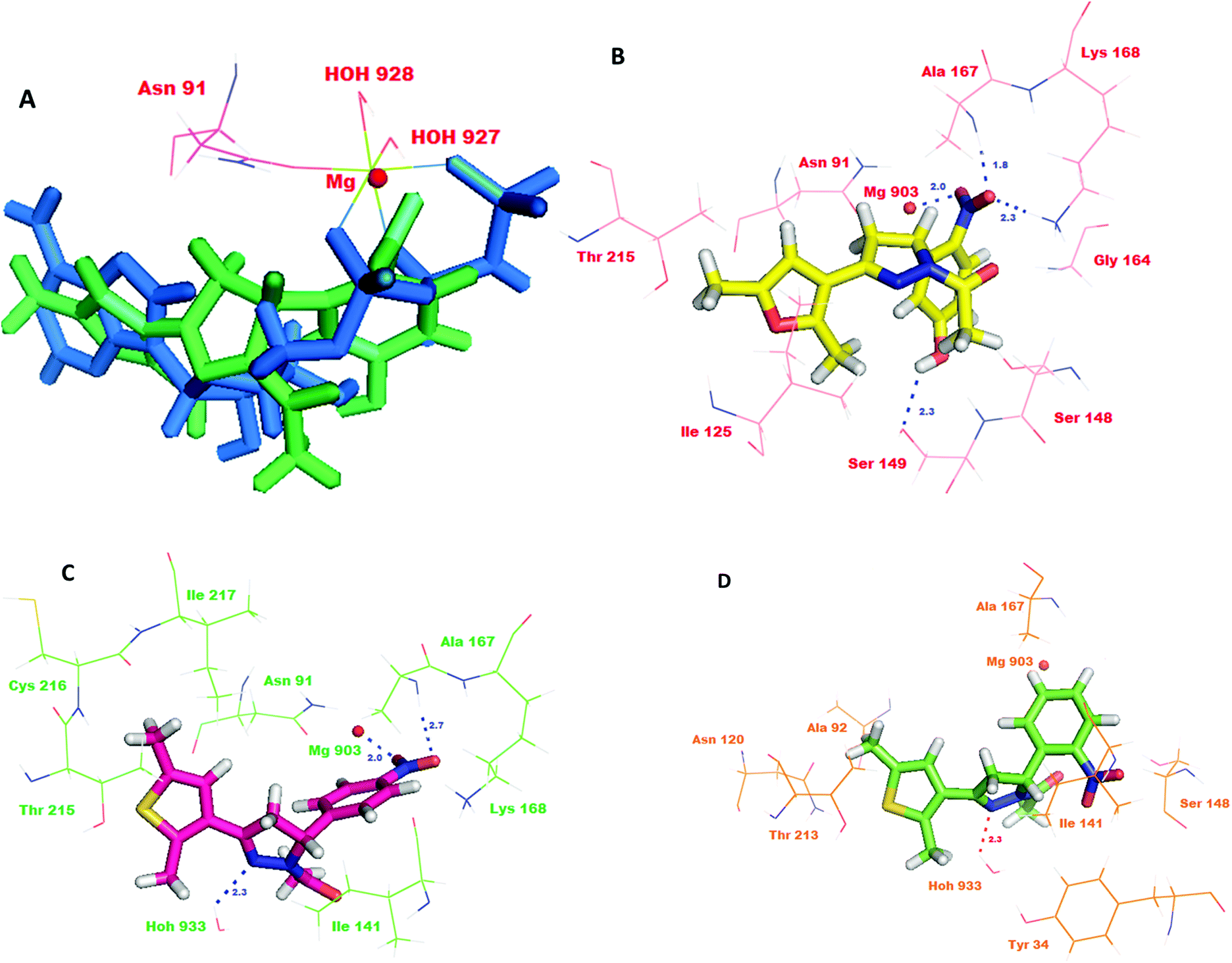 | ||
| Fig. 7 (A) Docking pose of 29R (green) at the binding site of AMPPNP (blue) in the ATPase domain of TopoIIα; (B), (C) and (D) docking poses of 29R (B), 11R (C) and 10R (D) showing the interactions with important amino acid residues in the ATPase domain of the TopoIIα compound. | ||
| Cd | Glide score | Interactions | Interactions with Mg2+ |
|---|---|---|---|
| 29R | −6.033 | Lys168, Ala167, Ser149 | Salt bridge |
| 11R | −4.931 | Ala167, HOH933 | Salt bridge |
| 10R | −4.252 | Lys168, HOH933 | No interaction |
| 29S | −4.157 | Lys168, Ser149, Phe142 | Metal coordination |
| 11S | −3.38 | Asn120, HOH931 | Arene–cation |
| 10S | −3.158 | Lys168, HOH924 | No interaction |
Antiproliferative activity
In order to determine the antiproliferative potential of compounds 10, 11 and 29 (which were the best selective Topo-IIα inhibitors), they were screened in vitro against a panel of five cancer cell lines, MCF-7 (breast), HCT-116 (colon) wild type as well as p53 null type, H460 (lung), and PC-3 (prostate). We tested the compounds on the p53 null type to ascertain whether the mode of inhibition is p53 independent or not. Overall the results indicate that the mode of action of the synthetic compounds is independent of p53. Since approximately 50% of cancers are mutated worldwide, the significance of the compounds increases further as their activity is independent of the p53 status in the cancer cells.The compounds 10, 11 and 29 showed significant antiproliferative effects in the aforesaid cancer lines. The results are summarized in Table 2. Etoposide (Etop)41 was used as the positive control. The compounds did not show significant cytotoxicity towards human peripheral blood cells (hPBMCs) that served as a normal control. This preferential cytotoxicity of the compounds to cancer cells could be due to intracellular accumulation of the compounds as well as their metabolites.42 Our results indicate that when the HCT-116 p53 null cell line was exposed to higher doses (25 μM) of our compounds, there is massive cell death. It has been shown earlier that DNA damage leads to mitotic catastrophe in p53 null cells.43 Considering our compounds as being topoisomerase inhibitors, it is assumed that the mode of apoptosis in p53 null cells at higher doses might be due to mitotic catastrophe.
| Cd | MCF-7 (breast) | HCT-116WT (colon) | HCT-116 (colon) (p53 null type) | H460 (lung) | PC-3 (prostate) |
|---|---|---|---|---|---|
| IC50a (μM) | |||||
| a Values are derived from averaging three independent experiments and each experiment was done in triplicate.b Not tested. | |||||
| 10 | 3.1 ± 0.3 | 3.5 ± 0.6 | 4.1 ± 0.6 | 5.0 ± 0.3 | 4.9 ± 0.5 |
| 11 | 2.5 ± 0.2 | 5.6 ± 0.2 | 4.3 ± 0.3 | 3.5 ± 0.4 | 6.1 ± 0.3 |
| 29 | 2.7 ± 0.5 | 8 ± 0.8 | 5.1 ± 0.3 | 3.9 ± 0.2 | 7.6 ± 0.2 |
| Etop | 20.933 | —b | —b | >30.33 | 18.233 |
Compounds induced reactive oxygen species (ROS) in cancer cells
Since many anticancer compounds bring about changes in the ROS status in the cells, it was hypothesized to check whether the anticancer effect of the compounds is mediated by free radicals or not? Compounds 10, 11 and 29 were used for the ROS study using a DHE based fluorescent detection system and utilising the MCF-7 cancer cell line. The results showed that there is a concentration dependent increase in ROS levels in the treated cells (Fig. 8). An increase in ROS levels is generally associated with changes in the mitochondrial membrane potential,28 which triggers its depolarization and leads to the release of cytochrome-c into the cytoplasm, thus inducing apoptosis. JC-1 dye was used to measure the membrane potential of the mitochondria (Fig. 9), and the results showed that there is a steady decrease in the OD590/OD527 ratio indicating increased mitochondrial membrane depolarization.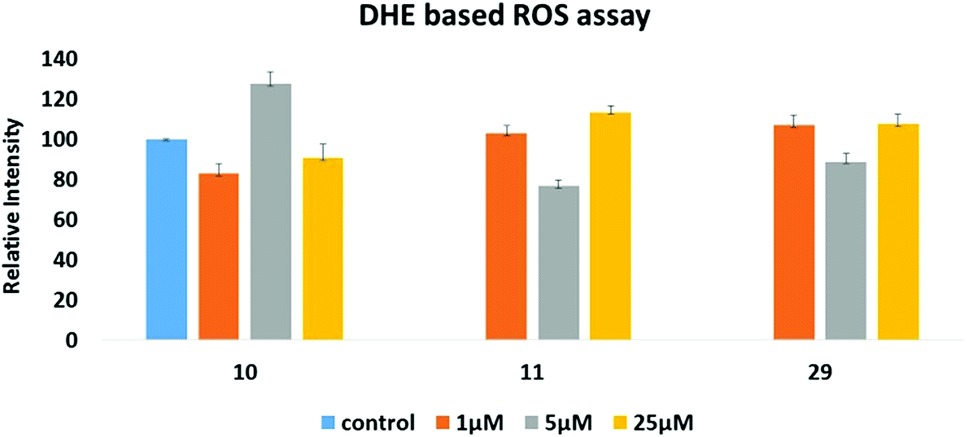 | ||
| Fig. 8 DHE based assay to measure intra-cellular reactive oxygen species (ROS) induced by 10, 11 and 29, respectively. | ||
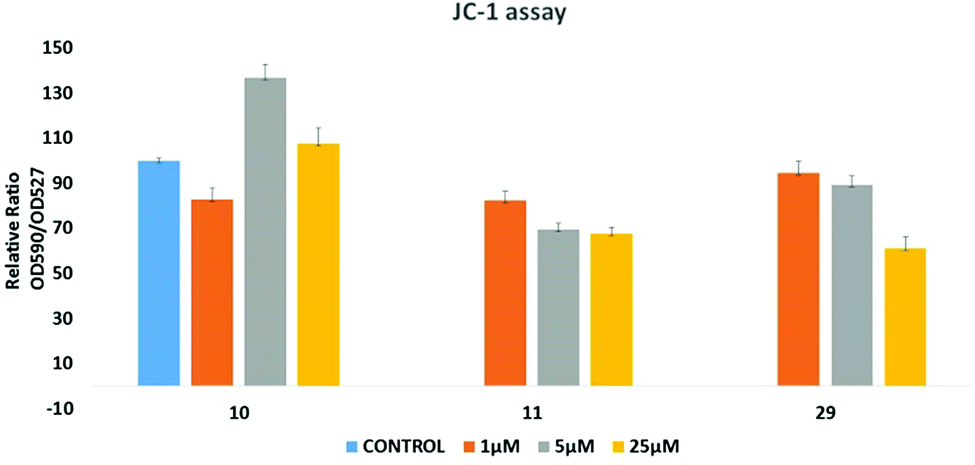 | ||
| Fig. 9 JC-1 dye based assay to measure the mitochondrial membrane potential altered by 10, 11 and 29, respectively. | ||
Cell cycle analysis
Cell cycle analysis was performed using flow cytometry following a procedure as described previously.44 The compound showed significant G1 phase arrest from 49.4% in the control to 71.8% and 76.8% at concentrations of 5 and 25 μL, respectively (Fig. 10). The predominant arrest of the G0/G1 phase is attributed to the selective TopoII catalytic inhibitors that might interfere with the binding between DNA and topoisomerase rendering inhibition of DNA replication, thus leading to cell cycle arrest at the G0/G1 phase.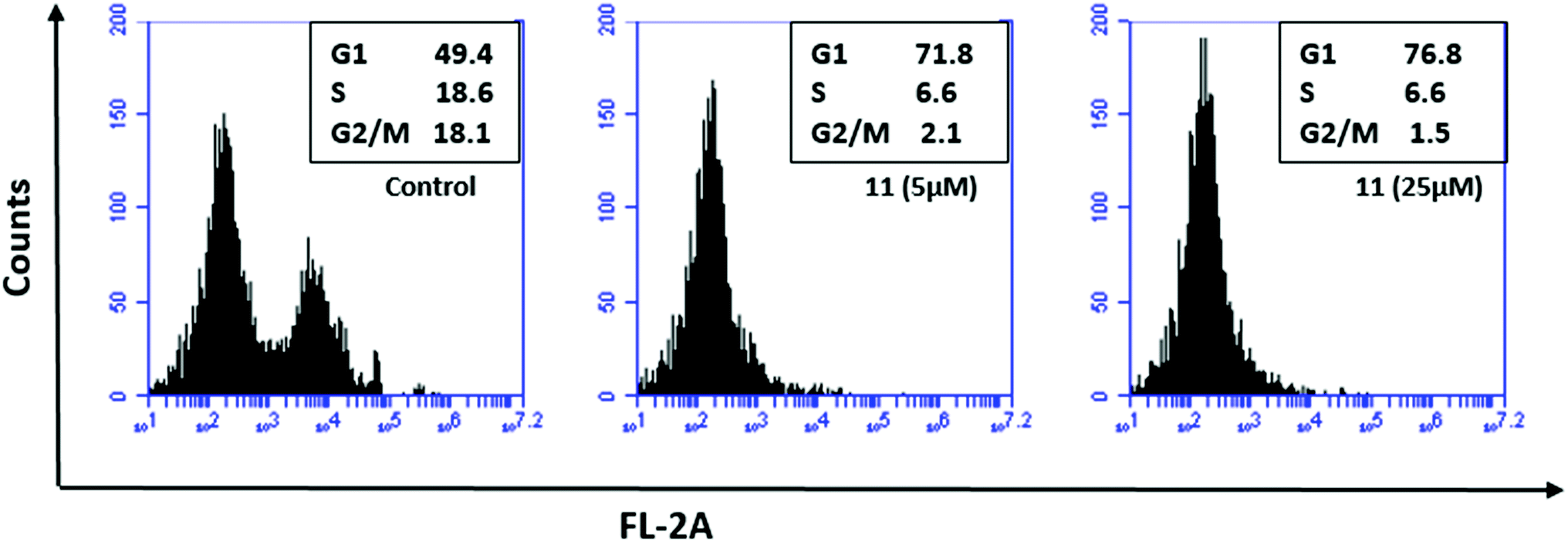 | ||
| Fig. 10 Cell cycle analysis using flow cytometry. The study was conducted for compound 11 using HCT-116 wild type cell line that showed profound G1 phase arrest. | ||
Conclusions
In summary, a series of new 2,5-dimethylthiophene/furan based N-acetyl pyrazolines were designed as topoisomerase inhibitors through a rational approach and were synthesised. The target compounds were evaluated by decatenation, relaxation, cleavage complex and DNA intercalation assays against human topoisomerases. Compounds 10, 11 and 29 exhibited potent, selective and catalytic topoisomerase II inhibitory activity with no intercalation with DNA, which was further supported by the molecular modeling study. In addition, these compounds proved their broad anticancer potential (low micromolar IC50) in vitro against breast, colon, lung and prostate cancer cell lines with almost no toxicity to normal cells. Furthermore, the compounds showed significant activity against HCT-116 colon cancer cells which are p53 null (HCT-116 p53 null type) indicating that the anticancer potential of these compounds is independent of p53. It thus signifies the importance of these compounds as almost 50% of human cancers are p53 mutant and these compounds can act in both p53 wild type as well as mutant type cancer cells. The compounds were able to induce cell cycle arrest at the G1 phase. In addition, the compounds were found to increase ROS levels as well as mitochondrial membrane depolarization, which might induce apoptosis, independent of their TopoII specific activity. These compounds could serve as promising leads for future antitumor drug discovery.Experimental
The reagents for the synthesis of the compounds were purchased from Sigma-Aldrich, Loba and CDH, India and used without further purification. All yields refer to isolated products after purification. Products were characterized by spectroscopic data (IR, 1H NMR, 13C NMR and MS spectra). NMR experiments were measured in CDCl3/DMSO-d6 relative to TMS (0.00 ppm). IR (KBr pellets) spectra were recorded on a Fourier transform infrared (FT-IR) Thermo spectrophotometer. Melting points were determined in open capillaries and were uncorrected. All of the target compounds were unreported and their physical data is presented below.Synthesis of the compounds
Synthesis of 1-[3-(2,5-dimethylthiophen-3-yl)-5-(3-nitro-phenyl)-4,5-dihydropyrazol-1H-1-yl]ethanone (5). Yellow solid; yield 76%; mp: 138–140 °C; IR (KBr cm−1): 2917 (C–H), 1665 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1645 (CN), 1560 (CC), 1524 (NO2 asymmetric), 1349 (NO2 symmetric), 1204 (C–N), 1141 (C–S). 1H NMR (300 MHz, CDCl3, TMS = 0): δ = 8.08–8.14 (2H, m), 7.48–7.59 (2H, m), 6.73 (1H, s), 5.58 (1H, dd, J = 4.8 and 12.00 Hz), 3.79 (1H, dd, J = 11.7 and 17.7 Hz), 3.09 (1H, dd, J = 4.8 and 17.7 Hz), 2.66 (3H, s), 2.40 (6H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.9, 21.9, 44.5, 58.4, 99.9, 120.8, 122.7, 125.3, 127.4, 129.9, 132.0, 136.3, 143.9, 144.1, 150.8, 168.3. MS (EI): 343.1 (m/z). Anal. calcd for C15H15N5O4: C, 54.71; H, 4.59; N, 21.27. Found: C, 54.67; H, 4.63; N, 21.32.
O), 1645 (CN), 1560 (CC), 1524 (NO2 asymmetric), 1349 (NO2 symmetric), 1204 (C–N), 1141 (C–S). 1H NMR (300 MHz, CDCl3, TMS = 0): δ = 8.08–8.14 (2H, m), 7.48–7.59 (2H, m), 6.73 (1H, s), 5.58 (1H, dd, J = 4.8 and 12.00 Hz), 3.79 (1H, dd, J = 11.7 and 17.7 Hz), 3.09 (1H, dd, J = 4.8 and 17.7 Hz), 2.66 (3H, s), 2.40 (6H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.9, 21.9, 44.5, 58.4, 99.9, 120.8, 122.7, 125.3, 127.4, 129.9, 132.0, 136.3, 143.9, 144.1, 150.8, 168.3. MS (EI): 343.1 (m/z). Anal. calcd for C15H15N5O4: C, 54.71; H, 4.59; N, 21.27. Found: C, 54.67; H, 4.63; N, 21.32.
Synthesis of 1-[5-(4-chlorophenyl)-3-(2,5-dimethyl-thiophen-3-yl)-4,5-dihydropyrazol-1H-1-yl]ethanone (6). Yellow solid; yield 71%; mp: 128–130 °C; IR (KBr cm−1): 3014 (C–H stretch), 1675 (C
O), 1635 (CN stretch), 1580 (CC aromatic), 1204 (C–N stretch), 1161 (C–S stretch), 769 (C–Cl stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.28 (2H, d, J = 8.7 Hz), 7.15 (2H, d, J = 8.4 Hz), 6.73 (1H, s), 5.46 (1H, dd, J = 4.5 and 11.17 Hz), 3.66 (1H, dd, J = 12.00 and 17.7 Hz), 3.06 (1H, dd, J = 4.5 and 17.7 Hz), 2.64 (3H, s), 2.40 (3H, s), 2.36 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.9, 21.9, 44.6, 58.4, 125.4, 127.1, 127.7, 129.0, 133.3, 136.1, 139.3, 140.4, 151.0, 168.7. MS (EI): 332.5 (m/z). Anal. calcd for C17H17ClN2OS: C, 61.34; H, 5.15; N, 8.42; S, 9.63. Found: C, 61.66; H, 5.23; N, 8.30; S, 9.86.
Synthesis of 1-[5-(3,4-dimethoxy-phenyl)-3-(2,5-dimethyl-thiophen-3-yl)-4,5-dihydropyrazol-1H-1-yl]ethanone (7). Yellow solid; yield 75%; mp: 115–118 °C; IR (KBr cm−1): 2860 (C–H stretch), 1667 (C
O), 1615 (CN stretch), 1562 (CC aromatic), 1250 (C–O stretch), 1219 (C–N stretch), 1181 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 6.75–6.81 (4H, m), 5.45 (1H, dd, J = 4.5 and 11.7 Hz), 3.86 (3H, s), 3.84 (3H, s), 3.68 (1H, dd, J = 12.00 and 17.7 Hz), 3.10 (1H, dd, J = 4.2 and 17.4 Hz), 2.65 (3H, s), 2.40 (3H, s), 2.37 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.8, 22.0, 44.8, 55.8, 55.9, 58.8, 109.0, 111.4, 117.5, 125.5, 128.0, 134.6, 136.0, 139.0, 148.0, 149.1, 151.2, 168.7. MS (EI): 328.1 (m/z). Anal. calcd for C19H22N2O3S: C, 63.66; H, 6.19; N, 7.82; S, 8.95. Found: C, 63.59; H, 6.33; N, 7.67; S, 8.84.
Synthesis of 1-[3-(2,5-dimethyl-thiophen-3-yl)-5-(3,4,5-trimethoxy-phenyl)-4,5-dihydro-pyrazol-1H-yl]-ethanone (8). Yellow solid; yield 79%; mp: 160–162 °C; IR (KBr cm−1): 3056 (C–H stretch), 1654 (C
O), 1624 (CN stretch), 1571 (CC aromatic), 1260 (C–O stretch), 1156 (C–N stretch), 1081 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 6.75 (1H, s), 6.41 (2H, s), 5.43 (1H, dd, J = 4.5 and 11.7 Hz), 3.83 (6H, s), 3.80 (3H, s), 3.70 (1H, dd, J = 12 and 17.7 Hz), 3.08 (1H, dd, J = 4.8 and 17.7 Hz), 2.65 (3H, s), 2.40 (6H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.8, 21.9, 44.0, 54.7, 56.2, 56.5, 56.7, 98.1, 110.6, 121.3, 125.5, 128.3, 135.9, 138.7, 143.1, 149.1, 150.4, 152.0, 168.6. MS (EI): 328.5 (m/z). Anal. calcd for C20H24N2O4S: C, 61.83; H, 6.23; N, 7.21; S, 8.25. Found: C, 61.77; H, 6.41; N, 7.19; S, 8.42.
Synthesis of 1-[3-(2,5-dimethyl-thiophen-3-yl)-5-(2,4,5-trimethoxy-phenyl)-4,5-dihydro-pyrazol-1H-1-yl]-ethanone (9). Light brown solid; yield 79%; mp: 132–135 °C; IR (KBr cm−1): 3090 (C–H stretch), 1670 (C
O), 1644 (CN stretch), 1551 (CC aromatic), 1250 (C–O stretch), 1148 (C–N stretch), 1050 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 6.74 (1H, s), 6.57 (1H, s), 6.52 (1H, s), 5.66 (1H, m), 3.86 (3H, s), 3.81 (3H, s), 3.78 (3H, s), 3.64 (1H, dd, J = 12 and 17.4 Hz), 2.98 (1H, dd, J = 4.2 and 17.4 Hz), 2.64 (3H, s), 2.39 (6H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.81, 21.9, 44.0, 54.7, 56.2, 56.4, 56.7, 98.17, 110.64, 121.31, 125.5, 128.2, 135.9, 138.7, 143.1, 149.1, 150.4, 152.0, 168.6. MS (EI): 358.4 (m/z). Anal. calcd for C20H24N2O4S: C, 61.83; H, 6.23; N, 7.21; S, 8.25. Found: C, 61.73; H, 6.32; N, 7.11; S, 8.10.
Synthesis of 1-[3-(2,5-dimethylthiophen-3-yl)-5-(2-nitro-phenyl)-4,5-dihydropyrazol-1H-1-yl]ethanone (10). Yellow solid; yield 82%; mp: 131–133 °C; IR (KBr cm−1): 2937 (C–H stretch), 1668 (C
O), 1639 (CN stretch), 1570 (CC aromatic), 1514 (NO2 asymmetric stretch), 1339 (NO2 symmetric stretch), 1219 (C–N stretch), 1147 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 8.10 (1H, d, J = 8.1 Hz), 7.59 (1H, t, J = 7.2 Hz), 7.43 (1H, t, J = 7.5 Hz), 7.24 (1H, m), 6.74 (1H, s), 6.04 (1H, dd, J = 4.5 and 11.4 Hz), 3.99 (1H, dd, J = 11.7 and 18.00 Hz), 3.08 (1H, dd, J = 4.8 and 18.00 Hz), 2.39 (3H, s), 2.21 (3H, s), 2.02 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.9, 15.7, 21.9, 43.4, 57.5, 110.1, 121.7, 122.7, 125.4, 128.4, 129.9, 132.1, 135.3, 141.9, 143.1, 150.1, 168.5. MS (EI): 343.2 (m/z). Anal. calcd for C17H17N3O3S: C, 59.46; H, 4.99; N, 12.24; S, 9.34. Found: C, 59.35; H, 5.12; N, 12.35; S, 9.39.
Synthesis of 1-[3-(2,5-dimethylthiophen-3-yl)-5-(4-nitro-phenyl)-4,5-dihydropyrazol-1-1H-yl]ethanone (11). Yellow solid; yield 77%; mp: 178–181 °C; IR (KBr cm−1): 2957 (C–H stretch), 1655 (C
O), 1635 (CN stretch), 1549 (CC aromatic), 1517 (NO2 asymmetric stretch), 1339 (NO2 symmetric stretch), 1219 (C–N stretch), 1157 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 8.18 (2H, d, J = 8.4 Hz), 7.39 (2H, d, J = 8.4 Hz), 6.73 (1H, s), 5.56 (1H, dd, J = 4.8 and 12.00 Hz), 3.77 (1H, dd, J = 12.00 and 17.4 Hz), 3.08 (1H, dd, J = 4.8 and 17.7 Hz), 2.65 (3H, s), 2.40 (3H, s), 2.39 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.0, 15.9, 21.8, 44.4, 58.5, 124.3, 125.2, 126.6, 127.3, 136.3, 139.6, 147.3, 148.9, 150.8, 168.8. MS (EI): 343.1 (m/z). Anal. calcd for C17H17N3O3S: C, 59.46; H, 4.99; N, 12.24; S, 9.34. Found: C, 59.39; H, 5.10; N, 12.41; S, 9.29.
Synthesis of 1-[3-(2,5-dimethyl-thiophen-3-yl)-5-(2-methoxy-phenyl)-4,5-dihydro-pyrazol-1H-1-yl]ethanone (12). Yellow solid; yield 72%; mp: 120–122 °C; IR (KBr cm−1): 2980 (C–H stretch), 1667 (C
O), 1625 (CN stretch), 1547 (CC aromatic), 1260 (C–O stretch), 1227 (C–N stretch), 1169 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.23 (1H, t, J = 7.5 Hz), 6.97 (1H, d, J = 7.5 Hz), 6.87–6.90 (2H, m), 6.72 (1H, s), 5.76 (1H, dd, J = 4.2 and 11.4 Hz), 3.85 (3H, s), 3.67 (1H, dd, J = 11.7 and 17.7 Hz), 2.95 (1H, dd, J = 4.2 and 17.4 Hz), 2.63 (3H, s), 2.42 (3H, s), 2.38 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.8, 15.7, 21.7, 44.3, 55.7, 58.5, 110.4, 116.5, 123.5, 127.0, 133.6, 136.5, 138.0, 147.0, 148.1, 151.0, 167.7. MS (EI): 328.1 (m/z). Anal. calcd for C18H20N2O2S: C, 65.83; H, 6.14; N, 8.53; S, 9.76. Found: C, 65.91; H, 6.20; N, 8.71; S, 9.62.
Synthesis of 1-[5-(2,5-dimethoxyphenyl)-3-(2,5-dimethyl-thiophen-3-yl)-4,5-dihydropyrazol-1H-1-yl]ethanone (13). Yellow solid; yield 78%; mp: 135–137 °C; IR (KBr cm−1): 2882 (C–H stretch), 1664 (C
O), 1627 (CN stretch), 1577 (CC aromatic), 1261 (C–O stretch), 1228 (C–N stretch), 1172 (C–S stretch); 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 6.71–6.79 (3H, m), 6.56 (1H, s) 5.73 (1H, d, J = 8.1 Hz), 3.81 (3H, s), 3.61–3.71 (4H, m), 2.93 (1H, d, J = 16.2 Hz), 2.62 (3H, s), 2.41 (3H, s), 2.38 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.9, 15.8, 21.9, 43.9, 54.5, 55.6, 56.0, 111.8, 111.9, 112.4, 125.5, 128.2, 130.6, 135.8, 138.8, 150.2, 151.9, 153.7, 168.6. MS (EI): 358.1 (m/z). Anal. calcd for C19H22N2O3S: C, 63.66; H, 6.19; N, 7.82; S, 8.95. Found: C, 63.59; H, 6.32; N, 7.63; S, 9.01.
Synthesis of 1-[3-(2,5-dimethylthiophen-3-yl)-5-(4-methoxy-phenyl)-4,5-dihydropyrazol-1H-1-yl]ethanone (14). Light brown solid; yield 75%; mp: 98–100 °C; IR (KBr cm−1): 2992 (C–H stretch), 1668 (C
O), 1632 (CN stretch), 1517 (CC aromatic), 1267 (C–O stretch), 1239 (C–N stretch), 1157 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.15 (2H, d, J = 8.1 Hz), 6.83 (2H, d, J 8.4 Hz), 6.75 (1H, s), 5.45 (1H, dd, J = 3.9 and 11.7 Hz), 3.77 (3H, s), 3.63–3.72 (1H, m), 3.09 (1H, dd, J = 3.9 and 17.4 Hz), 2.64 (3H, s), 2.40 (3H, s), 2.35 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.8, 22.0, 44.6, 55.5, 58.5, 114.2, 125.5, 126.9, 134.2, 136.0, 139.0, 140.1, 151.1, 158.0, 168.1. MS (EI): 328.3 (m/z). Anal. calcd for C18H20N2O2S: C, 65.83; H, 6.14; N, 8.53; S, 9.76. Found: C, 65.92; H, 6.23; N, 8.67; S, 9.53.
Synthesis of 1-[3-(2,5-dimethylthiophen-3-yl)-5-(2-hydroxy-phenyl)-4,5-dihydro-pyrazol-1H-1-yl]ethanone (15). Brown solid; yield 77%; mp: 139–141 °C; IR (KBr cm−1): 2995 (C–H stretch), 1656 (C
O), 1641 (CN stretch), 1569 (CC aromatic), 1257 (C–O stretch), 1212 (C–N stretch), 1161 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.21 (1H, m), 6.96–7.00 (2H, m), 6.84–6.89 (2H, m), 5.74 (1H, dd, J = 3 and 11.7 Hz), 3.64–3.76 (2H, m), 3.45 (1H, dd, J = 3 and 17.7 Hz), 2.66 (3H, s), 2.45 (3H, s), 2.38 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.8, 15.5, 21.6, 43.4, 57.3, 115.5, 121.3, 125.2, 126.4, 127.2, 136.3, 139.6, 147.3, 148.9, 150.8, 154.3, 167.4. MS (EI): 314.5 (m/z). Anal. calcd for C17H18N2O2S: C, 64.94; H, 5.77; N, 8.91; S, 10.20. Found: C, 64.76; H, 5.89; N, 8.71; S, 10.34.
Synthesis of 1-[5-(2,3-dimethoxyphenyl)-3-(2,5-dimethyl-thiophen-3-yl)-4,5-dihydro-pyrazol-1H-1-yl]ethanone (16). Yellow solid; yield 71%; mp: 127–129 °C; IR (KBr cm−1): 2991 (C–H stretch), 1672 (C
O), 1634 (CN stretch), 1551 (CC aromatic), 1256 (C–O stretch), 1239 (C–N stretch), 1172 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 6.97 (1H, d, J = 6.9 Hz), 6.81 (1H, d, J = 6.9 Hz) 6.73 (1H, s), 6.68 (1H, d, J = 7.2 Hz) 5.70 (1H, d, J = 8.1 Hz), 3.90 (3H, s), 3.85 (3H, s), 3.62–3.72 (1H, m), 3.03 (1H, d, J = 14.7 Hz), 2.63 (3H, s), 2.38 (6H, bs). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.8, 22.0, 44.2, 54.7, 55.7, 60.4, 111.5, 118.0, 124.4, 125.5, 128.1, 135.7, 135.9, 138.8, 145.7, 151.8, 152.8, 168.6. MS (EI): 328.6 (m/z). Anal. calcd for C19H22N2O3S: C, 63.66; H, 6.19; N, 7.82; S, 8.95. Found: C, 63.59; H, 6.33; N, 7.91; S, 8.71.
Synthesis of 1-[5-(3-bromophenyl)-3-(2,5-dimethyl-thiophen-3-yl)-4,5-dihydro-pyrazol-1H-1-yl]-ethanone (17). Yellow solid; yield 70%; mp: 103–105 °C; IR (KBr cm−1): 2968 (C–H stretch), 1667 (C
O), 1594 (CN stretch), 1567 (CC aromatic), 1229 (C–N stretch), 1199 (C–S stretch), 715 (C–Br stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.33–7.38 (2H, m), 7.13–7.21 (2H, m), 6.73 (1H, s), 5.44 (1H, dd, J = 4.5 and 12 Hz), 3.70 (1H, dd, J = 11.7 and 17.7 Hz), 3.06 (1H, dd, J = 4.5 and 17.7 Hz), 2.64 (3H, s), 2.40 (3H, s), 2.38 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.9, 21.9, 44.6, 58.5, 122.9, 124.3, 125.4, 127.7, 128.6, 130.5, 130.7, 136.1, 139.3, 144.2, 151.0, 168.7. MS (EI): 376.1, 378.3 (m/z). Anal. calcd for C17H17BrN2OS: C, 54.12; H, 4.54; N, 7.42; S, 8.50. Found: C, 54.09; H, 4.72; N, 7.38; S, 8.44.
Synthesis of 1-[3-(2,5-dimethylthiophen-3-yl)-5-(4-hydroxy-phenyl)-4,5-dihydro-pyrazol-1H-1-yl]ethanone (18). Yellow solid; yield 75%; mp: 153–155 °C; IR (KBr cm−1): 2987 (C–H stretch), 1661 (C
O), 1582 (CN stretch), 1577 (CC aromatic), 1254 (C–O stretch), 1235 (C–N stretch), 1179 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 6.95 (2H, d, J = 9 Hz), 6.71 (1H, m), 6.53 (2H, d, J = 9 Hz), 5.42 (1H, dd, J = 4.5 and 11.7 Hz), 3.66 (1H, dd, J = 11.7 and 17.7 Hz), 3.09 (1H, dd, J = 4.2 and 17.7 Hz), 2.62 (3H, s), 2.39 (6H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.1, 15.8, 21.6, 42.4, 59.5, 116.2, 126.3, 126.9, 127.1, 136.3, 139.6, 147.3, 150.8, 157.2, 168.9. MS (EI): 314.3 (m/z). Anal. calcd for C17H18N2O2S: C, 64.94; H, 5.77; N, 8.91; S, 10.20. Found: C, 64.81; H, 5.89; N, 8.70; S, 10.39.
Synthesis of 1-[3-(2,5-dimethylthiophen-3-yl)-5-(3-hydroxy-4-methoxy-phenyl)-4,5-dihydro-pyrazol-1H-1-yl]ethanone (19). Creamy solid; yield 76%; mp: 115–118 °C; IR (KBr cm−1): 3010 (C–H stretch), 1656 (C
O), 1627 (CN stretch), 1537 (CC aromatic), 1251 (C–O stretch), 1225 (C–N stretch), 1165 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 6.73–6.78 (4H, m), 5.40 (1H, dd, J = 4.2 and 11.7 Hz), 3.84 (3H, s), 3.65 (1H, dd, J = 12 and 18 Hz), 3.07 (1H, dd, J = 4.5 and 17.7 Hz), 2.63 (3H, s), 2.54 (3H, s), 2.36 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.1, 15.8, 21.6, 43.4, 56.3, 58.6, 112.5, 113.8, 119.5, 126.6, 127.3, 133.4, 135.8, 139.6, 146.3, 148.1, 151.8, 167.2. MS (EI): 344.4 (m/z). Anal. calcd for C18H20N2O3S: C, 62.77; H, 5.85; N, 8.13; S, 9.31. Found: C, 62.82; H, 5.71; N, 8.39; S, 9.41.
Synthesis of 1-[3-(2,5-dimethylthiophen-3-yl)-5-(3-methoxy-2-nitro-phenyl)-4,5-dihydro-pyrazol-1H-1-yl]-ethanone (20). Yellow solid; yield 68%; mp: 158–160 °C; IR (KBr cm−1): 2977 (C–H stretch), 1660 (C
O), 1639 (CN stretch), 1543 (CC aromatic), 1523 (NO2 asymmetric stretch), 1331 (NO2 symmetric stretch), 1253 (C–O stretch), 1229 (C–N stretch), 1173 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.36 (1H, t, J = 8.1 Hz), 6.95 (1H, d, J = 9 Hz), 6.72–6.76 (2H, m), 5.43 (1H, dd, J = 5.4 and 12.0 Hz), 3.89 (3H, s), 3.71 (1H, dd, J = 12.0 and 18.0 Hz), 3.13 (1H, dd, J = 5.4 and 18.0 Hz), 2.63 (3H, s), 2.39 (3H, s), 2.37 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.9, 15.9, 21.7, 44.5, 55.1, 56.5, 99.9, 111.0, 117.0, 125.4, 131.9, 135.5, 136.1, 138.5, 139.7, 141.1, 151.2, 168.7. MS (EI): 373.4 (m/z). Anal. calcd for C18H19N3O4S: C, 57.89; H, 5.13; N, 11.25; S, 8.59. Found: C, 57.92; H, 5.29; N, 11.12; S, 8.31.
Synthesis of 1-[3-(2,5-dimethylthiophen-3-yl)-5-(5-hydroxy-2-nitro-phenyl)-4,5-dihydropyrazol-1H-1-yl]-ethanone (21). Brown solid; yield 65%; mp: 210–212 °C; IR (KBr cm−1): 2996 (C–H stretch), 1651 (C
O), 1627 (CN stretch), 1531 (CC aromatic), 1523 (NO2 asymmetric stretch), 1337 (NO2 symmetric stretch), 1243 (C–O stretch), 1234 (C–N stretch), 1162 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.96 (1H, m), 6.55–6.76 (3H, s), 6.10 (1H, m), 3.97 (1H, m), 2.94 (1H, m), 2.64 (3H, s), 2.47 (6H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.5, 15.4, 21.5, 42.9, 54.0, 116.7, 122.7, 125.0, 125.5, 127.4, 127.7, 129.2, 135.1, 138.0, 151.5, 152.6, 167.9. MS (EI): 359.1 (m/z). Anal. calcd for C17H17N3O4S: C, 56.81; H, 4.77; N, 11.69; S, 8.92. Found: C, 56.72; H, 4.92; N, 11.71; S, 9.12.
Synthesis of 1-[3-(2,5-dimethyl-thiophen-3-yl)-5-naphthalen-2-yl-4,5-dihydro-pyrazol-1H-1-yl]ethanone (22). Dark brown semisolid; yield 67%; IR (KBr cm−1): 3010 (C–H stretch), 1665 (C
O), 1645 (CN stretch), 1580 (CC aromatic), 1202 (C–N stretch), 1135 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.67–7.78 (5H, m), 7.31–7.44 (2H, m), 6.75 (1H, s), 5.67 (1H, bs), 3.77 (1H, m), 3.15 (1H, m), 2.66 (3H, s), 2.40 (6H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.9, 22.0, 44.8, 59.2, 123.4, 124.4, 125.5, 125.9, 126.2, 127.6, 128.1, 129.0, 132.8, 133.3, 136.1, 139.17, 139.22, 149.1, 151.3, 168.8. MS (EI): 348.4 (m/z). Anal. calcd for C21H20N2OS: C, 72.38; H, 5.79; N, 8.04; S, 9.20. Found: C, 72.41; H, 5.81; N, 8.12; S, 9.31.
Synthesis of 1-[3-(2,5-dimethyl-thiophen-3-yl)-5-naphthalen-1-yl-4,5-dihydro-pyrazol-1H-1-yl]-ethanone (23). Brown semisolid; yield 65%; IR (KBr cm−1): 3015 (C–H stretch), 1677 (C
O), 1635 (CN stretch), 1561 (CC aromatic), 1223 (CN stretch), 1149 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.94 (1H, d, J = 7.8 Hz), 7.86 (1H, d, J = 6 Hz), 7.73 (1H, d, J = 9 Hz) 7.40–7.56 (3H, m), 7.18 (1H, d, J = 6.9 Hz), 6.62 (1H, s), 6.23 (1H, dd, J = 3.3 and 11.1 Hz), 3.87 (1H, dd, J = 12 and 17.4 Hz), 3.04 (1H, dd, J = 3.9 and 17.1 Hz), 2.64 (3H, s), 2.50 (3H, s), 2.33 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.9, 15.9, 22.0, 44.5, 56.5, 123.0, 125.5, 126.3, 127.9, 128.1, 129.1, 129.6, 134.3, 136.0, 136.1, 139.2, 143.6, 151.9, 168.9. MS (EI): 348.1 (m/z). Anal. calcd for C21H20N2OS: C, 72.38; H, 5.79; N, 8.04; S, 9.20. Found: C, 72.41; H, 5.81; N, 8.29; S, 9.17.
Synthesis of 1-(5-(benzo[d][1,3]dioxol-5-yl)-3-(2,5-dimethylthiophen-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)ethanone (24). Yellow solid; yield 68%; mp: 142–144 °C; IR (KBr cm−1): 2961 (C–H stretch), 1679 (C
O stretch), 1652 (CN stretch), 1559 (CC aromatic), 1259 (C–O stretch), 1229 (C–N stretch), 1127 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 6.70–6.76 (3H, m), 6.67 (1H, s), 5.91 (2H, s), 5.41 (1H, dd, J = 5.2 and 11.7 Hz), 3.66 (1H, dd, J = 11.7 and 17.4 Hz), 3.05 (1H, dd, J = 5.2 and 17.4 Hz), 2.64 (3H, s), 2.40 (3H, s), 2.36 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.9, 22.0, 44.8, 58.8, 101.1, 106.0, 108.4, 119.1, 125.4, 127.9, 136.0, 136.1, 139.1, 146.9, 148.0, 151.1, 168.7. MS (EI): 342.3 (m/z). Anal. calcd for C18H18N2O3S: C, 63.14; H, 5.30; N, 8.18; S, 9.36. Found: C, 63.11; H, 5.23; N, 8.18; S, 9.27.
Synthesis of 1-[3-(2,5-dimethylthiophen-3-yl)-5-styryl-4,5-dihydro-pyrazol-1H-1-yl]ethanone (25). Dark brown solid; yield 70%; mp: 108–110 °C; IR (KBr cm−1): 2980 (C–H stretch), 1660 (C
O), 1642 (CN stretch), 1595 (CC aromatic), 1211 (C–N stretch), 1142 (C–S stretch); 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.22–7.37 (6H, m), 6.87 (1H, s), 6.18 (1H, dd, J = 7.5 and 15.6 Hz), 5.49 (1H, m), 3.50 (1H, m), 3.42 (1H, m), 2.77 (3H, s), 2.41 (3H, s), 2.37 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.6, 15.7, 21.6, 43.6, 58.1, 126.1, 127.2, 127.9, 128.4, 128.8, 129.1, 133.8, 134.6, 136.7, 139.7, 151.8, 167.2. MS (EI): 324.3 (m/z). Anal. calcd for C19H20N2OS: C, 70.34; H, 6.21; N, 8.63; S, 9.88. Found: C, 70.21; H, 6.39; N, 8.74; S, 10.01.
Synthesis of 1-[3-(2,5-dimethyl-thiophen-3-yl)-5-furan-2-yl-4,5-dihydro-pyrazol-1H-1-yl]-ethanone (26). Brown solid; yield 67%; mp: 145–148 °C; IR (KBr cm−1): 3012 (C–H stretch), 1667 (C
O), 1639 (CN stretch), 1542 (CC aromatic), 1263 (C–O stretch), 1236 (C–N stretch), 1157 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.29 (1H, s), 6.78 (1H, s), 6.31–6.30 (2H, bs), 5.60 (1H, dd, J = 4.8 and 11.4 Hz), 3.53 (1H, dd, J = 11.7 and 17.4 Hz), 3.37 (1H, dd, J = 4.8 and 17.4 Hz), 2.64 (3H, s), 2.41 (3H, s), 2.34 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.02, 15.8, 21.9, 40.5, 52.4, 107.4, 110.5, 125.5, 127.8, 136.0, 139.1, 141.8, 151.3, 152.2, 168.81. MS (EI): 288.3 (m/z). Anal. calcd for C21H20N2O2: C, 75.88; H, 6.06; N, 8.43. Found: C, 75.92; H, 6.31; N, 8.24.
Synthesis of 1-[3-(2,5-dimethyl-furan-3-yl)-5-(3,4,5-trimethoxy-phenyl)-4,5-dihydro-pyrazol-1H-1-yl]-ethanone (27). Yellow solid; yield 70%; mp: 156–158 °C; IR (KBr cm−1): 3024 (C–H stretch), 1665 (C
O), 1617 (CN stretch), 1561 (CC aromatic), 1251 (C–O stretch), 1167 (C–N stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 6.40 (2H, s), 6.13 (1H, s), 5.43 (1H, dd, J = 4.5 and 11.7 Hz), 3.82 (6H, s), 3.80 (3H, s), 3.59 (1H, dd, J = 11.7 and 17.7 Hz), 2.99 (1H, dd, J = 4.8 and 17.7 Hz), 2.47 (3H, s), 2.38 (3H, s), 2.26 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.5, 15.4, 20.36, 21.4, 55.8, 55.9, 58.1, 128.2, 135.3, 138.3, 140.1, 151.54, 167.8. MS (EI): 372.1 (m/z). Anal. calcd for C20H24N2O5: C, 64.50; H, 6.50; N, 7.52. Found: C, 64.21; H, 6.61; N, 7.88.
Synthesis of 1-[3-(2,5-dimethylfuran-3-yl)-5-(2-methoxy-phenyl)-4,5-dihydro pyrazol-1H-1-yl]ethanone (28). Dark yellow solid; yield 79%; mp: 152–155 °C; IR (KBr cm−1): 3011 (C–H stretch), 1661 (C
O), 1629 (CN stretch), 1561 (CC aromatic), 1249 (C–O stretch), 1136 (C–N stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.25 (1H, m), 6.96 (1H, d, J = 7.5 Hz), 6.86–6.90 (2H, m), 6.10 (1H, s), 5.74 (1H, dd, J = 4.5 and 11.7 Hz), 3.84 (3H, s), 3.59 (1H, dd, J = 11.7 and 17.7 Hz), 2.85 (1H, dd, J = 4.5 and 17.4 Hz), 2.45 (3H, s), 2.39 (3H, s), 2.23 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.8, 15.8, 21.7, 44.1, 56.3, 58.4, 112.1, 115.1, 123.3, 124.2, 125.6, 127.2, 137.3, 139.2, 147.1, 145.9, 151.7, 168.6. MS (EI): 312.4 (m/z). Anal. calcd for C18H20N2O3: C, 69.21; H, 6.45; N, 8.97. Found: C, 69.30; H, 6.52; N, 9.02.
Synthesis of 1-[3-(2,5-dimethylfuran-3-yl)-5-(5-hydroxy-2-nitro-phenyl)-4,5-dihydro-pyrazol-1H-1-yl]ethanone (29). Black solid; yield 70%; mp: 212–215 °C; IR (KBr cm−1): 3015 (C–H stretch), 1659 (C
O), 1631 (CN stretch), 1529 (CC aromatic), 1531 (NO2 asymmetric stretch), 1342 (NO2 symmetric stretch), 1239 (C–O stretch), 1229 (C–N stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.78 (1H, d, J = 9 Hz), 6.44 (1H, s), 6.31 (1H, d, J = 8.7 Hz), 6.04–6.10 (2H, m), 3.92 (1H, dd, J = 11.4 and 17.7 Hz), 2.99 (1H, dd, J = 4.5 and 18 Hz), 2.53 (3H, s), 2.47 (3H, s), 2.24 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.5, 15.8, 21.7, 44.3, 58.3, 113.2, 114.6, 124.2, 125.3, 126.7, 128.3, 137.3, 139.6, 147.4, 148.9, 156.8, 168.8. MS (EI): 343.1 (m/z). Anal. calcd for C17H17N3O5: C, 59.47; H, 4.99; N, 12.24. Found: C, 59.39; H, 5.05; N, 12.38.
Synthesis of 1-[3-(2,5-dimethylfuran-3-yl)-5-furan-2-yl-4,5-dihydropyrazol-1H-1-yl]ethanone (30). Black solid; yield 75%; mp: 98–100 °C; IR (KBr cm−1): 3022 (C–H stretch), 1657 (C
O), 1629 (CN stretch), 1531 (CC aromatic), 1252 (C–O stretch), 1248 (C–N stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.29 (1H, s), 6.30 (2H, s), 6.14 (1H, s), 5.60 (1H, dd, J = 4.5 and 11.4 Hz), 3.45 (1H, dd, J = 11.7 and 17.4 Hz), 3.28 (1H, dd, J = 4.5 and 17.1 Hz), 2.49 (3H, s), 2.32 (3H, s), 2.26 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.3, 15.8, 21.7, 44.3, 58.1, 122.3, 125.3, 126.7, 127.3, 136.3, 139.6, 146.3, 150.9, 151.6, 167.9. MS (EI): 272.4 (m/z). Anal. calcd for C15H16N2O2S: C, 62.48; H, 5.59; N, 9.71; S, 11.12. Found: C, 62.33; H, 5.61; N, 9.62; S, 11.21.
Synthesis of 1-[3-(2,5-dimethyl-furan-3-yl)-5-naphthalen-1-yl-4,5-dihydro-pyrazol-1H-1-yl]ethanone (31). Brown semisolid; yield 65%; IR (KBr cm−1): 2998 (C–H stretch), 1671 (C
O), 1625 (CN stretch), 1553 (CC aromatic), 1272 (C–O stretch), 1235 (C–N stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 7.22–7.78 (7H, m), 6.37 (1H, s), 6.12 (1H, m), 3.79 (1H, dd, J = 11.7 and 17.1 Hz), 2.96 (1H, dd, J = 4.2 and 17.4 Hz), 2.49 (3H, s), 2.35 (3H, s), 2.28 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.1, 15.8, 21.9, 44.7, 59.1, 122.4, 125.4, 125.4, 125.8, 126.4, 127.7, 128.2, 129.1, 132.9, 134.3, 136.1, 138.1, 139.2, 148.1, 151.3, 168.8. MS (EI): 332.3 (m/z). Anal. calcd for C15H16N2O3: C, 66.16; H, 5.92; N, 10.29. Found: C, 66.22; H, 6.01; N, 10.38.
Synthesis of 1-(5-(benzo[d][1,3]dioxol-5-yl)-3-(2,5-dimethylfuran-3-yl)-4,5-dihydro-1H-pyrazol-1-yl)ethanone (32). Light yellow solid; yield 77%; mp: 145–148 °C; IR (KBr cm−1): 2982 (C–H stretch), 1671 (C
O stretch), 1639 (CN stretch), 1547 (CC aromatic), 1263 (C–O stretch), 1224 (C–N stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 6.71–6.72 (2H, m), 6.68 (1H, m), 6.12 (1H, s), 5.90 (2H, s), 5.41 (1H, dd, J = 4.5 and 11.7 Hz), 3.58 (1H, dd, J = 11.7 and 17.7 Hz), 2.96 (1H, dd, J = 4.5 and 17.4 Hz), 2.47 (3H, s), 2.34 (3H, s), 2.25 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 14.1, 14.9, 21.9, 44.8, 57.8, 108.1, 110.0, 115.4, 120.1, 125.5, 128.9, 136.2, 136.4, 139.3, 147.9, 148.2, 152.1, 168.8. MS (EI): 326.1 (m/z). Anal. calcd for C18H18N2O4: C, 66.25; H, 5.56; N, 8.58. Found: C, 66.18; H, 5.63; N, 8.49.
Synthesis of acetic acid [1-(2,5-dimethyl-thiophen-3-yl)-ethylidene]-hydrazide (33). Brown solid; yield 72%; mp: 146–148 °C; IR (KBr cm−1): 2981 (C–H stretch), 1665 (C
O), 1627 (CN stretch), 1597 (CC aromatic), 1260 (C–N stretch), 1187 (C–S stretch). 1H NMR (300 MHz, CDCl3, TMS = 0) δ = 8.91 (1H, NH, D2O exchangeable proton), 6.83 (1H, s), 2.54 (3H, s), 2.40 (3H, s), 2.32 (3H, s), 2.16 (3H, s). 13C NMR (100 MHz, CDCl3, TMS = 0) δ = 15.0, 15.2, 15.8, 20.7, 125.6, 134.2, 135.3, 136.4, 145.4, 173.6. MS (EI): 210.4 (m/z). Anal. calcd for C10H14N2OS: C, 57.11; H, 6.71; N, 13.32; S, 15.25. Found: C, 57.09; H, 6.88; N, 13.21; S, 15.39.
Topoisomerase assays
All of the reagents required for the testing of the investigational compounds were purchased from TopoGEN, Inc. (Columbus, OH). The testing of the compounds was performed using a commercially available hTopo-IIα drug screening kit. All of the synthesized compounds and etoposide were dissolved in DMSO at a concentration of 1 mM as a stock solution and stored at −20 °C.45,46Molecular docking
The molecular docking studies were executed using the GLIDE 6.1 module of the Schrödinger Suite. The co-crystal structure of human topoisomerase IIα with ANP (PDB code-1ZXM)6 was retrieved from the protein data bank. The R and S configurations of compounds 10, 11 and 29 were sketched in ChemBio3D Ultra 12.0 and the energy minimization was performed using the MM2 force field. The ligands were prepared and neutralized using the LIGPREP module. The protein preparation step involved pre-processing and filling up missing side chains and loops using PRIME. The metal binding states were generated and the protein was then optimized and minimized using the PREP WIZ module. The grid box was generated at the ATP binding site, around the centroid of the bound ligand. The docking was carried out by the GLIDE XP modules.Cell culture and treatment
All of the cell lines were acquired from the National cell repository, NCCS, Pune. Cell lines signifying different human cancers were grown in DMEM media with added 10% fetal bovine serum (FBS) and antibiotic solution (1% Penstrip, all of the reagents were from Invitrogen). Cells were cultured in DMEM media with 10% FBS, 50 U mL−1 penicillin G, 50 Ig mL−1 streptomycin sulfate and 1.25 lg mL−1 amphotericin B (fungizone). The cells were incubated at 37 °C with 5% CO2 and 95% humidity conditions. For experiments, cells were seeded in equal numbers after trypan blue cell counting (8000 cells per well of a 96-well plate). The compounds were dissolved in cell culture grade DMSO up to a concentration of 100 mM and further dilutions were done in serum free DMEM media. The total amount of media per well (100 μL per well of a 96-well plate) was kept constant and all the treatment volumes were accommodated within these ranges only.3-(4,5-Dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay
An MTT assay was carried out using a 96-well plate; each well was filled with 100 μL media to which cells were treated and subsequently washed with 1% PBS and were mixed with a 10 μL mL−1 well of MTT (5 mg in 10 mL of 1% PBS), and incubated at room temperature in the dark for 4 h to allow formation of formazan crystals. Each well was then mixed with 100 μL of DMSO to dissolve the crystals, followed by readings using microplate reader at 570 nm. The results were then represented as the mean ± S.D obtained from three independent experiments.Reactive oxygen species (ROS) and mitochondrial membrane integrity assays
MCF-7 cancer cells were seeded in a 96-well plate and the appropriate treatments of synthetics were given. 24 h post treatment, the cells were processed for JC-1 or DHE staining. For DHE staining, cells were washed with 1× PBS followed by staining with DHE at 37 °C for 30 minutes. Afterwards cells were washed again with ice-cold 1× PBS, and finally 100 μL of 1× PBS was added to each well, followed by measuring of the OD using a microplate reader. For the JC-1 assay, dye was directly added to the media and cells were incubated at 37 °C for 30 minutes, followed by washing with 1× PBS to remove extra dye. The OD was measured using a microplate reader (emission = 527 and 590).Cell cycle analysis
Cell cycle analysis was performed using flow cytometry (BD Accuri). Cellular samples (HCT-116 wild type previously treated with compound 11 for 24 h) were prepared for staining by transferring 1 × 105 to 1 × 106 cells to each tube. Then the cells were centrifuged at 1200 rpm for 5 min and washed with 1× PBS. The cells were then suspended using chilled ethanol and the cell concentration thereby raised to 5 × 105 to 1 × 106 cells per mL. The cells were then incubated for 3 h at −20 °C. After three hours the cells were treated with ribonuclease A (100 μg mL−1) to ensure that only DNA and not RNA gets stained, and were further incubated for 30 min. Then they were subjected to centrifugation at 2500 rpm and washed once with 1× PBS. Propidium iodide 200 μL (from 50 μg mL−1 stock solution) was added to each tube, incubated for 30 minutes at room temperature in the dark, and further analysis was done using the flow cytometry instrument.Acknowledgements
RK and SS thank DST and UGC, New Delhi, India for the financial assistance (F.No. SR/FT/CS-71/2011) and F.30-13/2013(BSR), respectively. SMA, ATB and UCB gratefully acknowledge the financial aid from DBT Government of India, New Delhi to carry out hTopoI and hTopoII activity evaluation studies. The great help during the synthetic work by Prof. K. L. Dhar and his team is well appreciated. The encouragement and support from Prof. P. Ramarao, Dean, and Prof. R. K. Kohli, Vice Chancellor Central University of Punjab, are gratefully acknowledged.References
- Y. Pommier, E. Leo, H. L. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- S.-y. N. Huang, S. Ghosh and Y. Pommier, J. Biol. Chem., 2015, 290, 14068–14076 CrossRef CAS PubMed.
- K. D. Corbett and J. M. Berger, Annu. Rev. Biophys. Biomol. Struct., 2004, 33, 95–118 CrossRef CAS PubMed.
- P. Chene, Nat. Rev. Drug Discovery, 2002, 1, 665–673 CrossRef CAS PubMed.
- J. C. Wang, Nat. Rev. Mol. Cell Biol., 2002, 3, 430–440 CrossRef CAS PubMed.
- H. Wei, A. J. Ruthenburg, S. K. Bechis and G. L. Verdine, J. Biol. Chem., 2005, 280, 37041–37047 CrossRef CAS PubMed.
- J. Roca, J. M. Berger, S. C. Harrison and J. C. Wang, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4057 CrossRef CAS.
- S. Classen, S. Olland and J. M. Berger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10629 CrossRef CAS PubMed.
- J. Roca, J. Biol. Chem., 2004, 279, 25783–25788 CrossRef CAS PubMed.
- M. Heck and W. C. Earnshaw, J. Cell Biol., 1986, 103, 2569–2581 CrossRef CAS PubMed.
- S. M. Cutts, A. Nudelman, A. Rephaeli and D. R. Phillips, IUBMB Life, 2005, 57, 73–81 CrossRef CAS PubMed.
- A. M. Wilstermann, R. P. Bender, M. Godfrey, S. Choi, C. Anklin, D. B. Berkowitz, N. Osheroff and D. E. Graves, Biochemistry, 2007, 46, 8217–8225 CrossRef CAS PubMed.
- S. J. Berger, S. Gupta, C. A. Belfi, D. M. Gosky and H. Mukhtar, Biochem. Biophys. Res. Commun., 2001, 288, 101–105 CrossRef CAS PubMed.
- O. J. Bandele and N. Osheroff, Chem. Res. Toxicol., 2008, 21, 936–943 CrossRef CAS PubMed.
- O. J. Bandele, S. J. Clawson and N. Osheroff, Chem. Res. Toxicol., 2008, 21, 1253–1260 CrossRef CAS PubMed.
- Y. Yoshinori, K. Sho-Zou and N. Hirofumi, Biochem. Pharmacol., 1990, 39, 737–744 CrossRef.
- J. Markovits, C. Linassier, P. Fosse, J. Couprie, J. Pierre, A. Jacquemin-Sablon, J. M. Saucier, J. B. L. Pecq and A. K. Larsen, Cancer Res., 1989, 49, 5111 CAS.
- A. M. Wilstermann and N. Osheroff, Curr. Top. Med. Chem., 2003, 3, 321–338 CrossRef CAS PubMed.
- P. Furet, J. Schoepfer, T. Radimerski and P. Chène, Bioorg. Med. Chem. Lett., 2009, 19, 4014–4017 CrossRef CAS PubMed.
- S. Jimenez-Alonso, H. C. Orellana, A. Estévez-Braun, A. G. Ravelo, E. Pérez-Sacau and F. Machín, J. Med. Chem., 2008, 51, 6761–6772 CrossRef CAS PubMed.
- A. T. Baviskar, C. Madaan, R. Preet, P. Mohapatra, V. Jain, A. Agarwal, S. K. Guchhait, C. N. Kundu, U. C. Banerjee and P. V. Bharatam, J. Med. Chem., 2011, 54, 5013–5030 CrossRef CAS PubMed.
- K. Shiomi, I. Kuriyama, H. Yoshida and Y. Mizushina, Food Chem., 2013, 139, 910–918 CrossRef CAS PubMed.
- J. Y. Jo, E. G. de Mejia and M. A. Lila, J. Agric. Food Chem., 2006, 54, 2083–2087 CrossRef CAS PubMed.
- R. D. Snyder and P. J. Gillies, Environ. Mol. Mutagen., 2002, 40, 266–276 CrossRef CAS PubMed.
- R. D. Snyder and P. J. Gillies, Food Chem. Toxicol., 2003, 41, 1291–1298 CrossRef CAS PubMed.
- A. K. Larsen, A. E. Escargueil and A. Skladanowski, Pharmacol. Ther., 2003, 99, 167–181 CrossRef CAS PubMed.
- A. Negi, J. M. Alex, S. M. Amrutkar, A. T. Baviskar, G. Joshi, S. Singh, U. C. Banerjee and R. Kumar, Bioorg. Med. Chem., 2015, 23, 5654–5661 CrossRef CAS PubMed.
- J. M. Alex, S. Singh and R. Kumar, Arch. Pharm., 2014, 347, 717–727 CrossRef CAS PubMed.
- M. Chauhan, A. Rana, J. M. Alex, A. Negi, S. Singh and R. Kumar, Bioorg. Chem., 2015, 58, 1–10 CrossRef CAS PubMed.
- A. Negi, P. Ramarao and R. Kumar, Mini-Rev. Med. Chem., 2013, 13, 653–681 CrossRef CAS PubMed.
- A. T. Baviskar, U. C. Banerjee, M. Gupta, R. Singh, S. Kumar, M. K. Gupta, S. Kumar, S. K. Raut, M. Khullar and S. Singh, Bioorg. Med. Chem., 2013, 21, 5782–5793 CrossRef CAS PubMed.
- G. Joshi, P. K. Singh, A. Negi, A. Rana, S. Singh and R. Kumar, Chem.-Biol. Interact., 2015, 240, 120–133 CrossRef CAS PubMed.
- M. Stefanisinova, V. Tomeckova, M. Kozurkova, A. Ostro and M. Marekova, Spectrochim. Acta, Part A, 2011, 81, 666–671 CrossRef CAS PubMed.
- Y. Na and J.-M. Nam, Bioorg. Med. Chem. Lett., 2011, 21, 211–214 CrossRef CAS PubMed.
- A. Basnet, P. Thapa, R. Karki, H. Choi, J. H. Choi, M. Yun, B.-S. Jeong, Y. Jahng, Y. Na and W.-J. Cho, Bioorg. Med. Chem. Lett., 2010, 20, 42–47 CrossRef CAS PubMed.
- X. Xu, H. J. Wang, P. A. Murphy, L. Cook and S. Hendrich, J. Nutr., 1994, 124, 825 CAS.
- P. C. H. Hollman, J. M. P. van Trijp, M. N. C. P. Buysman, M. J. B. Mengelers, J. H. M. de Vries and M. B. Katan, FEBS Lett., 1997, 418, 152–156 CrossRef CAS PubMed.
- M. J. Lee, Z. Y. Wang, H. Li, L. Chen, Y. Sun, S. Gobbo, D. A. Balentine and C. S. Yang, Cancer Epidemiol., Biomarkers Prev., 1995, 4, 393–399 CAS.
- C. H. Cottart, V. Nivet-Antoine, C. Laguillier-Morizot and J. L. Beaudeux, Mol. Nutr. Food Res., 2010, 54, 7–16 CAS.
- J. M. Fortune and N. Osheroff, J. Biol. Chem., 1998, 273, 17643–17650 CrossRef CAS PubMed.
- K. Hande, Eur. J. Cancer, 1998, 34, 1514–1521 CrossRef CAS PubMed.
- A. Rajapakse, C. Linder, R. D. Morrison, U. Sarkar, N. D. Leigh, C. L. Barnes, J. S. Daniels and K. S. Gates, Chem. Res. Toxicol., 2013, 26, 555–563 CrossRef CAS PubMed.
- M. Fragkos and P. Beard, PLoS One, 2011, 6, e22946 CAS.
- Z. Darzynkiewicz, S. Bruno, G. Del Bino, W. Gorczyca, M. Hotz, P. Lassota and F. Traganos, Cytometry, 1992, 13, 795 CrossRef CAS PubMed.
- A. T. Baviskar, S. M. Amrutkar, N. Trivedi, V. Chaudhary, A. Nayak, S. K. Guchhait, U. C. Banerjee, P. V. Bharatam and C. N. Kundu, ACS Med. Chem. Lett., 2015, 6, 481–485 CrossRef CAS PubMed.
- S. Jimenez-Alonso, H. C. Orellana, A. Estevez-Braun, A. G. Ravelo, E. Perez-Sacau and F. Machin, J. Med. Chem., 2008, 51, 6761–6772 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of selected compounds are provided. See DOI: 10.1039/c5ra25705k |
| This journal is © The Royal Society of Chemistry 2016 |