
Design, synthesis, biological evaluation and molecular docking of amide and sulfamide derivatives as Escherichia coli pyruvate dehydrogenase complex E1 inhibitors†
Haifeng He‡
a,
Jiangtao Feng‡a,
Junbo Heb,
Qin Xiaa,
Yanliang Rena,
Fang Wanga,
Hao Penga,
Hongwu He*a and
Lingling Feng*a
aKey Laboratory of Pesticide and Chemical Biology of Ministry of Education, College of Chemistry, Central China Normal University, 152 Luoyu Road, Wuhan 430079, P. R. China. E-mail: he1208@mail.ccnu.edu.cn; fll708@mail.ccnu.edu.cn; Fax: +86-27-67867960
bCollege of Food Science & Engineering, Wuhan Polytechnic University, Wuhan 430023, China
First published on 4th January 2016
Abstract
In this study, a series of novel amide derivatives and sulfamide derivatives as potential E. coli PDHc E1 inhibitors were designed and synthesized by optimizing the linker between triazole and benzene ring moieties based on the structure of lead compound I as thiamin diphosphate (ThDP) analogs. Their inhibitory activity against E. coli PDHc E1 were examined in vitro and their inhibitory activity against microbial diseases were further evaluated. Most of these compounds exhibit good inhibitory activity against E. coli PHDc E1 (IC50 1.99 to 25.66 μM) and obvious antibacterial activity. 5a, 5c and 9i showed 90–100% antibacterial activity against Xanthomonas oryzae pv. oryzae (Xoo), Acidovorax avenae subsp. avenae (Aaa) and cyanobacteria. Sulfamide derivatives 9 showed more potent inhibitory activity against E. coli PDHc E1 (IC50 < 14 μM) than that of amide derivatives 5 or lead compound I. Especially 9d (IC50 = 2.95 μM) and 9k (IC50 = 1.99 μM) exhibited not only the most powerful inhibitory potency against E. coli PDHc E1, but also 9k showed 99% antibacterial activity against Aaa at 500 μg mL−1 and almost the best inhibition of 97% against cyanobacteria at 20 μg mL−1. Furthermore, the binding mode of 5d and 9d to E. coli PDHc E1 was analyzed by a molecular docking method. The possible interactions of 9d with the important residues of E. coli PDHc E1 were further verified via site-directed mutagenesis enzymatic assays, and fluorescence spectral analysis. Both theoretical and experimental results revealed that 9d could display a more powerful interaction than that of 5d or I by forming a hydrogen bond between a sulfamide linkage and residues Lsy392, Tyr599 and His106 at active site of E. coli PDHc E1. 9k, 9d and 9i with both potent enzyme inhibition and significant antibacterial activity, could be used as novel lead compounds for further optimization. These results proved that a series of compounds with potential antibacterial activity could be obtained by the biorational design of E. coli PDHc E1 inhibitors.
Introduction
Bactericides play an important role in modern agriculture due to the harm of bacteria in agriculture. Although there are many bactericides can control these bacterium infections, the repeated use of the same bactericides or repeated treatment with bactericides having the same mode of action, has resulted in the widespread evolution of resistance.1,2 It is essential to develop efficient microbicides or bactericides with novel structures or modes of action to overcome microbial disease and bactericide resistance.The pyruvate dehydrogenase complex (PDHc) plays a key regulatory role in cellular metabolism catalyzing the oxidative decarboxylation of pyruvate and the subsequent acetylation of coenzyme A (CoA) to acetyl-CoA.3,4 The overall reaction of oxidative decarboxylation can be simply shown in Fig. 1.
 | ||
| Fig. 1 Oxidative decarboxylation catalyzed by PDHc. | ||
The complex is comprised of three different enzymes components including pyruvate dehydrogenase (E1), dihydrolipoamide acetyltransferase (E2), dihydrolipoamide dehydrogenase (E3) and a number of cofactors.5 Pyruvate dehydrogenase complex E1 component (PDHc E1, EC 1.2.4.1) is the initial member of PDHc, which catalyzes the first step of multistep process, using thiamine diphosphate (ThDP) and Mg2+ as cofactors.6–8 Especially this PDHc E1 catalyzed process is the rate limiting step among multistep process which is catalyzed by PDHc. Therefore, blocking the activity of PDHc E1 will be the best way to inactivate the PDHc. In our study, E. coli PDHc E1 was selected as the target, which should be an interesting site of action for bactericide design. Some ThDP analogs as inhibitors of PDHc E1 in E. coli have been studied and reported (such as triazole ThDP in Fig. 2) because of the important role of ThDP in oxidative decarboxylation catalyzed by PDHc.9–13 However there was little report about their bactericidal or fungicidal activity. In fact some reported ThDP analogs are unsuitable for the usage as agricultural chemicals due to their complex structure with highly charged pyrophosphate and poor bioavailability. Aiming at the above-mentioned problems, both the thiazolium ring and the pyrophosphate moiety in ThDP were replaced by 1,2,3-triazole ring and substituted benzene ring, respectively. It has been verified that some hit compounds with aminopyrimidine, triazole and benzene ring moiety could be effective in occupying the ThDP-binding pocket of PDHc E1 by using structure-based molecular docking.14
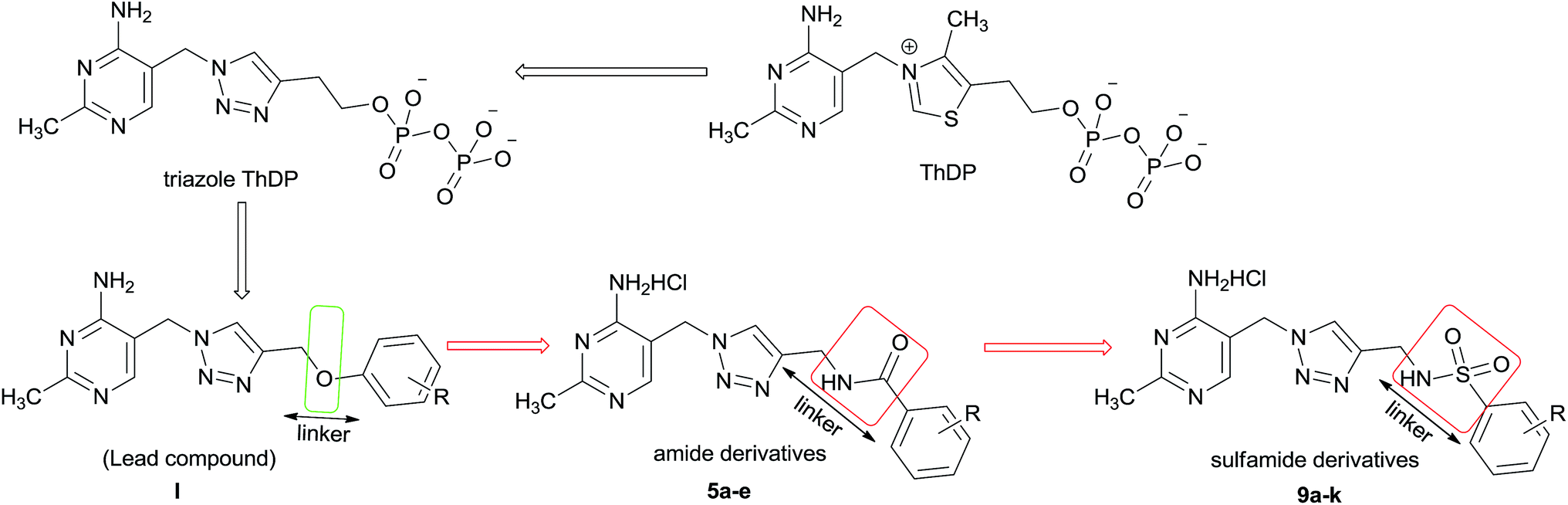 | ||
| Fig. 2 ThDP analogue and design of new amide and sulfamide derivatives as E. coli PDHc E1 inhibitors. | ||
In order to obtain potential fungicide or bactericide by designing PHDc E1 inhibitor, series of 2-methylpyrimidine-4-ylamine derivatives I containing 1,2,3-triazole ring and substituted benzene ring (Fig. 2) as ThDP analogs had been firstly chemically synthesized and demonstrated to be effective inhibitors against E. coli PDHc E1.15,16 The structure skeleton of I avoided the high charge of the pyrophosphate moiety and some of these compounds exhibited antifungal activity. These preliminary progresses encouraged us to choose I as lead compound for further optimization. As a distinguishing feature, ether (C–O–C) group was used as a linker to connect the triazole and benzene ring in lead compound I. The result of molecular docking showed that there was no interaction between oxygen atom of ether moiety and amino acid residue in the active site of PHDc-E1.16 It was thought that the formation of hydrogen bond between the structural part of linker and amino acid residues in the active site should be much beneficial for enhancing inhibition against E. coli PDHc E1. In order to increase the inhibitory potency against E. coli PDHc E1, the scaffold of 2-methylpyrimidine-4-ylamine derivatives containing 1,2,3-triazole and benzene ring were kept, further optimization focused on “linker part” between the triazole and benzene ring. Considering amide or sulfamide group containing NH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or OSO structure unit, which could be used as hydrogen donor (NH) or hydrogen receptor (CO, OSO). Therefore, amide or sulfamide group as a “linker part” was introduced into the parent structure respectively to design novel series of N-((1-((4-amino-2-methylpyrimidin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)-substituted-benzamide hydrochloride 5, and N-((1-((4-amino-2-methylpyrimidin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)-substituted-benzenesulfonamide hydrochloride 9.
O or OSO structure unit, which could be used as hydrogen donor (NH) or hydrogen receptor (CO, OSO). Therefore, amide or sulfamide group as a “linker part” was introduced into the parent structure respectively to design novel series of N-((1-((4-amino-2-methylpyrimidin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)-substituted-benzamide hydrochloride 5, and N-((1-((4-amino-2-methylpyrimidin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)-substituted-benzenesulfonamide hydrochloride 9.
Herein, we report the design and synthesis of series of new amide and sulfamide derivatives 5a–e and 9a–k by incorporating the active amide or sulfamide pharmacophore as a “linker” to form novel ThDP analog as potential inhibitors against E. coli PDHc E1 (Fig. 2). Some amide and sulfamide derivatives have attracted our attention due to their excellent antibacterial activity.17,18 Therefore, these title compounds are expected to be good E. coli PDHc E1 inhibitors with bactericidal activity. In order to make sure the effect of benzene ring in parent structure on inhibitory activity, N-((1-((4-amino-2-methylpyrimidin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl) methanesulfonamide hydrochloride 13 also was designed and synthesized.
In this work, both inhibitory activity against E. coli PDHc E1 and antibacterial activity of title compounds 5, 9 and 13 were examined. The interaction mode of the important residues of E. coli PDHc E1 with some title compounds was also studied by molecular docking method and site-directed mutagenesis, probable inhibition mechanism was discussed.
Results and discussion
Synthesis
The synthetic route of title compounds 5a–e, 9a–k and 13 is depicted in Scheme 1. Vitamin B or thiamine hydrochloride as starting material was used to prepare 5-azido-methyl-2-methylpyrimidine-4-ylamine 1, according to the literature method.19 1 is the key intermediate for the preparation of title compounds 5, 9 and 13. The title compounds 5a–e, 9a–k and 13 could be synthesized by a four or five-step sequence starting from substituted benzoic acid, substituted sulfonyl chloride or methyl sulfonyl chloride respectively. Various substituted benzoic acid reacted with oxalyl chloride in the presence of DMF to produce substituted benzoyl chlorides 2a–e, which reacted with propargylamine to form N-(prop-2-yn-1-yl)-sub-benzamide 3a–e using Et3N as base. Under same condition, substituted sulfonyl chloride 6a–k or methyl sulfonyl chloride 10 could be converted into corresponding N-(prop-2-yn-1-yl)-sub-benzenesulfonamide 7a–k or N-(prop-2-yn-1-yl)methanesulfonamide 11, respectively.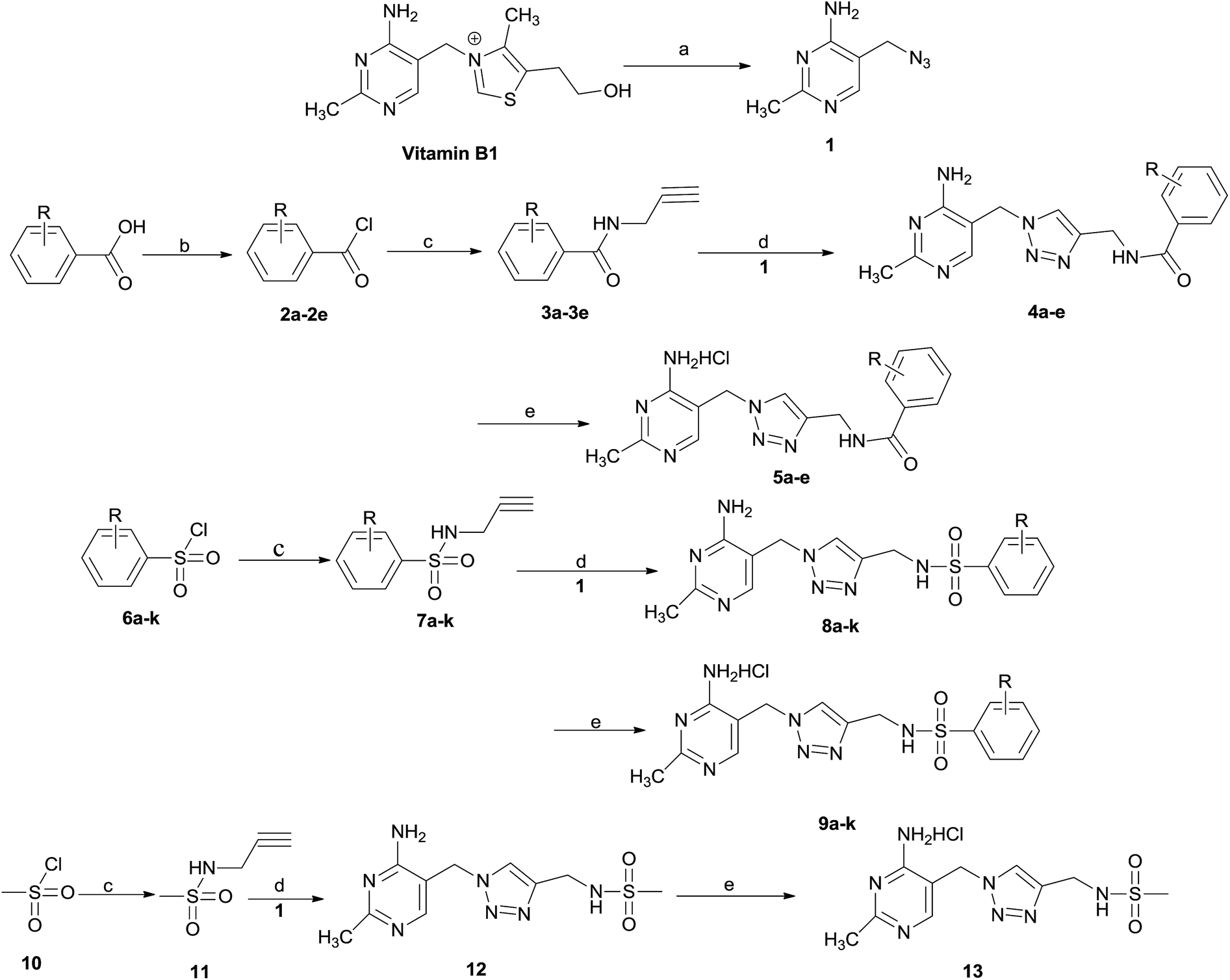 | ||
| Scheme 1 Reagents and conditions (a) NaN3, Na2SO3, H2O, 60–65 °C 56%; (b) C2O2Cl2 DCM/DMF, 0 °C; (c) CHCCH2NH2, Et3N, DCM 0 °C 70–85%; (d) CuI, Et3N, THF, rt, 10–15 h; (e) HCl. | ||
The 1,2,3-triazol ring in the parent skeleton of title compounds could be constructed by a synthetic method of ‘click chemistry’. Important intermediates, 4a–e, 8a–k or 12 with key skeleton containing 1,2,3-triazol ring were prepared by the Cu-catalyzed 1,3-dipolar cycloaddition of 1 with N-substituted-prop-2-yn-1-amines including 3a–e, 7a–k, or 11 respectively using CuI and Et3N in the presence of THF.
In order to increases the solubility of compounds, 4a–e, 8a–k and 12 were further converted into corresponding hydrochloride 5a–e, 9a–k and 13 by using hydrochloric acid. All title compounds, 5a–e, 9a–k and 13 were characterized by 1H NMR, 13C NMR, mass spectrometry (MS), and confirmed by elementary analysis.
Inhibitory potency against E. coli PDHc E1
In order to enhance the inhibitory potency against E. coli PDHc E1, lead structure I was modified by replacing ether bond (C–O–C) in the structure skeleton I with amide linkage.Several amide derivatives 5a–e were firstly synthesized and evaluated for their inhibitory activity against E. coli PDHc E1. The IC50 values of 5a–e and some I16 were summarized in Table 1. The results showed that the inhibitory activity of 5a–e against E. coli PDHc E1 could be improved by introducing amide bond into the “linker part”. Some compounds 5 exhibited better inhibitory activity than that of lead compound I, such as inhibitory activity, 5a > Ia; 5e > Ie, especially 5e showed 7-folds activity higher than that of Ie. However 5c showed lower inhibitory activity than that of Ic. On the basis of this work, the structure skeleton of 5 was further optimized by introducing sulfamide bond as a linker to form a series of 9a–k.
|
|||||
|---|---|---|---|---|---|
| Compd | R | IC50a (μM) | Compd | R | IC50a (μM) |
| a IC50 (μM) value is defined as the micromolar concentration required for 50% inhibition on PDHc E1 from E. coli in vitro. | |||||
| Ia | H | 55.15 ± 4.65 | 5e | 4-Cl | 3.16 ± 0.46 |
| 5a | H | 25.66 ± 2.34 | 9e | 4-Cl | 10.53 ± 0.70 |
| 9a | H | 4.01 ± 0.17 | If | 4-OCH3 | 81.62 ± 5.85 |
| 5b | 4-Me | 13.69 ± 1.56 | 9f | 4-OCH3 | 7.53 ± 0.31 |
| 9b | 4-Me | 12.93 ± 0.30 | 9g | 4-Br | 11.64 ± 0.39 |
| Ic | 4-NO2 | 8.8 ± 0.35 | 9h | 4-F | 9.11 ± 0.40 |
| 5c | 4-NO2 | 24.27 ± 1.84 | 9i | 2,4,6-Me3 | 6.60 ± 0.16 |
| 9c | 4-NO2 | 5.99 ± 0.37 | 9j | 2-NO2 | 5.31 ± 0.31 |
| 5d | 3-NO2 | 13.52 ± 1.48 | 9k | 3-NO2-4-Cl | 1.99 ± 0.08 |
| 9d | 3-NO2 | 2.95 ± 0.14 | 13 | 7.40 ± 0.64 | |
| Ie | 4-Cl | 26.44 ± 1.68 | |||
It was found that the inhibitory activity was further enhanced by changing amide into sulfamide linkage. When R kept same, 9a, 9c, and 9d (IC50 = 4.01 ± 0.17, 5.99 ± 0.37, and 2.95 ± 0.14 μM) with sulfamide linkage displayed higher inhibitory activity than that of corresponding 5a, 5c, and 5d (IC50 = 25.66 ± 1.34, 24.27 ± 1.84, and 13.52 ± 1.48 μM) with amide linkage. As an example, 9a (R = H) showed 6-folds activity higher than that of 5a (R = H). Although 9e showed weaker inhibitory activity than that of 5e, it was still higher than that of Ie. It was also observed that 9a, 9c, 5e, 9f (IC50 = 4.01 ± 0.17, 5.99 ± 0.37, 3.16 ± 0.46 and 7.53 ± 0.31 μM) showed higher inhibitory activity than that of corresponding compounds (IC50 = 75.5 ± 0.02, 6.7 ± 0.48, 6.9 ± 1.19 and 30.2 ± 3.45 μM), which had been reported20 with oxime ether linkage.
Above observation showed the “linker part” between the triazole and benzene ring in parent structure played a very important role in inhibitory potency against E. coli PDHc E1. Compared with ether bond or amide bond, the sulfamide bond as linker was much beneficial to inhibitory activity. These results suggest that the structure skeleton of 9 is better than both 5 and lead structure I for finding more powerful PDHc E1 inhibitor.
As shown in Table 1, R on the benzene ring also had great influence on the inhibitory activity base on the structure skeleton of 9. The inhibitory activity of 9 could be greatly enhanced by optimizing R on the benzene ring. The effects of R on inhibitory activity can be shown by following activity sequence, 9b (R = 4-CH3, IC50 = 12.93 μM) < 9g (R = 4-Br, IC50 = 11.64 μM) < 9e (R = 4-Cl, IC50 = 10.53 μM) < 9h (R = 4-F, IC50 = 9.11 μM) < 9f (R = 4-OCH3, IC50 = 7.53 μM) < 9i (R = 2,4,6-Me3, IC50 = 6.60 μM) < 9c (R = 4-NO2, IC50 = 5.99 μM) < 9j (R = 2-NO2, IC50 = 5.31 μM) < 9a (R = H, IC50 = 4.01 μM) < 9d (R = 3-NO2, IC50 = 2.95 μM) < 9k (R = 3-NO2-4-Cl, IC50 = 1.99 μM). These result showed all compounds 9 with NO2 as R, such as 9c, 9j, 9d, 9k exhibited better inhibitory activity than that of compounds 9 with other substituents, and NO2 at 3-position on the benzene ring seems to be favorable to inhibitory activity. Compound with electron-withdrawing group as R was promotive for inhibitory activity against E. coli PDHc E1. Especially, 9k with 3-NO2, 4-Cl as R was found to be most effective compound against E. coli PDHc E1. It exhibited 6-folds activity higher than that of 9b with 4-CH3 as R. Above results indicated that the inhibitory activity of 9 was also dependent upon the structure and position of R on the benzene ring.
In order to examine the effect of benzene ring in structure 9 on inhibitory activity, the benzene ring was replaced with a methyl group to prepare compound 13 and its inhibitory activity against E. coli PDHc E1 was tested. The result showed that the lost of benzene ring led to a decrease in inhibitory activity against E. coli PDHc E1 compared with 13 (IC50 = 7.40 μM) with 9a (R = H, IC50 = 4.01 μM) or 9k (R = 3-NO2-4-Cl, IC50 = 1.99 μM). It indicated that substituted benzene ring also an important pharmacophore for structure 9.
Antibacterial activity
Most reported ThDP analogs as E. coli PDHc E1 inhibitors did not show antibacterial activity or antifungal activity. In order to find useful PDHc E1 inhibitors with antibacterial activity or antifungal activity, amide derivatives 6a–e, sulfamide derivatives 9a–k and 13 as new E. coli PDHc E1 inhibitors were evaluated for their antifungal activity against Gibberella zeae (G. zeae), Rhizoctonia solani (R. solani), Botrytis cinerea (B. cinerea), Alternaria solani (A. solani), and antibacterial activity against Xanthomonas oryzae pv. oryzae (Xoo) Acidovorax avenae subsp. avenae (Aaa) and cyanobacteria.It is very interesting to find all the title compounds displayed good antibacterial activity, but very weak fungicidal activity. As shown in Table 2, all the title compounds 5 and 9 exhibited obvious antibacterial activity against cyanobacteria, Xoo or Aaa. All 5 and 9 could 90–99% control cyanobacteria at 20 μg mL−1, except 5d, 5e and 9g. Moreover 5a, 5c, 9c, 9i, and 9k could 90–99% control Aaa, and 5a–e, 9a, 9e, 9f, 9i and 9j could 90–100% control Xoo at 500 μg mL−1, but compounds I only showed <20% inhibitory potency against cyanobacteria, Xoo and Aaa. The effect of these compounds against cyanobacteria, Xoo and Aaa were comparable to commercial bactericide, CuSO4 or streptomycin sulfate as a positive control. Especially 5a, 5c and 9i showed 90–100% antibacterial activity against cyanobacteria, Xoo and Aaa.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | R | Inhibitory potencyb (%) 20 μg mL−1 | Inhibitory potencyb (%) 500 μg mL−1 | Compd | R | Inhibitory potencyb (%) 20 μg mL−1 | Inhibitory potencyb (%) 500 μg mL−1 | ||
| Cyanobacteria | Aaaa | Xooa | Cyanobacteria | Aaaa | Xooa | ||||
| a Xoo, Xanthomonas oryzae pv. oryzae; Aaa, Acidovorax avenae subsp. avenae.b Inhibitory potency (%) against the growth of pathogenic fungi at 100 mg mL−1, 0 (no effect), 100% (completely kill). | |||||||||
| Ia | H | <20 | <10 | <10 | 9d | 3-NO2 | 97 | 84 | 76 |
| Ic | 4-NO2 | <20 | <10 | <10 | 9e | 4-Cl | 94 | 80 | 100 |
| Ie | 4-Cl | <20 | <10 | <10 | 9f | 4-OCH3 | 94 | 84 | 100 |
| If | 4-OCH3 | <20 | <10 | <10 | 9g | 4-Br | 77 | 60 | 81 |
| 5a | H | 92 | 99 | 95 | 9h | 4-F | 96 | 80 | 75 |
| 5b | 4-Me | 90 | 60 | 93 | 9i | 2,4,6-Me3 | 99 | 90 | 100 |
| 5c | 4-NO2 | 94 | 99 | 90 | 9j | 2-NO2 | 94 | 79 | 99 |
| 5d | 3-NO2 | 76 | 39 | 96 | 9k | 3-NO2-4-Cl | 97 | 99 | 28 |
| 5e | 4-Cl | 59 | 71 | 95 | 13 | 99 | 89 | 85 | |
| 9a | H | 95 | 50 | 99 | CuSO4 | 95 | |||
| 9b | 4-Me | 94 | 78 | 74 | Streptomycin sulfate | 97 | 98 | ||
| 9c | 4-NO2 | 97 | 90 | 80 | |||||
According to the data in Tables 1 and 2, all title compounds 5 and 9 with the IC50 values ranging from 1.99 to 25.66 μM against E. coli PHDc E1 could exhibit moderate to good antibacterial activity against Xoo and Aaa at 500 μg mL−1, and showed obviously inhibition against cyanobacteria at 20 μg mL−1. However I with much weaker inhibitory potency against E. coli PHDc E1 showed much weaker or no inhibition against cyanobacteria, Xoo and Aaa. When the ether (C–O–C) group as a linker in lead structure I was replaced by an amide or sulfamide group, both enzyme inhibitory potency and antibacterial activity could be greatly enhanced. Most title compounds 5 and 9 with amide or sulfamide linkage exhibited higher inhibitory potency and antibacterial activity than that of lead compounds I with ether linkage. And all of the reported compounds with oxime ether linkage had no antibacterial activity reported.20 When R was kept same, 9d, or 9e with sulfamide linkage seemed to exhibit better inhibition against cyanobacteria than that of corresponding amide derivatives 5d or 5e. The results showed that R on the benzene ring also had significant influence on the inhibition against cyanobacteria, Xoo and Aaa, but there was not clear regularity to establish the relationship of the structure and position of R on antibacterial activity.
The bioassay showed that 9d and 9k with sulfamide linkage were the most two effective compounds (IC50 = 2.95 and 1.99 μM) against E. coli PDHc E1. 9d and 9k with 3-NO2 and 3-NO2-4-Cl as R exhibited much higher inhibitory potency against E. coli PDHc E1 than that of corresponding 5d (R = 3-NO2, IC50 = 13.52 μM). It was found that 9d and 9k with 97% inhibitory potency also displayed higher inhibitory activity against cyanobacteria than that of 5d with 76% inhibitory potency at 20 μg mL−1. 9k, displaying the most potent inhibitory activity against E. coli PDHc E1, showed almost best inhibitory activity against cyanobacteria at 20 μg mL−1 and Aaa at 500 μg mL−1. It was noted that 9i with potent inhibitory potency against E. coli PDHc E1 displayed 90–100% inhibition against cyanobacteria, Xoo and Aaa at the test concentration.
Above observation showed that the degree of antibacterial activity of most tested compounds positively correlated with that of their inhibition against E. coli PDHc E1. Both amide and sulfamide group, especial sulfamide linkage is much favorable to both enzyme inhibition against E. coli PDHc E1 and antibacterial activity against Xoo, Aaa and cyanobacteria. Although 5a–e and 9a–e exhibited obvious antibacterial activity, they had no significant fungicidal activity against tested fungus. As shown in Table 3, all amide derivatives 5a–e and sulfamide derivatives 9a–e only showed <50% (0–43%) inhibitory potency against G. zeae, R. solani, B. cinerea and A. solani. According to our previous results, Ia, Ic, Ie also had similar weak inhibitory potency (0–73%) against G. zeae, R. solani, B. cinerea and A. solani.15 These results suggested that these title compounds 5 and 9 could selectively inhibit bacterium due to their good inhibition against PDHc E1 from E. coli. Their inhibition against cyanobacteria is worth further examination.
| Compd | Inhibitory potencyb (%) | |||
|---|---|---|---|---|
| G. zeaea | R. solania | B. cinereaa | A. solania | |
| a G. zeae, Gibberella zeae; R. solani, Rhizoctonia solani; B. cinerea, Botrytis cinerea; A. solani, Alternaria solani.b Inhibitory potency (%) against the growth of pathogenic fungi at 100 mg mL−1, 0 (no effect), 100% (completely kill). | ||||
| 5a | 0 | 0 | 33 | 0 |
| 5b | 0 | 0 | 20 | 0 |
| 5c | 0 | 0 | 26 | 0 |
| 5d | 23 | 43 | 64 | 26 |
| 5e | 0 | 39 | 39 | 22 |
| 9a | 19 | 19 | 7 | 18 |
| 9b | 14 | 3 | 46 | 9 |
| 9c | 23 | 18 | 7 | 18 |
| 9d | 14 | 3 | 7 | 18 |
| 9e | 23 | 9 | 14 | 18 |
| 9f | 28 | 28 | 10 | 27 |
| 9g | 19 | 9 | 17 | 9 |
| 9h | 9 | 31 | 7 | 18 |
| 9i | 23 | 12 | 32 | 9 |
| 9j | 23 | 21 | 35 | 9 |
| 9k | 19 | 25 | 7 | 27 |
| 13 | 28 | 15 | 14 | 18 |
Analyses of the interaction between inhibitors and E. coli PDHc E1
In order to understand the mechanism of antibacterial activity, the interaction mode of amide derivative 5 or sulfamide derivative 9 with active site of E. coli PDHc E1 was explored. Several molecular docking simulation studies were carried out by using the SURFLEX module of SYBYL package.14 In this study, both 5d and 9d with 3-NO2 as R displaying significant inhibitory activity against E. coli PDHc E1, were selected as hit compounds for molecular docking.The binding mode of 5d and 9d is shown in Fig. 3A and B, respectively. 5d and 9d with the ‘V’ conformation occupies the ThDP-binding pocket and bind in the active site of E. coli PDHc E1. On the right side of the ‘V’ conformation, the aminopyrimidine ring of 5d and 9d can form hydrogen bonds with amino acid residues Met194, Glu571 and Val192 (Fig. 3A and B), which is similar to the interactions of ThDP or lead structure I with amino acid residues. The nitryl group as R on the benzene ring of 5d or 9d respectively has an interaction with Asn260 and Ser109 in the active site of E. coli PDHc E1 by forming hydrogen bonds. In order to validate the prediction of molecular docking, site-directed mutagenesis and enzymatic assays were performed. These results showed the IC50 values of 9d against mutants M194A (20.2 μM), G571A (21.67 μM), V192A (42.13 μM) and S109A (13.78 μM) were about 5.8-fold, 6.3-fold, 13.3-fold and 3.7-fold higher than its value against wild-type PDHc E1 enzyme (2.95 μM), respectively (Fig. 4). These results suggest that the hydrogen bonding interaction between 9d and amino acid residues Met194, Glu571, Val192 or Ser109 plays an important role in the binding of 9d with E. coli PDHc E1.
 | ||
| Fig. 3 Binding modes of compound 5d (A) and 9d (B) target into active site of E. coli PDHc E1, in which PDHc E1 is shown in ribbon, ligands and some key residues are shown in stick, both coordination bonds and hydrogen bonds are shown in dashed lines. | ||
 | ||
| Fig. 4 The IC50 values of compound 9d against the wild type (WT) and mutants of E. coli PDHc E1. | ||
It is not possible to confirm the interaction between 9d and Asn260 through the enzymatic assay of the N260A mutant directly due to this mutant exhibits much less enzymatic activity. Therefore the binding constant (Kb) values of N260A were investigated using fluorescence spectral. As shown in Fig. 5, the Kb value of wild type enzyme (3100 M−1) is over 9-fold higher than the Kb value of N260A (330 M−1), suggesting that there is a stronger interaction between 9d and Asn260. These results further confirm and explain the binding-mode between the inhibitors and the active site of PDHc E1. Above observation showed that the binding mode of 4-aminopyrimidine and substituted benzene ring in 5d and 9d with amino acid residues in the active site of E. coli PDHc E1 were very similar to the binding mode of lead compound I.
 | ||
| Fig. 5 Binding constants (Kb) determined by fluorescence spectral analyses for the binding of compound 9d to the wild type (WT) and mutants of E. coli PDHc E1. | ||
It was very interesting to explore the different of the binding mode of I, 5 and 9 by comparing the linker in their parent structures. No hydrogen bond between any amino acid residues and the oxygen atom of ether bond (C–O–C) as a linker at the middle of the ‘V’ conformation of structure I was observed according to previous molecular docking study on structure I.14–16 However the oxygen atom of amide (CO) bond as a linker at the middle of the ‘V’ conformation of compound 5d could form a strong hydrogen bond with residue Lsy392 (Fig. 3A), which was very important for stabilizing the bound of 5d with the enzyme. As shown in Fig. 3B, one of the oxygen atoms of sulfamide linkage in 9d can form two strong hydrogen bonds with Lsy392, the other oxygen atom of sulfamide linkage can form a strong hydrogen bond with His106. Meanwhile, the 1,2,3-triazole moiety of 9d also form a hydrogen bond with Tyr599. It showed that the binding mode of 9d with sulfamide linkage displayed much powerful interaction than that of 5d with amide linkage or lead compound I. Site-directed mutagenesis and enzymatic assays showed that the IC50 values of compound 9d against the mutants K392A (22.19 μM), H106A (21.14 μM) and Y599A (9.61 μM) were about 7.5-fold, 6-fold and 2.3-fold higher than its value against wild-type PDHc E1 enzyme (2.95 μM) (Fig. 4). This suggests that the interaction between 9d and residue Lsy392, Tyr599 or His106 by forming hydrogen bond has a significant contribution for its inhibitory activity against E. coli PDHc E1.
It showed that the prediction of molecular docking correlated well to the results of site-directed mutagenesis. These results provided us a reasonable explanation for why sulfamide derivative had more potent inhibitory activity against E. coli PDHc E1 than that of amide derivative 5d or lead compound I. It indicated that sulfamide linkage was most favorable to enzyme inhibition against E. coli PDHc E1 due to more binding position and stronger interaction with the active site of E. coli PDHc E1 then that of 5d or I.
Conclusions
In summary, two series of amide and sulfamide derivatives 5a–e, 9a–k and 13 were synthesized as potential E. coli PDHc E1 inhibitors. Their inhibition against both E. coli PDHc E1 and microbial disease were evaluated. SAR analyses indicated that the inhibitory potency of compounds against both E. coli PDHc E1 and bacterium could be greatly increased by replacing ether bond (C–O–C) linkage in lead structure I with amide or sulfamide linkage. All 5a–e or 9a–k and 13 with the IC50 values ranging from 1.99 to 25.66 μM against E. coli PHDc E1 could exhibit moderate to good antibacterial activity against cyanobacteria, Xoo and Aaa. However I with much weaker inhibitory potency against E. coli PHDc E1 showed no antibacterial activity against cyanobacteria, Xoo and Aaa. Sulfamide derivatives 9 showed more potent inhibitory activity against E. coli PDHc E1 (IC50 < 14 μM) than that of amide derivatives 5 or lead compound I. 9d (R = 3-NO2, IC50 = 2.95 μM) and 9k (R = 3-NO2-4-Cl IC50 = 1.99 μM) with sulfamide linkage exhibited much higher inhibitory potency against E. coli PDHc E1 than that of corresponding 5d (R = 3-NO2 IC50 = 13.52 μM). Meanwhlie, 9d and 9k also displayed 97% inhibition against cyanobacteria at 20 μg mL−1, much higher than that of 5d (with 76% inhibitory potency). Especially, 9k, displaying the most potent inhibitory activity against E. coli PDHc E1, showed almost best inhibitory activity against cyanobacteria or Aaa at 20 μg mL−1 or 500 μg mL−1 respectively. The above findings showed that there was some correlation between enzymatic inhibition and antibacterial activity.The interaction mode of amide derivatives 5 or sulfamide derivatives 9 with active site of E. coli PDHc E1 was explored by molecular docking to understand the mechanism of antibacterial activity. Binding mode analysis revealed that 9d displayed much powerful interaction by forming hydrogen bond between sulfamide linkage and residue Lsy392, Tyr599 and His106 at active site of E. coli PDHc E1.
These possible binding modes of 9d with important residues of PDHc E1 were further verified via site-directed mutagenesis, enzymatic assays, and fluorescence spectral analysis. It suggested 9d had more potent inhibitory activity against E. coli PDHc E1 or bacterium than that of 5d or lead compound I due to sulfamide group as a “linker part” with more binding position and stronger interaction in the active site of E. coli PDHc E1 than 5d or I. These results proved that sulfamide group as a “linker part” of triazole and benzene ring in the parent structure was much favorable to both inhibition against E. coli PDHc E1 and bacterium. To the best of our knowledge, 9k, 9d and 9i seem to be the first ThDP analogs as PDHc E1 inhibitors with both potent enzyme inhibition and significant antibacterial activity, and they could be used as lead compound for further optimization. These results proved that antibacterial activity compounds could be obtained by the biorational design of E. coli PDHc E1 inhibitors.
Experimental procedures
General procedures
Melting points (mp) were measured on an electrothermal melting point apparatus and were uncorrected.1H NMR and 13C NMR spectra were recorded at 400 MHz, in DMSO-d6 solution on a Varian Mercury-Plus 400 spectrometer and chemical shifts were recorded in parts per million (ppm) with TMS as the internal reference. Mass spectra (MS) were obtained on a QTRAP LC/MS/MS system (API2000; Applied Biosystems, Foster City, CA, USA), and signals were given in m/z. Elemental analysis (EA) was measured on a Vario ELIII CHNSO elemental analyzer. Unless otherwise noted, reagents were purchased from commercial suppliers and used without further purification. Intermediate 5-(azidomethyl)-2-methylpyrimidin-4-amine 1 was synthesized according to the existing methods.19General procedure for preparation of N-(prop-2-yn-1-yl)-(substituent)benzamide (3a–e)
The reaction of substituted benzoic acid (10 mmol) with oxalyl chloride (1.89 g, 15 mmol) gave substituted benzoyl chloride. A solution of the substituted benzoyl chloride was added dropwise to the solution of prop-2-yn-1-amine (0.55 g, 10 mmol) and trimethylamine (1.21 g, 12 mmol) in dichloromethane (15 mL) at 0 °C. Then the reaction mixture was stirred for 3.5 h at room temperature. Dichloromethane (20 mL) was added to the mixture and the mixture was washed with water, a solution of sodium hydroxide (1 M), a solution of diluted hydrochloric acid (1 M) and brine, dried over sodium sulfate and filtered. The solvent was evaporated under reduced pressure to get crude products. The crude products were recrystallized with dichloromethane to give the pure compounds 3a–e, which were used directly for the next step.General procedure for preparation of N-(prop-2-yn-1-yl)-(substituent)benzenesulfonamide (7a–k) and N-(prop-2-yn-1-yl)methanesulfonamide 11
A solution of the substituted sulfonyl chloride (10 mmol) was added dropwise to the solution of prop-2-yn-1-amine (0.55 g, 10 mmol) and trimethylamine (1.21 g, 12 mmol) in dichloromethane (15 mL) at 0 °C. Then the reaction mixture was stirred for 3.5 h at room temperature. Dichloromethane (20 mL) was added to the mixture and the mixture was washed with water, a solution of sodium hydroxide (1 M), a solution of diluted hydrochloric acid (1 M) and brine, dried over sodium sulfate and filtered. The solvent was evaporated under reduced pressure to get crude products. The crude products were recrystallized with dichloromethane to give the pure compounds 7a–k and 11, which were used directly for the next step.General procedure for preparation of N-((1-((4-amino-2-methylpyrimidin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)-(substituent)benzamide hydrochloride (5a–e); N-((1-((4-amino-2-methylpyr-imidin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)-(substituent)benzenesulfonamide hydrochloride (9a–k) and N-((1-((4-amino-2-methylpyrimidin-5-yl)methyl)-1H-,2,3-triazol-4-yl)methyl)methanesulfonamide hydrochloride 13
We added CuI (0.04 g, 0.2 mmol) to a stirred solution of 5-azidomethyl-2-methylpyrimidine-4-ylamine 1 (0.33 g, 2 mmol) and 3a–e, 7a–k or 11 (2 mmol) in THF (10 mL) followed by Et3N (0.24 g, 2.4 mmol). After over night stirring at room temperature, the reaction mixture was poured into water (50 mL), and the precipitate was collected by filtration and dried under atmospheric pressure obtained 4a–e, 8a–k and 12. Then they reacted with 36% hydrogen chloride afforded title compounds 5a–e, 9a–k and 13.Molecular docking
For docking purposes, the crystallographic coordinates of the PDHc-E1 with bound ThDP from E. coli (PDB code: 1L8A) were obtained from Brookhaven Data Bank. Hydrogen atoms were added to the structure allowing for appropriate ionization at physiological pH. The protonated state of several important residues, such as His142, Tyr177, Glu751, His640 and Met 194, were adjusted by using SYBYL7.3 (Tripos, St. Louis, USA) in favor of forming reasonable hydrogen bond with the ligand. Molecular docking analysis was carried out by the SURFLEX module of SYBYL package to explore the interaction model for the active site of PDHc-E1 with its ligand. All atoms located within the range of 6.5 Å from any atom of the cofactor ThDP were selected into the active site, and the corresponding amino acid residue was, therefore, involved into the active site if only one of its atoms was selected. Other default parameters were adopted in the SURFLEX-docking calculations. All calculations were performed on a CCNU Grid-based computational environment (CCNU Grid website http://www.202.114.32.71:8090/ccnu/chem/platform.xml).Acknowledgements
The work was supported in part by the National Basic Research Program of China (No. 2010CB126100); the National Natural Science Foundation of China (No. 20772042, 21172090, 21272089, 21472061 and 21472062); Excellent doctorial dissertation cultivation grant from Central China Normal University.References
- H. Y. Sun, H. C. Wang, Y. Chen, H. X. Li, C. J. Chen and M. G. Zhou, Plant Dis., 2010, 94, 551–556 CrossRef CAS.
- A. S. Puertoa, J. G. Fernandeza, J. D. L. Castillob, M. J. S. Pinoa and G. P. Anguloa, Diagn. Microbiol. Infect. Dis., 2006, 54, 135–139 CrossRef PubMed.
- D. L. Bates, M. J. Danson, G. Hale, E. A. Hopper and R. N. Perham, Nature, 1977, 268, 313–316 CrossRef CAS PubMed.
- R. N. Perham, Annu. Rev. Biochem., 2000, 69, 961–1004 CrossRef CAS PubMed.
- W. Wei, H. Li, N. Nemeria and F. Jordan, Protein Expression Purif., 2003, 28, 140–150 CrossRef CAS PubMed.
- D. Dobritzsch, S. König, G. Schneider and G. Lu, J. Biol. Chem., 1998, 273, 20196–20204 CrossRef CAS PubMed.
- R. Kluger and D. C. Pike, J. Am. Chem. Soc., 1977, 99, 4504–4506 CrossRef CAS PubMed.
- A. C. Baillie, K. Wright, B. J. Wright and C. G. Earnshaw, Pestic. Biochem. Physiol., 1988, 30, 103–112 CrossRef CAS.
- P. N. Lowe, F. J. Leeper and R. N. Perham, Biochemistry, 1983, 22, 150–157 CrossRef CAS PubMed.
- N. Nemeria, Y. Yan, Z. Zhang, A. M. Brown, P. Arjunan, W. Furey, J. R. Guest and F. Jordan, J. Biol. Chem., 2001, 276, 45969–45978 CrossRef CAS PubMed.
- P. Arjunan, K. Chandrasekhar, M. Sax, A. Brunskill, N. Nemeria, F. Jordan and W. Furey, Biochemistry, 2004, 43, 2405–2411 CrossRef CAS PubMed.
- P. Arjunan, M. Sax, A. Brunskill, K. Chandrasekhar, N. Nemeria, S. Zhang, F. Jordan and W. Furey, J. Biol. Chem., 2006, 281, 15296–15303 CrossRef CAS PubMed.
- K. M. Erixon, C. L. Dabalos and F. J. Leeper, Org. Biomol. Chem., 2008, 6, 3561–3572 CAS.
- Y. L. Ren, J. B. He, L. L. Feng, X. Liao, J. Jin, Y. J. Li, Y. Cao, J. Wan and H. W. He, Bioorg. Med. Chem., 2011, 19, 7501–7506 CrossRef CAS PubMed.
- J. B. He, L. L. Feng, J. Li, R. J. Tao, F. Wang, X. Liao, Q. S. Sun, Q. W. Long, Y. L. Ren, J. Wan and H. W. He, Bioorg. Med. Chem., 2012, 20, 1665–1670 CrossRef CAS PubMed.
- J. B. He, H. F. He, L. L. Zhao, L. Zhang, G. Y. You, L. L. Feng, J. Wan and H. W. He, Bioorg. Med. Chem., 2015, 23, 1395–1401 CrossRef CAS PubMed.
- M. T. Labro, Clin. Microbiol. Rev., 2000, 13, 615–650 CrossRef CAS PubMed.
- S. S. Printsevskaya Svetlana, E. S. Solovieva Svetlana, N. E. Olsufyeva Eugenia, P. E. Mirchink Elena, B. E. Isakova Elena, E. D. Clercq Erik, J. Balzarini Jan and N. M. Preobrazhenskaya Maria, J. Med. Chem., 2005, 48, 3885–3890 CrossRef CAS PubMed.
- K. M. Erixon, C. L. Dabalos and F. J. Leeper, Org. Biomol. Chem., 2008, 6, 3561–3572 CAS.
- L. L. Feng, J. B. He, H. F. He, L. L. Zhao, L. F. Deng, L. Zhang, L. Zhang, Y. L. Ren, J. Wan and H. W. He, Org. Biomol. Chem., 2014, 12, 8911–8918 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Assay of E. coli PDHc E1 (in vitro) and site-directed mutagenesis of PDHc E1; molecular docking; fluorescence spectral analyses; inhibitory bacterial activity and fungal activity evaluation of compounds; characterization datas mentioned in the paper. See DOI: 10.1039/c5ra22573f |
| ‡ Those authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |