DOI:
10.1039/C5RA25365A
(Paper)
RSC Adv., 2016,
6, 11344-11352
Conformational flexibility influences structure–function relationships in tyrosyl protein sulfotransferase-2†
Received
29th November 2015
, Accepted 8th January 2016
First published on 18th January 2016
Abstract
Tyrosine sulfation is a very important posttranslational modification of proteins. It is catalyzed by tyrosylprotein sulfotransferase and recently became increasingly important for biomedicine and pharmacy. An important insight about structure–activity relationships of human tyrosylprotein sulfotransferase has been received by elucidating the crystal structure, but there is still no understanding about how conformational flexibility and dynamics which are fundamental protein properties influence structure–function relationships of the enzyme. In order to provide this missing but crucially important knowledge we performed a comprehensive atomistic molecular dynamics study which revealed that (i) the conformational flexibility influences sensitively key structural determinants and interactions between the enzyme, the substrate and the cofactor; (ii) a more open conformation adopted by the substrate for binding in TPST 2; (iii) the mutations of key residues related with catalysis and binding change alter the enzyme structure and influence important interactions between the enzyme, the cofactor and the substrate.
Introduction
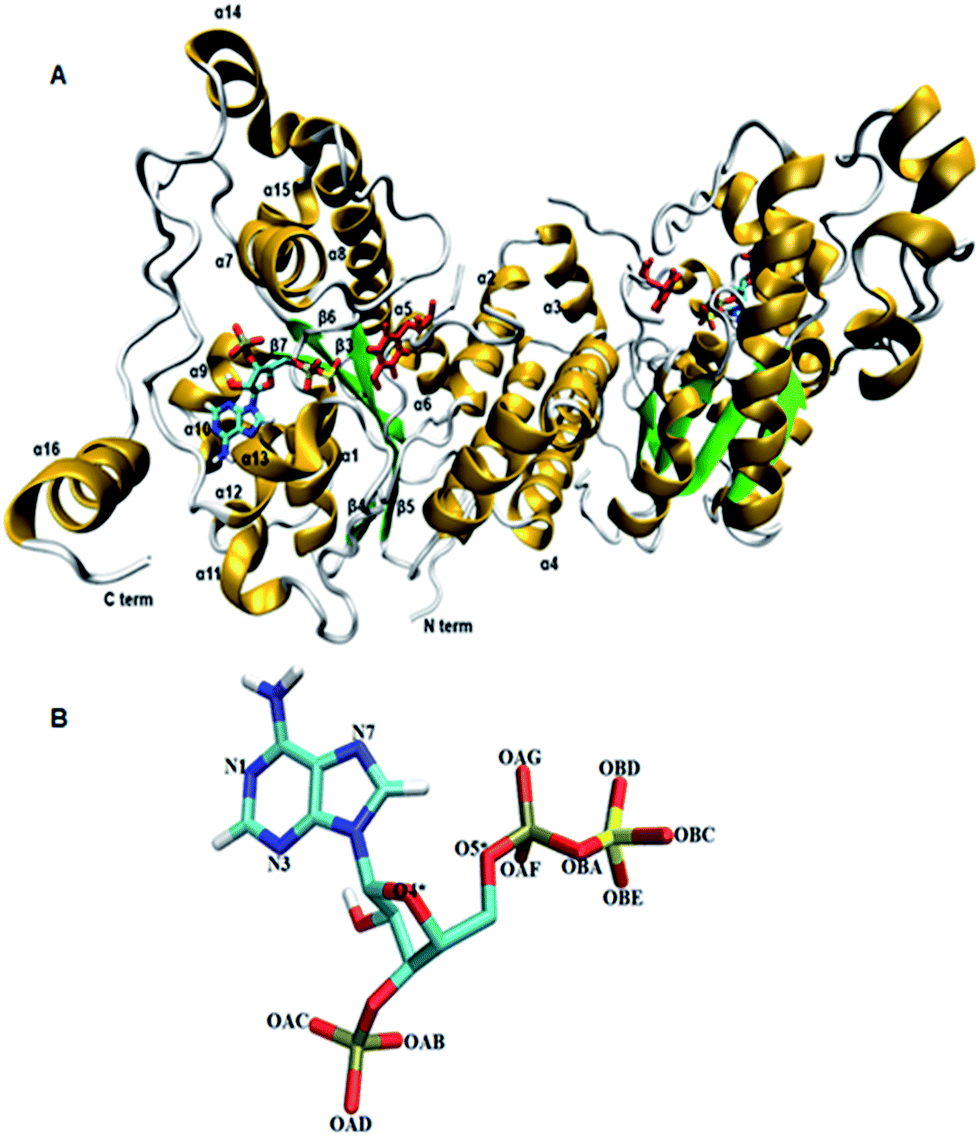
Tyrosine sulfated proteins are the product of post translational modification (PTM), where a sulfate group is covalently added to the hydroxyl group of tyrosine residues of the polypeptide chain.1 The trans-Golgi networks in the cell harbor tyrosylprotein sulfotransferase (TPST) which is an enzyme responsible for the tyrosine sulfation reaction.1,2 The tyrosine sulfation is very well characterized in eukaryotes and it has been predicted that up to 1% of tyrosine residues in the eukaryotic proteome have the potential to be sulfated.3 Tyrosine sulfation plays a crucial role in protein–protein interactions in the extracellular environment and recently became increasingly important for biomedicine and as a target for drug design.4–8 For example, the interaction of HIV's gp120 protein with CCR5 in order to get entry to CD4+ T-lymphocytes requires sulfated tyrosine residues.9 The enterovirus71 virus responsible for neurological diseases in children depends on the tyrosine sulfation of PSGL-1 on leukocytes in order to gain entry to cells and cause infection.10 In fact, approximately sixty immune system proteins have been shown to contain tyrosine sulfated residues.11,12 TPSTs catalyze the transfer of a negatively charged sulfate group from the universal sulfate donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to the hydroxyl group of a tyrosine residue of polypeptide to form a tyrosine O4-sulfate ester and adenosine 3′,5′-diphosphate.1 In humans, two isoforms TPTST-1, and TPST-2, are encoded by the TPST gene have been identified.13 The molecular weight of TPST-1 (370 residues) and TPST-2 (377 residues) isoforms are 42.2 and 41.9 kDa respectively.4 TPST has a type II transmembrane topology i.e. a short N terminal cytoplasmic domain, 17 residue transmembrane domain, and a luminal catalytic domain.14 The enzyme has two N-glycosylation sites, that are four cysteine residues on the luminal oriented side of enzyme.7 The first crystal structure of core domain of human TPST-2 (designated TPST2ΔC18 encompassing from G43 to L359), complex with PAP and the substrate peptide C4P5Y3 was solved at a resolution of 1.9 Å (ref. 15) (Fig. 1A). C4PY3 contains only one tyrosine sulfate acceptor site and consists of six acidic residues, thus giving the substrate an overall charge of minus six.15 The catalytic domain of TPTS-2 comprises a single α/β motif with a five-stranded parallel β-sheet, flanked on both sides by α helices.15 The TPST-2 exists as a homodimer and the two subunits of the dimer are designated as protomer A and B.15 The crystal structure of human TPST-2 reveals the important atomistic details of the enzyme and the ligand binding and possible mechanism, but shows no information about how conformational flexibility and dynamics influence protein structure, its structural determinants, key interactions with substrate and cofactor and the effects of mutations on the enzyme structure and ligand binding. Proteins are large flexible molecules and conformational dynamics is their fundamental property which correlates proteins structure and functions.16,17 In order to understand how the flexibility influences structure–function relationships of TPST-2, we performed 100 ns atomistic (AT) molecular dynamics (MD) simulations on the wild-type full complex TPST-2, containing the apoenzyme, the cofactor and the substrate (WT FC), and its mutant forms. The mutant forms of TPST-2 were studied experimentally15 and contains residues involved in active site (R78, E99, K158, S285) substrate binding (P77, E99, T198, R101, R105, R122) and cofactor binding (R78, S285).
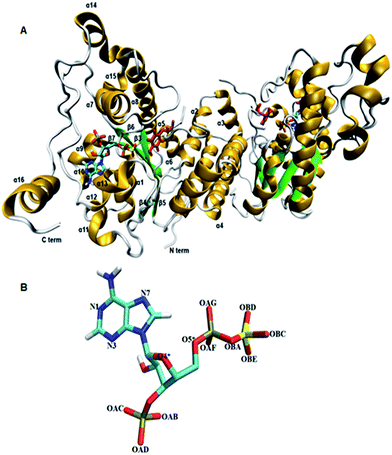 |
| | Fig. 1 The 3D structure of human TPTS-2(PDB code: 3AP1)15 enzyme using Visual Molecular Dynamics (VMD). (A) The homodimer TPST-2 is represented in new cartoon model with beta sheets colored in green and alpha helices in gold color. The cofactor (PAPS) and substrate (acceptor tyrosine; color red) are represented using licorice representation. (B) The cofactor PAPS is represented in licorice representation and labeled according to the nomenclature as it appeared in the MD simulation. The OBA represents the bridge oxygen between the phosphate and sulfate group of the 5′-PBS region of cofactor. | |
Methods
Initial structure preparation
The coordinates of the wild type TPST-2 were obtained from Protein Data Bank (PDB)18 (PDB ID 3AP1).15 The SwissPDBViewer was used for adding missing atoms and selecting one from the alternative side chain orientations.19 The sulfate group was added to the cofactor molecule PAP using GaussView 5.0.20 The resulting PAPS molecule (Fig. 1B) was used as a cofactor for the molecular dynamics simulation of TPST-2. There were eight single amino acid mutants used in this study (Table S1†). These mutants were prepared by using What IF server.21 The single amino acid mutations were prepared in both protomer A and B of the TPST-2 enzyme. The force field parameters for cofactor were calculated using PRODRG online server22 and were fitted to quantum chemical calculations and other published parameters of PAPS.23
Molecular dynamics simulations
In order to explore the dynamic properties of homodimer TPST-2, we performed extensive sets of Molecular Dynamics simulations for 100 ns using Gromacs 4.5.5 package24–26 with GROMOS96 43a1 (ref. 27) force field (Table S1†). The hydrogen atoms were added to the protein molecule by using pdb2gmx utility in Gromacs. The protonation states of histidine residues in the protein molecule were assigned based upon the optimal hydrogen bonding conformation performed in Gromacs using pdb2gmx. In order to remove the bad contact or clashes in the protein structure which might have occurred during the crystallization, in vacuum energy minimization was performed first by using the steepest descent28 and then by using conjugate gradient algorithm.29 The editconf command was used to define the dimension of the cubical box and the protein molecule was placed in the box. The periodic boundary conditions were then applied to treat all the parts of the system equally both at its interior and edges. The box size was set to ensure a distance of at least 1.0 nm between the protein and the box boundaries. The energy minimized protein structure was then solvated by using Single Point Charge (SPC)30 water model in the cubical simulation box by using Genbox command. The system was neutralized by adding the Na+ to various mutants and the wild-type setups (Table S1†). In order to relax the solvent molecules and remove constrains from the entire system, the energy minimization of the whole system was performed using first the steepest descent followed by the conjugate gradient algorithm until the maximum force on the atoms was smaller than 100 kJ mol−1 nm−1. The energy minimized structure was then subjected to position restrain dynamics for 50 ps. The simulation was performed in NVT ensemble (constant number (N) of particles, volume (V), and temperature (T))31 at constant temperature of 300 K with time step of 0.002 ps. The productive 100 ns MD was carried out using NPT (constant number of particles (N), system pressure (P) and temperature (T)) ensemble at constant temperature of 300 K and the initial velocities for MD simulation were drawn from Maxwell velocity distribution at 300 K. The Berendsen temperature coupling and Parrinello–Rahman pressure coupling were used to keep the system at 300 K, time constant (τT) of 0.1 ps and 1 bar pressure, time constant (τP) of 0.5 ps during the simulation procedure. The MD was performed with an integration time step of 0.002 ps. The Particle Mesh Ewald (PME)32 method was used for electrostatic interactions with Coulomb cut off of 1.0 nm, Fourier spacing of 0.135 nm tolerance of 1 × 10−5 and an interpolation order of 4. The Lennard Jones potential was employed for the treatment of van der Waals interaction with cut off distance set to 1.4 nm (rvdw) applying switching function. The LINCS algorithm33 was utilized to keep all the covalent bonds involving hydrogen atom rigid. The coordinates were saved after every 20 ps from multiple MD trajectories.
Analysis of molecular dynamics simulations
The analyses of the trajectories obtained from the simulations were performed using tools from the Gromacs software package. The Root Mean Square Deviation (RMSD) of Cα atoms of the protein with respect to minimized crystal structure, Root Mean Square Fluctuations (RMSF), Radius of gyration (Rg), electrostatic interactions, hydrogen bonds, and cluster analysis were also performed using Gromacs. The visualization of MD trajectories and the structures were performed using VMD34 software. The Free energy landscape analysis was performed by using g_sham in Gromacs. The graph of free energy landscape was produced by in-house scripts. The Bio3D package35 in R was used to produce domain cross correlation analysis. Dynamic cross correlation analysis was performed on the wild type TPST-2 in order to understand extends of the correlated motion. The cross correlation between the ith and jth atoms are represented by Cij matrix which range from −1 to +1. The Cα atoms of the protein, substrate and all atoms of the cofactor were used to create cross correlation Cij matrix. The positive value represents the correlated motion and the negative values represent the anticorrelated motion.
Results and discussion
Overall stability and flexibility of TPST-2 structures
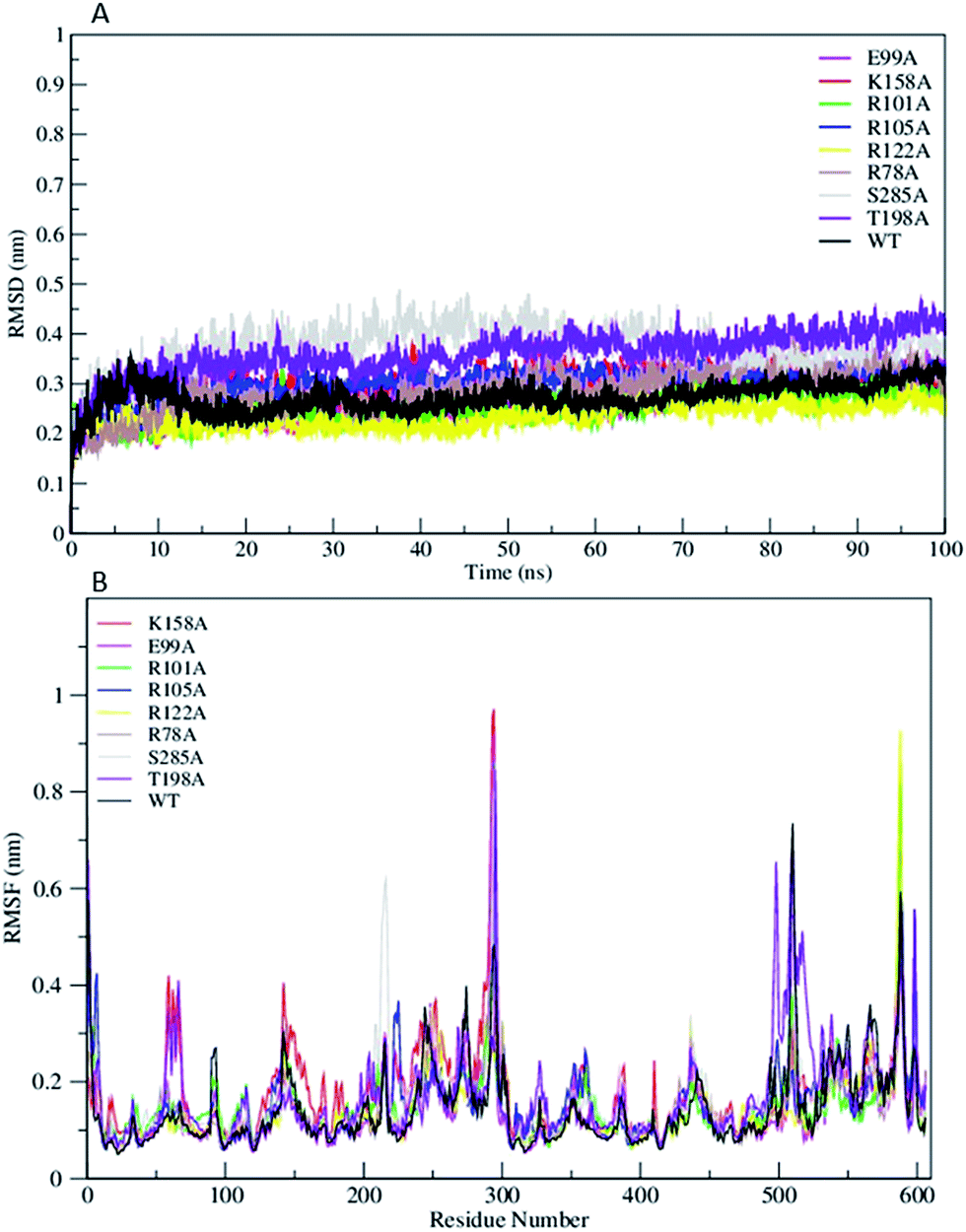
The Root Mean Square Deviation (RMSDs) of WT FC and its mutants are represented in Fig. 2A. The WT FC shows that system has equilibrated after 15 ns. The RMSD profiles of substrate binding mutants (R101A, R105A, R122A, and T198A) equilibrated roughly after 10 ns (Fig. 2A). The active site mutants (R78A, E99A, K158A and S285A) showed equilibration after 15 ns and R78A and E99A after 20 ns. The RMSD profile of the S285A and T198A mutants out of all the mutants showed the greatest structural deviation from the WT FC trajectory. The average RMSD value of all Cα atoms of WT FC is ∼2.7 Å, whereas in mutants its ranges from 3.7 Å in S285A to 2.3 Å in R122A mutant (Table S2†). The RMSDs of Cα atoms from the secondary structural elements (α-helices, β sheets) of WT FC shows that beta sheets have lowest RMSD value of 1.7 Å along the 100 ns trajectory compared to alpha helix 2.2 Å and loops 3.2 Å (Fig. S1†). Similar trends were seen in the mutants as well. The RMSDs of secondary structures for the mutants ranges from 3.8 Å (S285A) to 2.6 Å (R112A) for alpha helix, 2.6 Å (T198A) to 1.8 Å (R122A) for beta sheets and 4.7 Å (S285A) to 2.8 Å (R122A) for loops (Table S2†). The RMSF profiles of WT FC and the mutants are shown in Fig. 2B. The detailed analysis of the Root Mean Square Fluctuations (RMSF) plot of the WT FC is shown in (Fig. S2†). The basal level fluctuation of the WT FC was considered to be 1 Å, 43% of RMSFs in WT FC are below 1 Å and 57% of residue's RMSF are above 1 Å. All the mutants have shown significant increase in the number of residues with RMSF > 1 Å apart from E99A mutant which shows slightly decrease in this percentage (Table S2†). The most significant effect on the RMSFs was seen with K158A mutant. The cofactor binding region of the protein (5′ PBS and 3′ PBS), substrate binding region βe and many secondary core structural elements were increased in the K158A mutant relative to WT FC (Table S3†). The S285A mutant also showed an increase in the 5′ PBS cofactor binding site. The S285A mutant showed a significant increase in α11, α12 alpha helix and the loop connecting α12 and α13. This loop contains the S285 residue in the WT FC. The mutant E99A also showed an increase in the α2 helix. In the substrate binding residues, the most significant increase of RMSFs was seen in R101A, R105A and T198A mutants in contrast to WT FC. The results indicate that mutations affect the flexibility not only the nearby residues around the mutation site, but also have more complex structural effect on different parts of the enzyme molecule.
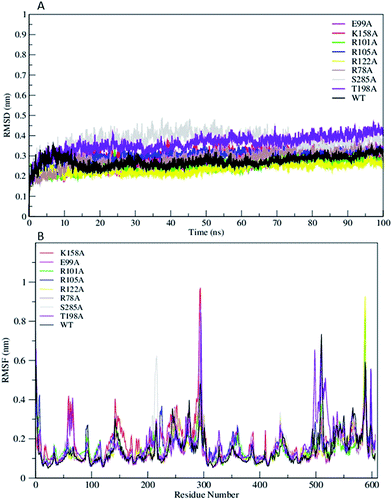 |
| | Fig. 2 (A) The RMSD trajectory of all C α atoms in of wild type TPST-2 (WT FC) and mutants as function of simulation time in nanoseconds (ns), (B) RMSF of all C α atoms of residues of WT FC and mutants for 100 ns trajectory. | |
Extending the time scale and multiple simulations
In the scientific community there is missing consensus whether is better to run one longer simulation which allows exploring conformational effects at longer time scales or instead to run several shorter runs in order to clean the statistic noise. In order to explore this issue we did additional simulations. We extended therefore the trajectory of the Wild type TPST-2 to 200 ns (Fig. S3†). The RMSD profile remain stable and to there is not considerable difference between the averaged values to 100 ns (2.7 Å) and from 100 ns to 200 ns (2.9 Å) (Table S14†). The mean values of three important geometric determinants discussed later in the manuscript (the distance between carboxyl group of E99 and the hydroxyl group of the acceptor Y1006 from the substrate, the distance between Y1006 and the sulfur atom of PAPS and the angle of bending of substrate) were compared. In all cases the average values remain very close in the trajectory up to 100 ns and from 100 ns to 200 ns (Table S14†). Therefore for TPST2 we would be able to use 100 ns trajectory for comparative atomistic analysis of the interactions in TPST2. In order to evaluate the effect of the statistical noise, two additional simulations for the WTFC of TPST-2 were run for 100 ns with different initial velocities; trajectory analysis was done and compared to original simulation (Fig. S4 and Table S14 in ESI†). The RMSDs and the three geometric determinants mentioned above show very similar values and trends in the three simulations, e.g., the standard deviations between the mean RMSD values are within 0.22 Å. In order additionally to account for the statistical errors in mutants simulations, we run second simulation for 100 ns (Fig. S5†). The averaged values of the RMSDs and three geometric determinants, specific for the E99A mutant (discussed in details later in the manuscript) indicate about good convergence between the two runs (Table S15†) and the plausibility to use single simulations further on in this comparative MD study. In order to evaluate the equilibration using more comprehensive criteria we monitored the fluctuations of multiple parameters along the time. The time fluctuations of the Radius of gyration (Rg) in the three runs of WTFC (Fig. S6†) showed very similar trends, indicating that the protein structure equilibrated and remains compact with the time. In order further to explore how multiple parameters changes together with the time, we monitored the changes of the free energy landscapes as 2D plots from (i) RMSD and Rg; (ii) Rg and the number of protein–protein hydrogen bonds (Fig. S7†) as in ref. 38. The free energy plot of Rg vs. RMSD shows the presence of local minima which represents the stable state in WTFC. In addition the total energy and potential energy of the WTFC was also stable through the simulation and (Fig. S8†). These results indicating towards the structural stability and convergence of simulation which could be used to carry out further structural analysis.
Conformational effects on the binding the cofactor
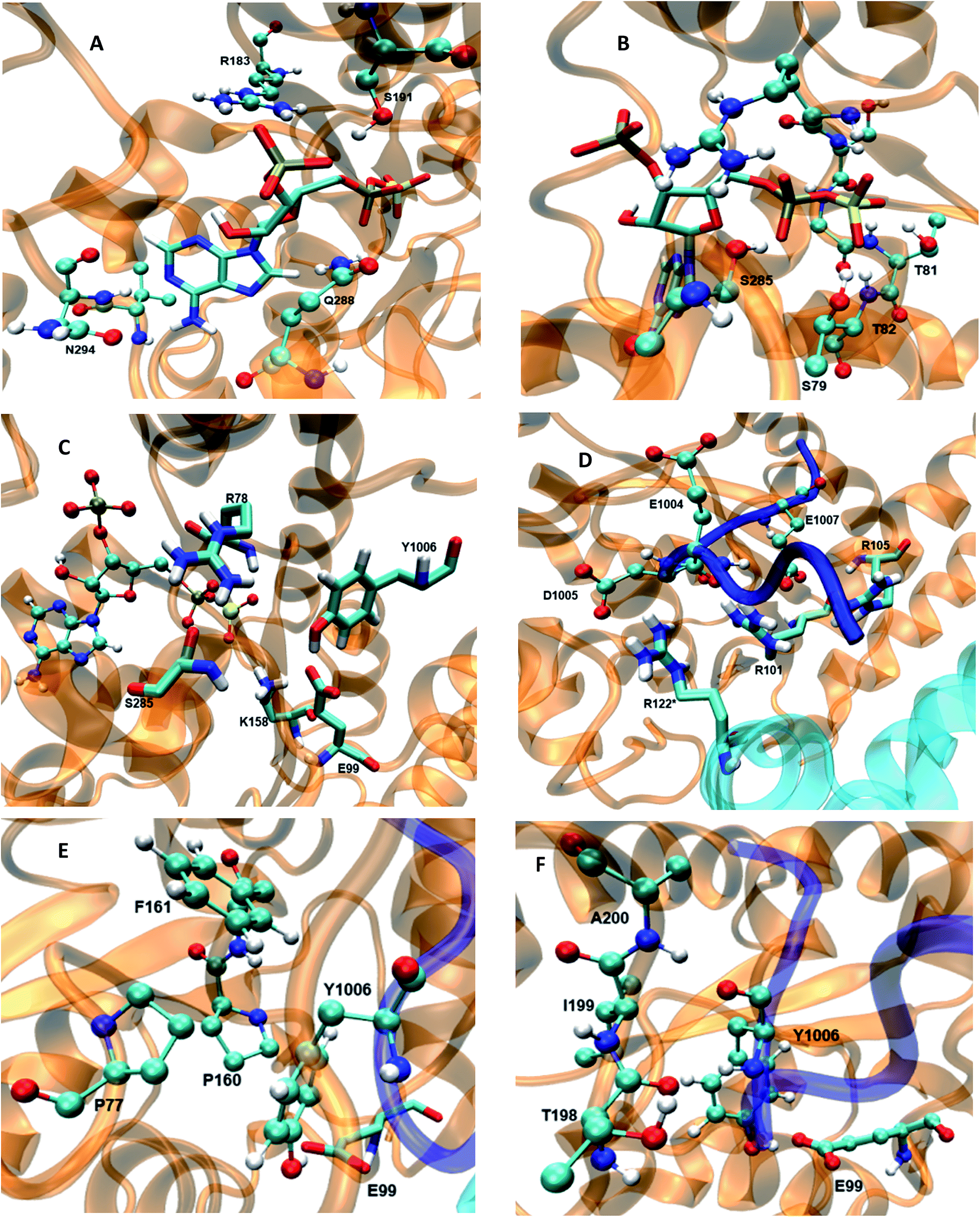
The 5′-phosphosulfate binding motif (5′-PBS) (residues 75–83) (Fig. 3B) is located between the β3 and α1 of WT FC. The crystal structure suggests extensive interactions of the 5′-PBS with the PAPS cofactor. The average value and time evolution of RMSDs and RMSFs of the 5′-PBS region show this region to be stable (Fig. S9†). R78, which is proposed to act as a catalytic residue in the crystal structure15 stabilizes the 5′-phosphate group of PAPS by electrostatic interactions. The hydrogen bonding profile of the 5′-PBS region of the WT FC is shown in Table S4.† The sulfate group is not present in the crystal structure and was added to PAP which allows analysis of the specific interactions which stabilize and orient it for reaction with the acceptor tyrosine of the substrate. Residues S79, G80, T81 and T82 are mainly involved in hydrogen bonds with sulfate group of PAPS. The NH group of L83 makes hydrogen bonds with the OAG oxygen of the phosphate of 5′-PBS region. The 3′-phosphate binding motif (3′-PBS) (residues 180–195) (Fig. 3A) spans β6 to α7. This region is next to the βe region (Fig. 3F) which is involved in the substrate binding. The RMSDs and RMSFs (Fig. S10†) indicate that the 3′-PBS motif is also stable during the simulation. The side chain of R183 makes electrostatic interactions with the 3′-phosphate group of the PAPS (Fig. S11†) with an average distance 3.7 Å. The side chains of S191 also make hydrogen bonds with the oxygen of the 3′-phosphate group which is stable in 67% of the 100 ns trajectory. The side chain of R195 make hydrogen bonds in the crystal structure, but it is not preserved in MD studies due to the high flexibility of the R195 side chain. The K300 side chain in the crystal structure makes close contact with 3′-phosphate group of PAPS, but in the MD simulations the average distance is larger (6.03 Å). The backbone of N294 gets closer to the N7 of the adenine ring of the cofactor during the simulation and interacts by hydrogen bond during 89% of the trajectory, which is not present in the crystal structure where the distance is 6 Å. In contrast to the crystal structure, the side chain of V293 makes hydrophobic interaction with C8 atom of the adenine ring (4.2 Å averaged distance in the simulation against 7.8 Å in the crystal structure). The Y283 in crystal structure stabilizes the nitrogen atom of the adenine ring, however in the MD trajectory the distance between these two residues is 9.6 Å. The cofactor binding motifs (5′-PBS, 3′-PBS) are highly conserved among several families of sulfotransferases such as estrogen sulfotransferase etc.36 The simulations reveal that the sulfate group (which is not presented in the crystal structure) is stabilized by hydrogen bonds with S79, G80, T81 and T82, the vast majority of interactions in the crystal structure are stable and also that there are two new hydrophobic interactions stabilizing the adenine ring of the cofactor.
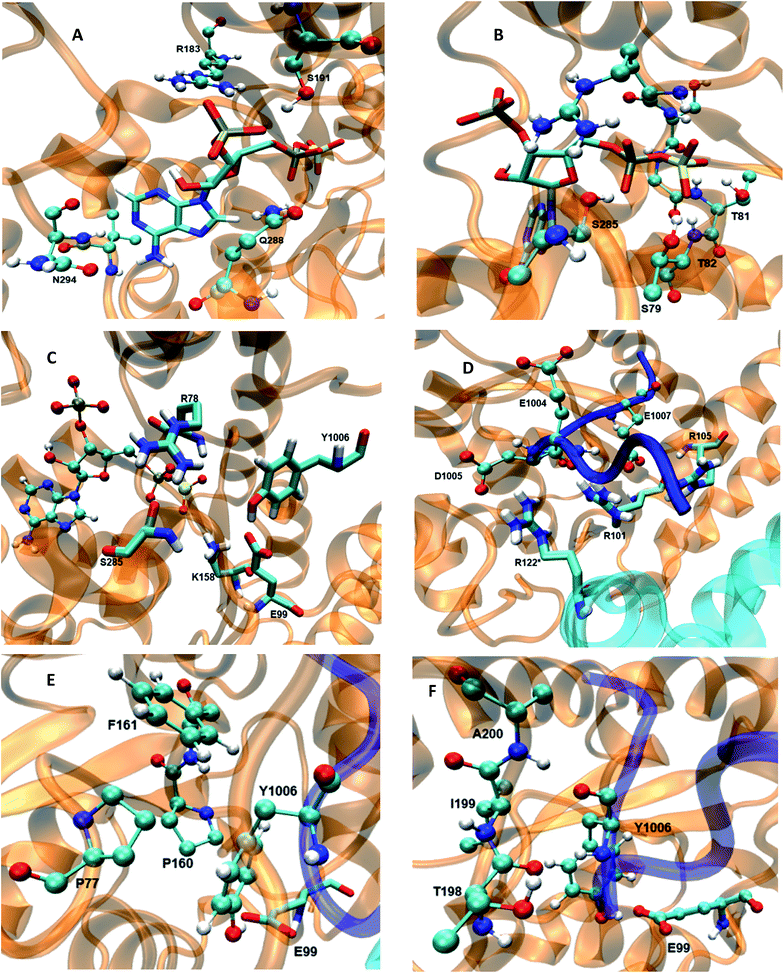 |
| | Fig. 3 The wild type TPST-2 enzyme and important residues obtained from the most populated cluster from the 100 ns trajectory, (A) 3′-PBS cofactor binding, (B) 5′-PBS cofactor binding, (C) active site residue, (D) substrate binding site and residues, (E) hydrophobic interactions of Y1006 acceptor tyrosine (F) backbone interactions βe (198-A200) of acceptor tyrosine residue. R122* residue of protomer B interacts with substrate peptide and residues of protomer. | |
Flexibility influences the binding of the substrate
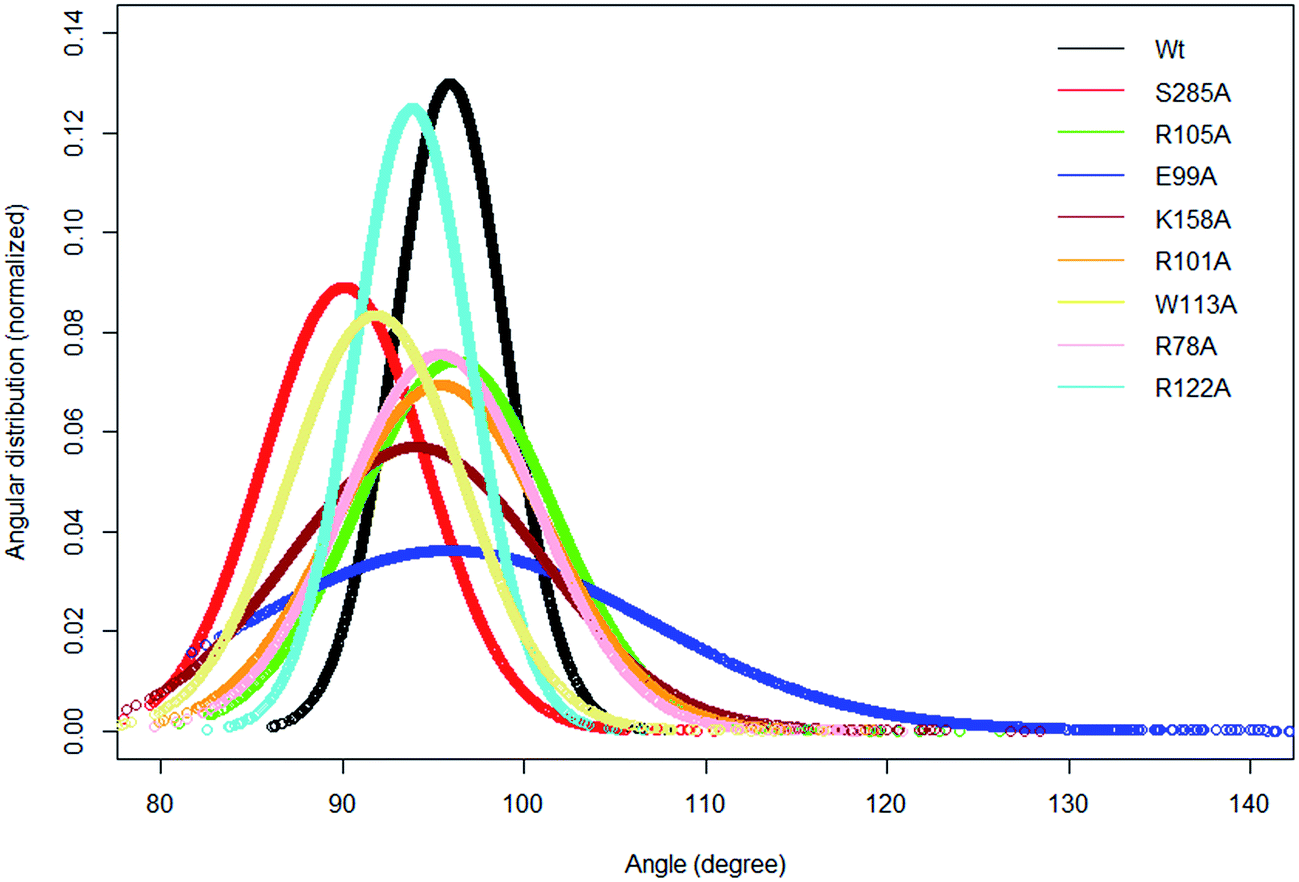
In the crystal structure the phenol hydroxyl group of the acceptor tyrosine Y1006 of the substrate is recognized by E99 and its aromatic ring by hydrophobic interactions with P77, whilst the backbone of Y1006 is recognized by the backbone of the short β-strand, βe (Thr198–Ala200).15 The bending of the substrate in the crystal structure (the L-shaped conformation) is confirmed by measurement of the angle between the Cα of Y1006, D1005 and E1004 which is 86° in the crystal structure. The same angle as a function of time shows an average value 95.9° in WT FC which indicates that there is a more open conformation of the peptide substrate and more open binding site than in the crystal structure.15 The substrate angle for the mutant forms ranges from 90° to 103.5° (Table S6†). The most significant effect on the angle was observed in T198, E99 and S285 mutants. The normalized angular distributions of angle for the mutants and the WT FC are plotted in Fig. 4. The average angle for E99A mutant is 103.5° which influences the orientation of acceptor tyrosine and provides computationally derived structural explanation of the experimental finding that the E99A mutant has no enzyme activity.15 The backbones of T198 and A200 interact with the backbone of the substrate tyrosine (Fig. S12, Table S7† and Fig. 3F). The side chain of I199 makes hydrophobic interactions with the side chain of the Y1006 acceptor tyrosine during the simulation with an averaged distance of 4.1 Å. The crystal structure suggests that the P77 makes hydrophobic interactions with the aromatic ring of Y1006; however in the simulation this interaction is stable in only 41% of the trajectory. Importantly, two other residues (P160 and P161), not mentioned in the crystal structure are also involved in hydrophobic interactions with the substrate tyrosine. The P160 side chain makes hydrophobic contact with the Y1006 aromatic ring during 66.4% of the trajectory and the F161 ring during 24% of the simulation time. These results indicate that the aromatic ring is properly stabilized and oriented for catalysis by hydrophobic cluster containing P77, P160, and P161 residues, rather than single interaction as in the crystal structure (Fig. 3E and S13†). The hydroxyl group of Y1006 and E99 interact by stable hydrogen bonds as in the crystal structure (Fig. S14 and Table S7†). The analysis of the two most populated clusters of WT FC and the E99A mutant (Fig. S15 and Table S13†) showed that the distance between the A99 and Y1006 hydroxyl group to be 10.6 Å as compared to 2.8 Å in WT FC which explains the lack of activity of E99A. The crystal structure suggests that the backbones of D1005 and E1004 are recognized by the side chains of T198 and R101. The MD studies of WT FC show that T198 side chain makes stable hydrogen bonds with the backbone NH group of D1005 of the substrate, whilst in the T198A mutant this interaction is weaker with an average distance of 5.2 Å (Fig. S16 and Table S9†). These results explain why T198A shows reduced enzyme activity in vitro15 and confirms the role of T198 in the proper orientation of the substrate. The MD trajectory analysis reveals there is electrostatic interaction between the side chains of R122 and D1005 during the simulations, however, it is stronger up to 40 ns (4.6 Å up to 40 ns and ∼5.8 Å after 40 ns) (Fig. S17†). Importantly, the simulations reveal that the side chain of R122 interacts electrostatically with the side chain of the catalytic base E99 and both interactions are lost in the trajectory of the R122A mutant (Fig. S18†). The side chain of R101 makes hydrogen bonding with the backbone of E1004 and D1005 from substrate for 70 ns and 90 ns respectively (Fig. S19 and Table S7†) and the backbone of R101 interacts with side chain of D159 for entire 100 ns trajectory which does not exist in the crystal structure. D159 is located next to the catalytically important residue K158 and this interaction might be important for the proper orientation of K158 for catalysis. During the simulation the side chains of R101 and aromatic ring of F1003 interact by cation-π interactions (with average distance 4.3 Å) which most stable during the first 70 ns and were not presented in the crystal structure. The simulations of R101A mutant show the complete loss of interactions mentioned above which gives molecular modelling explanation of the experimental studies showing that the monomeric R101A mutant15 exhibits nearly complete loss of enzyme activity (Fig. S20 and Table S10†). There is stabilizing hydrogen bonding between the R105 of the enzyme and E1007 of the substrate in the crystal structure. The MD studies confirm that the both residues interact closely with averaged distance of 3.5 Å (Fig. S21†). In the R105A mutant the average distance between A105 and E1007 was increased to 14 Å (Fig. S22†) which is in agreement with the finding that this mutant shows reduction in enzyme activity.15 The MD simulations provide complementary structural insight to the experimentally measured effects of the mutations of key binding residues, for which experimentally determined structures are not available. In addition the simulations suggest that conformational flexibility leads to a more open conformation of the substrate than indicated in the crystal structure and indicate the appearance of a small hydrophobic cluster which stabilizes the aromatic ring of the acceptor Y1006.
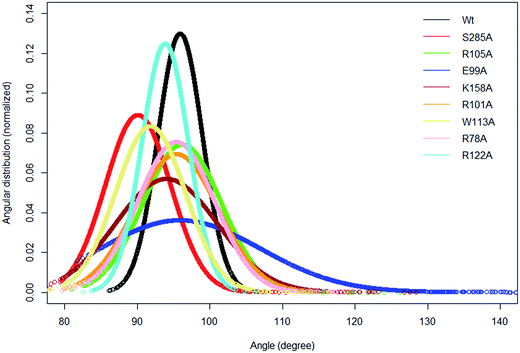 |
| | Fig. 4 The normalized distribution of the substrate angle of the wild type TPST-2 and the mutants. The angle was evaluated using C α atoms of Y1006, D1005 and E1004 substrate peptide residues. The plot was prepared using R by using normal distribution function available in R. | |
Dynamical effects on the catalytic site and interactions
The catalytically important residues are R78, E99, K158 and S285A (Table S5† and Fig. 3C). R78 which is proposed to act as the catalytic acid interacts with the bridge oxygen of the leaving phosphate group as in the crystal structure (Fig. S23†). In the simulations the R78 side chain also interacts with the side chain of Q288 and with the hydroxyl group of S285. The backbone of R78 makes stable interaction with the hydroxyl group of the acceptor Y1006 of the substrate. In the R78A mutant (Fig. S24†) the interactions between the PAPS and the A78 side chain weaken and the distance between the methyl groups of A78 and the bridge oxygen are at an average distance of 6 Å, as compared to 3 Å in the crystal structure. Importantly, in the R78A mutant E99 does not make hydrogen bond with the hydroxyl group of the acceptor tyrosine. The computationally modelled structural effect of R78 explains the experiments which demonstrate that R78A reduces the enzyme activity by 95% with respect to crystal structure.15 S285 which might be involved in the stabilization of the transition state interacts with the oxygen of leaving phosphate group in the crystal structure, an interaction which is also conserved during the simulation (Fig. S25†). The RMSF plot of the S285A mutant and WT FC indicates that the region spanning residues 265–280 (α11, α12 and loop between α12 and α13) shows higher fluctuations in the S285A mutant compared to WT FC. The S285 residue is located in between the loop α12 and α13. The RMSF of PAPS in the S285A mutant also showed increased fluctuations compared to WT FC equivalent, indicating the stabilizing effect of this residue on the cofactor (Fig. S26†). S285A mutant shows weaker contact between the OAF oxygen of the 5′ phosphate group of the cofactor, than WT FC (Fig. S27 and Table S11†). K158, which is also proposed to be involved in the transition state stabilization, interacts with both substrate and the cofactor in the crystal structure. The distance of side chain of K158 from the oxygen (OBE) of the sulfate group of PAPS and the hydroxyl group of acceptor tyrosine (Y1006) is 3.6 and 4.6 respectively during the simulation (Fig. S28†). The distances between the methyl group of A158 in the K158A mutant and hydroxyl group of tyrosine acceptor (Y1006) and to the sulfate group of cofactor shows considerable increase with respect to the WT FC (Fig. S29 and Table S12†). The structural effect of this mutation is in agreement with the significantly reduced enzyme activity of WT FC. Interestingly, the RMSFs of the PAPS cofactor in the K158A mutant are higher than the WT FC, which supports its stabilizing effect for PAPS (Fig. S30†). E99, which is proposed to act as a catalytic base, makes stable electrostatic interactions with the side chain of K158. In the E99A mutant the hydroxyl group of Y1006 no longer makes hydrogen bonds with the A99 side chain (Fig. S31 and Table S13†) and the 5′-PBS region of the cofactor. Additionally, the residues close to E99A show higher fluctuations demonstrating the stabilizing effects of E99 on the binding site and the cofactor. The sulfate group of PAPS is on average 6.0 Å away from the hydroxyl group of the acceptor tyrosine and this distance is stable during the simulation.
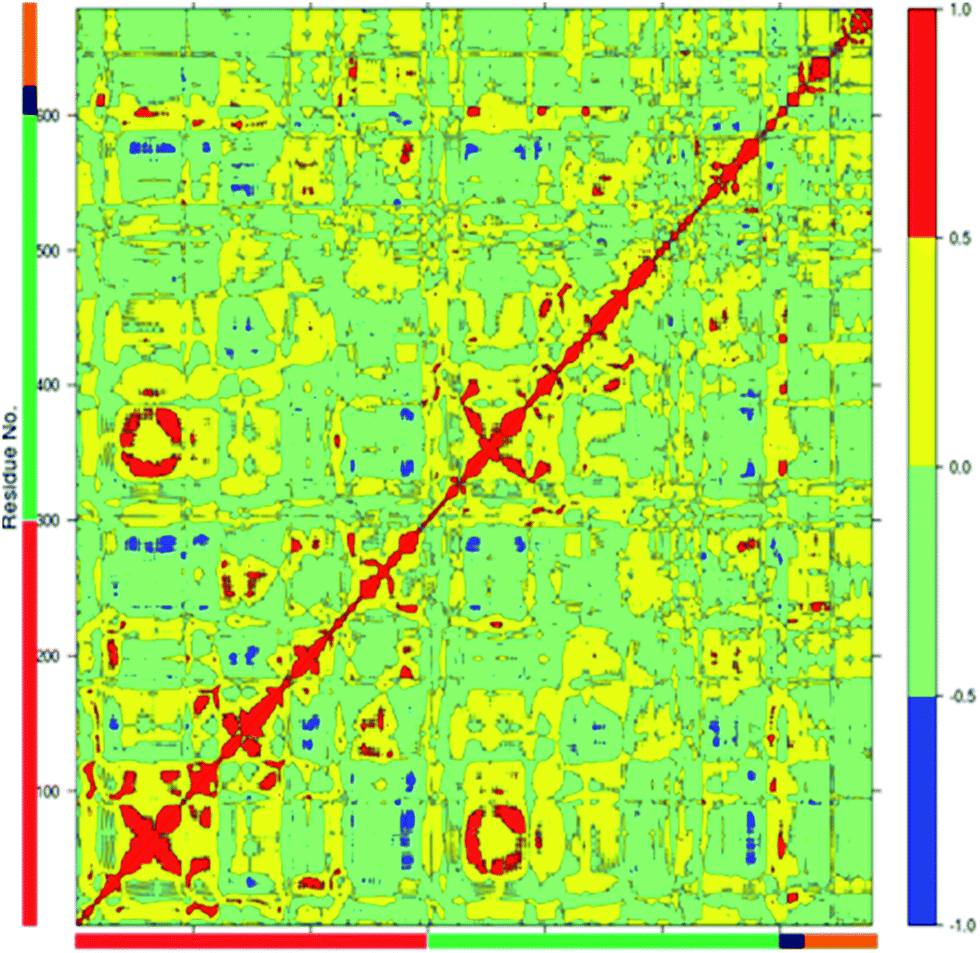
Correlated motions in the wild type full complex TPST2
The dynamics of proteins can be realized through complex network of correlated motions between different parts of the protein molecules which could have important mechanistic and functional role. An insight into the correlated motions between different parts of the enzyme molecule can be provided by Dynamic Cross Correlation Analysis (DCCA).37 The DCCA of WT FC from the 100 ns trajectory is shown in Fig. 5. The positive regions in the map are represented with red and yellow colour scheme is associated with correlated motion whereas anti-correlated motions (negative regions) are shown in light green and blue. Residues 78–82 (part of 5′-PBS region) of WT FC show correlated motion with the sulfate group of the PAPS cofactor. The residues 70–80 (part of 5′ PBS region) also showed correlation with the residues from α5–α6 and β5 (160–180, close to 3′-PBS site and catalytic site). The residues 240–250, which belong to the loop between α13 and α14 (loop close to adenine ring of PAPS) move in correlation with the adenine ring of the cofactor. The key residues from the substrate binding site (R101, R105 and E99) show strong correlation with the substrate peptide residues in particular E1004, D1005 and Y1006 residues respectively. The residues from 99–110 (from substrate binding region α2) also showed correlation with the residues of α5–α6 helixes (160–170) which are close to 3′-PBS. Interesting K158 residue showed strong correlation to the substrate peptide (Y1006) and weak correlation to the 5'-sulfate group of the PAPS. The βe region showed correlation with the substrate residues D1005 and Y1006. There was also strong correlation between the residues encompassing α1, β4 (90–101) and α10, α11, α12 (250–270) of WT FC. The residues between α3 and α4 (115–130), involved in dimerization near the N terminal of protein show anti-correlated motion in respect to residues 324–333 which are far away at C terminal of protein. The analysis reveals that there are important correlated motions between the PBS and the cofactor and between the substrate binding site and the substrate which might underline the mechanics of binding the cofactor and the substrate respectively. Importantly, there are correlated motions between the PBS and the substrate binding site which suggest that the biding of the cofactor and substrate might be correlated and mutually influence each other.
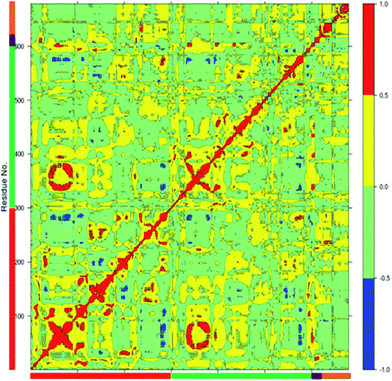 |
| | Fig. 5 The Dynamic Cross Correlation Analysis (DCCA) of wild type TPST-2. Protomer A and B, substrate peptide and cofactor are represented in red, green, black and orange colours respectively. The positive correlated motion is shown in red colour and the negative correlated motion in blue colour. The analysis was done using R Bio3D package.35 | |
Conclusions
The present MD study reveals that the conformational flexibility and dynamics influences the structure–functions relationships of TPST-2. The simulations show that the sulfate group (which is not presented in the crystal structure) is stabilized by hydrogen bonds with S79, G80, T81 and T82, that the vast majority of interactions in the crystal structure are stable during the simulation and also that there are two new hydrophobic interactions stabilizing the adenine ring. The mutations influence the flexibility not only of the nearby residues around the site, but also have effect on the different closer or distant parts of the enzyme molecule. The MD results provide computational structural insight, which in absence of experimentally determined structures of mutants explains the kinetic measurements of the effects of the mutations of key residues. The modelling results complement the crystallographic studies of the wild type full complex, suggesting about more open conformation of the substrate and the appearance of small hydrophobic cluster which stabilize the aromatic ring of the acceptor Y1006. The MD studies suggest about the key importance of the conformational flexibility for understanding the TPST-2 interactions with the substrate and cofactor and the effects of mutations which can be further used in protein engineering and drug design.
Author contributions
W. S wrote the manuscript and carried out molecular dynamics simulations analyzed and discussed the results. C. C and T. K designed the experiments, analyzed results, wrote, discussed and revised manuscript. G. W and O. S discussed the results and revised manuscript.
Acknowledgements
TK and CC acknowledge Marie Curie International Career Development Fellowships, NSCCS grants and HEC-Biosim grants. WS thanks for Northumbria University Studentship. The authors acknowledge Departmental HPC-Cluster “Pasteur”.
Notes and references
- C. Niehrs and W. B. Huttner, Purification and characterization of tyrosylprotein sulfotransferase, Eur. Mol. Biol. Organ. J., 1990, 9, 35 CAS.
- C. Niehrs, J. Stinchcombe and W. Huttner, Two membrane-bound forms of tyrosylprotein sulfotransferase as revealed by phase partitioning in Triton X-114, Eur. J. Cell Biol., 1992, 58, 35–43 Search PubMed.
- W. B. Huttner, Tyrosine sulfation and the secretory pathway, Annu. Rev. Physiol., 1988, 50, 363–376 CrossRef CAS PubMed.
- K. L. Moore, The biology and enzymology of protein tyrosine O-sulfation, J. Biol. Chem., 2003, 278, 24243–24246 CrossRef CAS PubMed.
- J. W. Kehoe and C. R. Bertozzi, Tyrosine sulfation: a modulator of extracellular protein–protein interactions, Chem. Biol., 2000, 7, R57–R61 CrossRef CAS PubMed.
- J. Z. Zhu, C. J. Millard, J. P. Ludeman, L. S. Simpson, D. J. Clayton, R. J. Payne, T. S. Widlanski and M. J. Stone, Tyrosine sulfation influences the chemokine binding selectivity of peptides derived from chemokine receptor CCR3, J. Biochem., 2011, 50, 1524–1534 CrossRef CAS PubMed.
- M. J. Stone, S. Chuang, X. Hou, M. Shoham and J. Z. Zhu, Tyrosine sulfation: an increasingly recognised post-translational modification of secreted proteins, New Biotechnol., 2009, 25, 299–317 CrossRef CAS.
- S. Costagliola, V. Panneels, M. Bonomi, J. Koch, M.-C. Many, G. Smits and G. Vassart, Tyrosine sulfation is required for agonist recognition by glycoprotein hormone receptors, Eur. Mol. Biol. Organ. J., 2002, 21, 504–513 CrossRef CAS.
- H. Choe and M. Farzan, Chapter 7. Tyrosine sulfation of HIV-1 coreceptors and other chemokine receptors, Methods Enzymol., 2009, 461, 147–170 CAS.
- Y. Nishimura, T. Wakita and H. Shimizu, Tyrosine sulfation of the amino terminus of PSGL-1 is critical for enterovirus 71 infection, PLoS Pathog., 2010, 6, e1001174 Search PubMed.
- S. Kraemer, S. Alampour-Rajabi, O. E. Bounkari and J. Bernhagen, Hetero-oligomerization of chemokine receptors: diversity and relevance for function, Curr. Med. Chem., 2013, 20, 2524–2536 CrossRef CAS PubMed.
- L. Zhu, Q. Zhao and B. Wu, Structure-based studies of chemokine receptors, Curr. Opin. Struct. Biol., 2013, 23, 539–546 CrossRef CAS PubMed.
- R. Beisswanger, D. Corbeil, C. Vannier, C. Thiele, U. Dohrmann, R. Kellner, K. Ashman, C. Niehrs and W. B. Huttner, Existence of distinct tyrosylprotein sulfotransferase genes: molecular characterization of tyrosylprotein sulfotransferase-2, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11134–11139 CrossRef CAS.
- Y.-B. Ouyang and K. L. Moore, Molecular cloning and expression of human and mouse tyrosylprotein sulfotransferase-2 and a tyrosylprotein sulfotransferase homologue in Caenorhabditis elegans, J. Biol. Chem., 1998, 273, 24770–24774 CrossRef CAS PubMed.
- T. Teramoto, Y. Fujikawa, Y. Kawaguchi, K. Kurogi, M. Soejima, R. Adachi, Y. Nakanishi, E. Mishiro-Sato, M.-C. Liu and Y. Sakakibara, Crystal structure of human tyrosylprotein sulfotransferase-2 reveals the mechanism of protein tyrosine sulfation reaction, Nat. Commun., 2013, 4, 1572 CrossRef PubMed.
- M. Karplus and J. A. McCammon, Molecular dynamics simulations of biomolecules, Nat. Struct. Biol., 2002, 9, 646–652 CrossRef CAS PubMed.
- T. G. Karabencheva-Christova, U. Carlsson, K. Balali-Mood, G. W. Black and C. Z. Christov, Conformational effects on the circular dichroism of Human Carbonic Anhydrase II: a multilevel computational study, PLoS One, 2013, 8, e56874 CAS.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The Protein Data Bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- N. Guex and M. C. Peitsch, SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling, Electrophoresis, 1997, 18, 2714–2723 CrossRef CAS PubMed.
-
R. K. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem Inc, Shawnee Mission, KS, 2009 Search PubMed.
- G. Vriend, WHAT IF: a molecular modeling and drug design program, J. Mol. Graphics, 1990, 8, 52–56 CrossRef CAS PubMed.
- A. W. Schuttelkopf and D. M. van Aalten, PRODRG: a tool for high-throughput crystallography of protein-ligand complexes, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- T. F. Gesteira, L. Pol-Fachin, V. J. Coulson-Thomas, M. A. Lima, H. Verli and H. B. Nader, Insights into the N-sulfation mechanism: molecular dynamics simulations of the N-sulfotransferase domain of ndst1 and mutants, PLoS One, 2013, 8, e70880 CAS.
- S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- H. J. Berendsen, D. van der Spoel and R. van Drunen, GROMACS: A message-passing parallel molecular dynamics implementation, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, GROMACS: fast, flexible, and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- L. D. Schuler, X. Daura and W. F. Van Gunsteren, An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase, J. Comput. Chem., 2001, 22, 1205–1218 CrossRef CAS.
- R. Fletcher and M. J. Powell, A rapidly convergent descent method for minimization, Comput. J., 1963, 6, 163–168 CrossRef.
- M. R. Hestenes and E. Stiefel, Methods of conjugate gradients for solving linear systems, J. Res. Natl. Bur. Stand., 1952, 49, 409–436 CrossRef.
-
H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, and J. Hermans, Interaction models for water in relation to protein hydration, Intermolecular forces, ed. B. Pullman, 1981, pp. 331–342 Search PubMed.
- I. R. McDonald, NpT-ensemble Monte Carlo calculations for binary liquid mixtures, Mol. Phys., 1972, 23, 41–58 CrossRef CAS.
- P. P. Ewald, Die Berechnung optischer und elektrostatischer Gitterpotentiale, Ann. Phys., 1921, 369, 253–287 CrossRef.
- B. Hess, P-LINCS:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A Parallel Linear Constraint Solver for Molecular Simulation, J. Chem. Theory Comput., 2007, 4, 116–122 CrossRef PubMed.
A Parallel Linear Constraint Solver for Molecular Simulation, J. Chem. Theory Comput., 2007, 4, 116–122 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics., 1996, 14, 33–38 CrossRef CAS PubMed , 27-8.
- B. J. Grant, A. P. Rodrigues, K. M. ElSawy, J. A. McCammon and L. S. Caves, Bio3d: an R package for the comparative analysis of protein structures, Bioinformatics, 2006, 22, 2695–2696 CrossRef CAS PubMed.
- Y. Kakuta, L. G. Pedersen, C. W. Carter, M. Negishi and L. C. Pedersen, Crystal structure of estrogen sulphotransferase, Nat. Struct. Biol., 1997, 4, 904–908 CrossRef CAS PubMed.
- K. Kasahara, I. Fukuda and H. Nakamura, A Novel Approach of Dynamic Cross Correlation Analysis on Molecular Dynamics Simulations and Its Application to Ets1 Dimer–DNA Complex, PLoS One, 2014, 9, e112419 Search PubMed.
- J. E. Shea and C. L. Brooks III, From folding theories to folding proteins: a review and assessment of simulation studies of protein folding and unfolding, Annu. Rev. Phys. Chem., 2001, 52, 499–535 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra25365a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a