
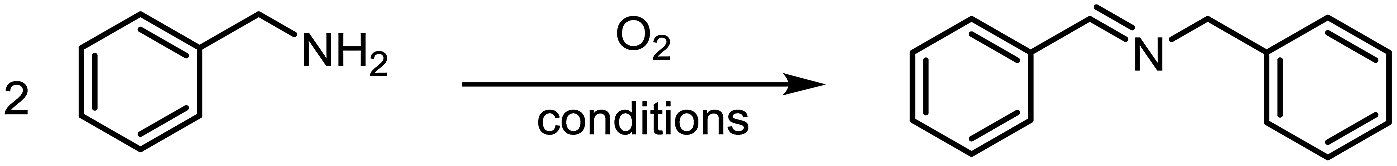
Metal-free aerobic oxidative coupling of amines in dimethyl sulfoxide via a radical pathway†
Mu Lin‡
,
Zikuan Wang‡,
Huayi Fang,
Lianghui Liu,
Haolin Yin,
Chun-Hua Yan* and
Xuefeng Fu*
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: fuxf@pku.edu.cn; yan@pku.edu.cn; Fax: +86-10-62756787; Tel: +86-10-62756787
First published on 18th January 2016
Abstract
Metal-free oxidative coupling of amines is achieved simply by heating their dimethyl sulfoxide (DMSO) solution under oxygen as oxidant without any other catalysts or additives, accompanied by the formation of an equimolar amount of dimethyl sulfone (DMSO2). EPR experiments indicate that the reaction proceeds via a radical pathway. DMSO may play a triple role as solvent, radical initiator and co-reductant.
Introduction
Amines are among the most widely used synthetic precursors in a variety of organic transformations and industry as important and ubiquitous building blocks. Among them, imines and azo compounds are popular synthetic intermediates for pharmaceuticals, valuable chemicals, molecular motors and biomolecules.1 As an alternative strategy for the synthesis of both imines and azo compounds, oxidative coupling of amines has attracted increasing attention in recent years.2 However, most reported catalytic systems on imines were based on metal catalysts, including Fe,3 Au,4 Cu,5 Ru,6 V,7 Pt/Ir,8 and TiO2.9 The introduction of metals incurs cost problems and metal residue contamination issues, which are particularly important in the pharmaceutical industry. Thus, great endeavors are highly required from the synthetic community to develop metal-free oxidative coupling reactions of amines.DMSO is not only an important polar aprotic solvent but also a good reactant for many novel transformations, like Kornblum10 and Swern oxidation,11 in which DMSO functions as an oxidant for the oxidation of alcohols. Recently, metal-free oxidative radical cyclization12 and skeletal rearrangment13 reactions were reported to proceed smoothly in DMSO under aerobic conditions.
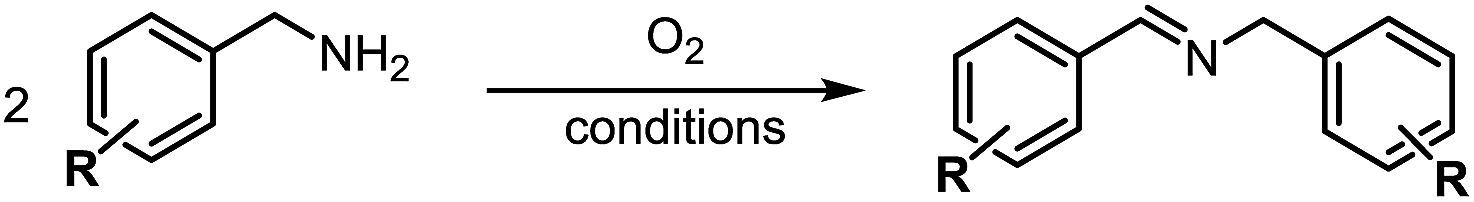
Herein by taking advantage of the redox-active features of DMSO, the aerobic oxidative coupling of amines under metal-free conditions was achieved (Scheme 1). In this case, DMSO acted not only as a solvent, but also a radical initiator and a reducing agent.
 | ||
| Scheme 1 Aerobic oxidative coupling of amines in DMSO. | ||
Results and discussion
Formation of imine was observed when heating a 0.6 M DMSO solution of benzylamine under an atmosphere of oxygen. A sufficient concentration of oxygen was found to be necessary for the reaction (Table 1, entry 1–2), with the reaction rate retarded under air and no reaction was observed under N2 (Table 1, entry 3–4). Hydrogen peroxide also functioned as an effective oxidant, but the conversion and the yield were lower and most of the hydrogen peroxide was consumed in oxidizing the solvent DMSO to dimethyl sulfone (DMSO2, Table 1, entry 5). A very interesting phenomenon was that almost an equal amount of DMSO2 was also formed in all aerobic conditions as that of the imine product (Table 1, entry 1–2), in contrast to that no DMSO2 formed in the absence of amine (Table 1, entry 6). Consequently, hydrogen peroxide is probably generated during the oxidation of amine to imine, which subsequently oxidized DMSO to DMSO2. The use of other sulfoxide solvents like DMSO-d6 and tetrahydrothiophene 1-oxide (THTO) gave similar yields (Table 1, entry 7–8) and the byproducts DMSO2-d6 and sulfolane from oxidation of the solvents were also observed by GC and GC-MS. Moreover, we observed 24% of imine product using DMF as the solvent as well (Table 1, entry 9). When other solvents were employed, little imine product was observed under the same reaction condition (Table 1S, ESI†).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | Gas | Cb (%) | Yb (%) | Rc |
| a Reaction conditions: 0.6 mmol of benzylamine in the solvent of 1 mL. Gas pressure: 1 atm. Reaction time: 24 h.b C = conversion of substrate, Y = yield of GC results.c R (ratio) = n (sulfone)/n (imine).d No DMSO2 was observed.e 1.8 mmol of H2O2 was added.f No benzylamine was added.g 42% of PhCH2NHCHO was detected. | ||||||
| 1 | DMSO | 105 | O2 | 100 | 84 | 0.9 |
| 2 | DMSO | 110 | O2 | 100 | 78 | 1.0 |
| 3 | DMSO | 105 | Air | 76 | 63 | 0.5 |
| 4 | DMSO | 105 | N2 | 1 | 1 | Nd |
| 5e | DMSO | 105 | N2 | 80 | 62 | 5.8 |
| 6f | DMSO | 105 | O2 | — | — | Nd |
| 7 | DMSO-D6 | 100 | O2 | 88 | 82 | — |
| 8 | THTO | 100 | O2 | 94 | 78 | — |
| 9 | DMF | 100 | O2 | 66 | 24g | — |
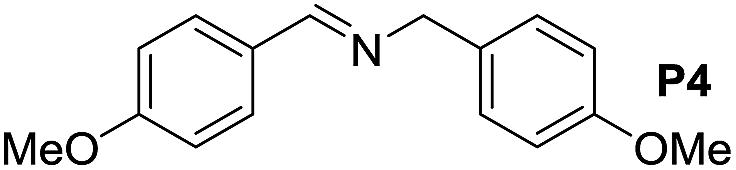
A variety of arylmethylamines were oxidized to imines in good to excellent yields (Table 2). Benzylamines having either electron rich or electron withdrawing substituents on the phenyl ring gave good yields, except for 4-chlorobenzylamine giving a moderate yield (Table 2, entry 3). Sterically hindered imine P7 was obtained in a good yield (Table 2, entry 7). It's worth noting that excess dehydrating agent and an acid catalyst are usually needed for the condensation between benzophenone and diphenylmethanamine.14 Naphthalene- and heterocycle-substituted amines were also tolerated (Table 2, entry 8–11). The oxidation of furfurylamine was rarely reported, and the yields were relatively low;7a,9 however in our system, 52% yield of P10 could be achieved (Table 2, entry 10). Because of its stronger alkalinity and higher reactivity,15 oxidation of pyridine-2-ylmethylamine was accomplished at a very low temperature 40 °C with a moderate yield (Table 2, entry 11).
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | T (°C) | Cb (%) | Yb (%) |
| a Reaction conditions: 0.6 mmol of amine, 1 mL of DMSO, 1 atm oxygen, 24 h.b C = conversion of substrate, Y = yield of GC results. Isolated yields are given in parentheses. | |||||
| 1 |  |
 |
105 | 100 | 84 (51) |
| 2 |  |
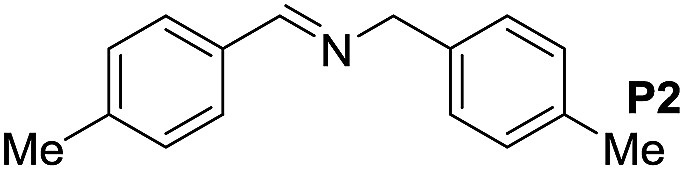 |
105 | 93 | 85 (60) |
| 3 | 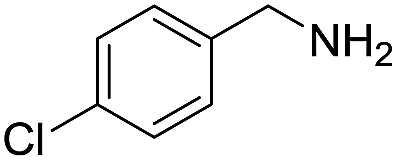 |
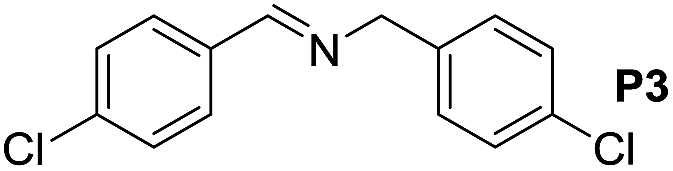 |
80 | 100 | 66 (58) |
| 4 |  |
 |
110 | 93 | 80 (76) |
| 5 | 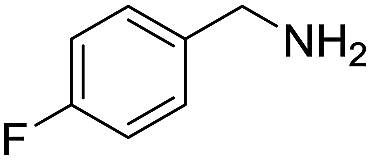 |
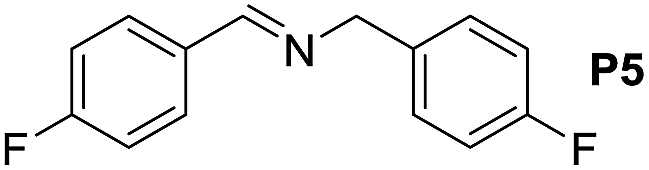 |
110 | 95 | 91 (85) |
| 6 | 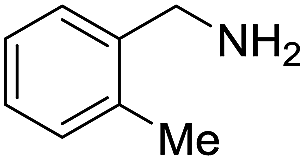 |
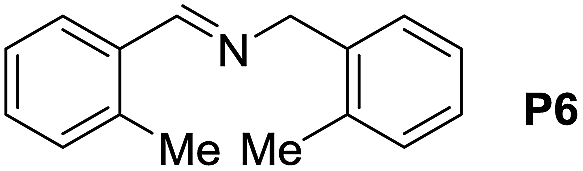 |
105 | 98 | 88 (71) |
| 7 | 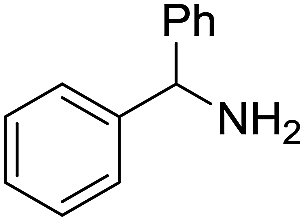 |
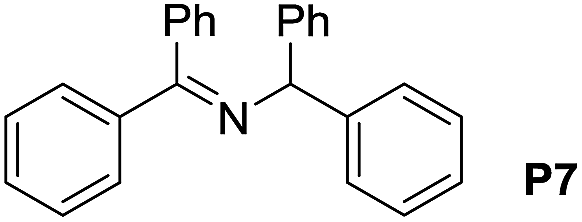 |
105 | 92 | 81 (71) |
| 8 | 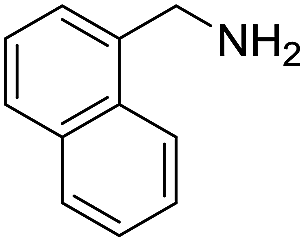 |
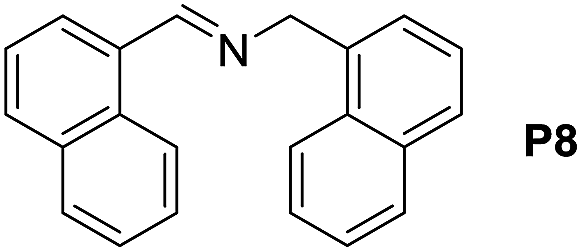 |
100 | 100 | 84 (69) |
| 9 |  |
 |
85 | 92 | 81 (73) |
| 10 |  |
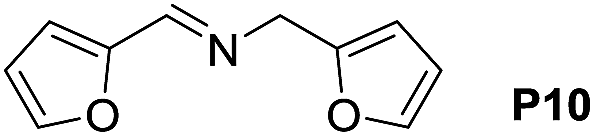 |
80 | 100 | 52 (43) |
| 11 | 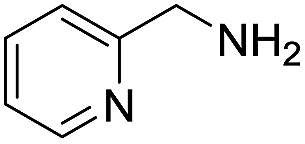 |
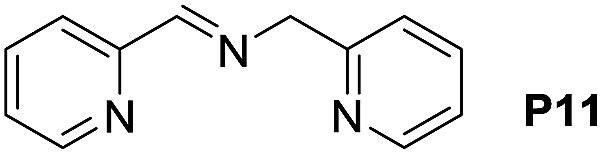 |
40 | 91 | 53 (47) |
Although the homo-coupling of aliphatic amines failed due to their low reactivity, the oxidative cross-coupling between benzylamine and aliphatic amines was successful (Table 3). Heating a DMSO solution containing benzylamine and linear aliphatic amines at 120 °C for 24 hours, 54–61% yields of cross-coupling products were formed (Table 3, entry 1–2). At a higher temperature (135 °C), 72% yield of N-benzylidene-tert-butylamine was accomplished (Table 3, entry 3). The reaction between benzylamine and cyclic aliphatic amines was also feasible with satisfying yields (Table 3, entry 4–5).
|
|||||
|---|---|---|---|---|---|
| Entry | Amine 1 | Amine 2 | Product | T (°C) | Yb (%) |
| a Reaction condition: 0.6 mmol of Amine 1, 1.8 mmol of Amine 2, 1 mL of DMSO, 1 atm oxygen, 24 h.b C = conversion of substrate, Y = yield of GC results. Isolated yields are given in parentheses. The conversions of benzylamine were over 95% and the yields of N-benzylidene-1-benzylamine were less than 5%. | |||||
| 1 |  |
 |
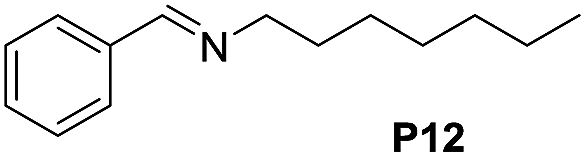 |
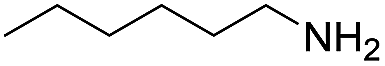
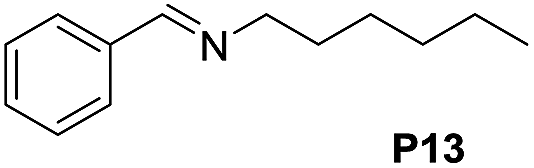
120 | 61 (53) |
| 2 | 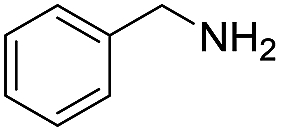 |
 |
 |
120 | 54 (36) |
| 3 | 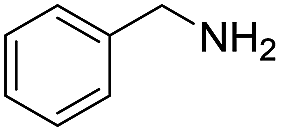 |
 |
 |
135 | 72 (54) |
| 4 |  |
 |
 |
100 | 70 (51) |
| 5 |  |
 |
 |
95 | 68 (62) |

To gain insight into the mechanism, we added one equivalent of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) to the reaction mixture, which resulted in complete inhibition of the oxidation reaction. A weak radical signal was observed in situ by EPR after heating a mixture of benzylamine and DMSO under 1 atmosphere of oxygen at 110 °C (Figure 1S, ESI†).
When the spin trapping reagent N-tert-butyl-α-phenylnitrone (PBN) was present in the oxidation of 4-methoxybenzylamine, an EPR signal with a g-value of 2.0060 (AN = 15.1 G and AH = 3.1 G) was observed, typical of a PBN radical adduct (Fig. 1). Thus an oxygen- or carbon-centered radical (e.g. HO˙, O2−, (4-methoxy-Ph) (H)(NH2)C˙, etc.) is most likely formed during the reaction. Further identification of the PBN-trapped radical is still on the way.
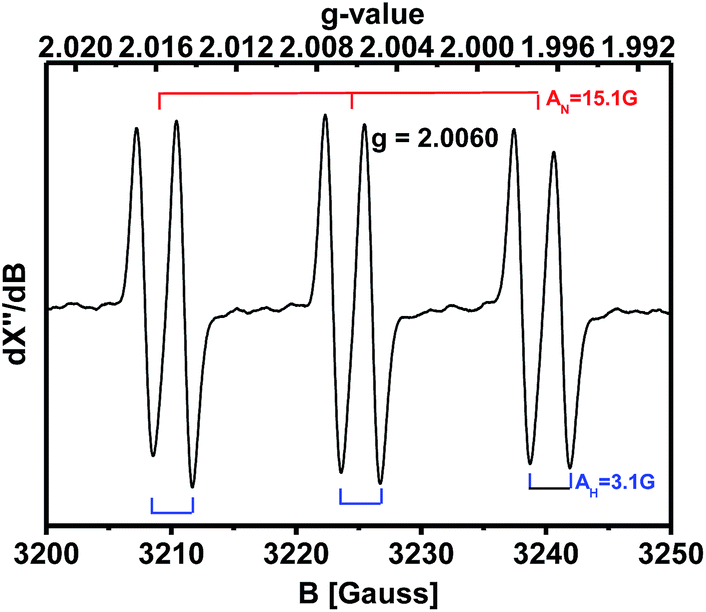 | ||
| Fig. 1 X-band EPR spectrum of radicals formed after heating a 0.6 M 4-methoxybenzylamine DMSO solution containing 1 mg of PBN at 110 °C under 1 atm O2 for 1 h. | ||
The aforementioned preliminary mechanistic studies indicate a radical chain pathway (Scheme 2) proposed to occur through (a) the reaction of DMSO with O2 which leads to the formation of DMSO cation and the very active superoxide radical; (b) deprotonation of the newly formed DMSO cation, in which the acidity of the C–H bond is stronger than that in DMSO, by the Lewis base, which is benzylamine in this system, producing the DMSO radical; (c) generation of aminomethyl radical by the reaction of the superoxide radical O2− with the amine substrates accompanied with the formation of H2O2 which can then oxidize the DMSO solvent; (d) the key imine intermediate forms via the reaction of the aminomethyl radical with DMSO radical; (e) conversion of the imine intermediate to the final coupling product. DMSO functions as solvent, radical initiator and reductant simultaneously throughout the whole reaction process.
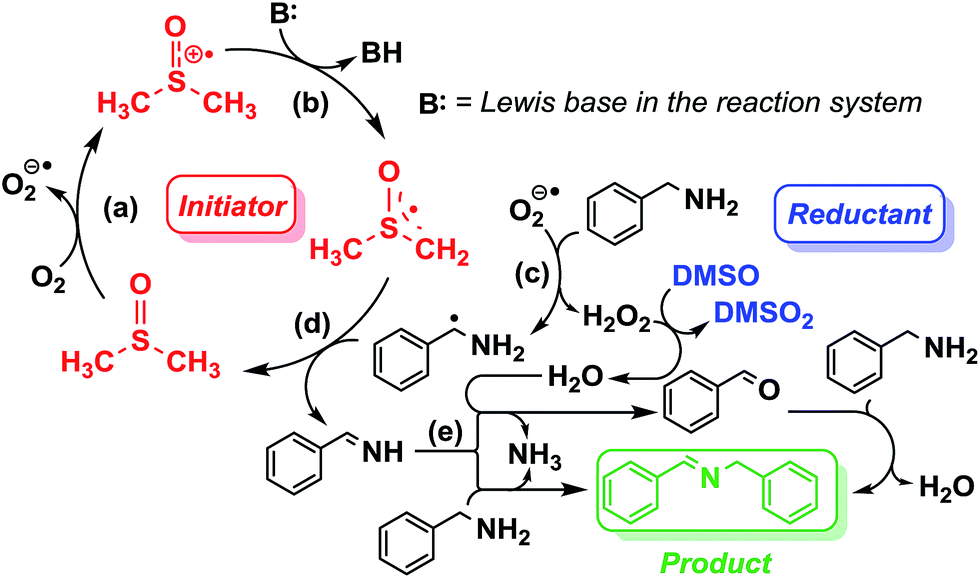 | ||
| Scheme 2 Proposed radical chain mechanism. | ||
Inspired by the mechanistic insights of amine oxidation by DMSO and oxygen, we also introduced aromatic amines into this DMSO based system. However, no reaction occurred under the standard conditions, possibly because the basicity of aromatic amines acting as Lewis base is lower than that of benzylamines and the process (b) of deprotonation in Scheme 2 could not happen. Thus, KOH was added to take the deprotonation and azo compounds could be achieved under similar conditions as shown in Table 4. Proposed mechanism for azo compounds is shown in Scheme S1 (see ESI†), in which the function of DMSO is similar as shown in Scheme 2.
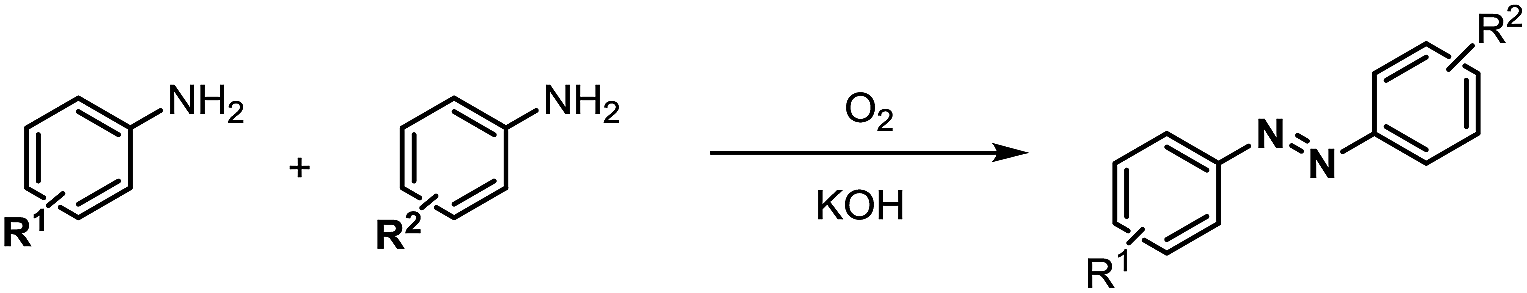
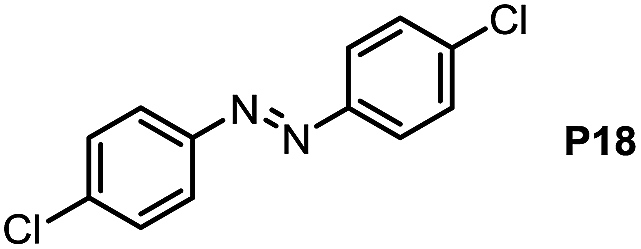
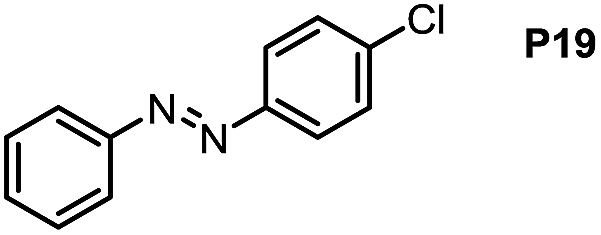
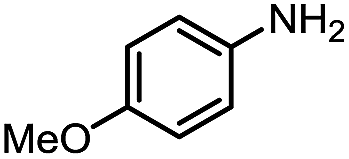
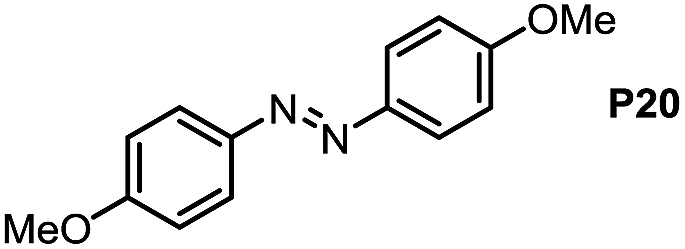
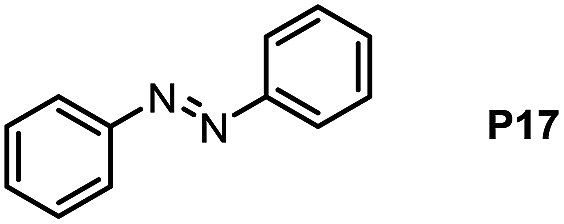
Conclusion
In this work, a simple but efficient protocol for aerobic oxidative coupling of amines to form imines and azo compounds, just by heating the mixture of amines and DMSO under aerobic conditions, was reported. This green and low-cost system using O2 as the sole oxidant may find a wide range of application in green oxidation chemistry. Detailed investigation on the mechanism and further extension of this system to more practical reactions are currently under investigation.Acknowledgements
This work was supported by National Science Foundation of China (Grant. 21171012) and the 7th China Postdoctoral Science Foundation Funded Project (2014T70009). M. Lin was also supported in part by the Postdoctoral Fellowship of Peking-Tsinghua Center for Life Sciences.Notes and references
- For reviews on the formation of imines and typical reactions: (a) S.-I. Murahashi, Angew. Chem., Int. Ed. Engl., 1995, 34, 2443–2465 CrossRef CAS; (b) J. P. Adams, J. Chem. Soc., Perkin Trans. 1, 2000, 125–139 RSC; (c) J. S. M. Samec, A. H. Éll and J.-E. Bäckvall, Chem.–Eur. J., 2005, 11, 2327–2334 CrossRef CAS PubMed.
- M. T. Schümperli, C. Hammond and I. Hermans, ACS Catal., 2012, 2, 1108–1117 CrossRef.
- F. Su, S. C. Mathew, L. Möhlmann, M. Antonietti, X. Wang and S. Blechert, Angew. Chem., Int. Ed., 2011, 50, 657–660 CrossRef CAS PubMed.
- (a) B. Zhu and R. J. Angelici, Chem. Commun., 2007, 2157–2159 RSC; (b) B. Zhu, M. Lazar, B. G. Trewyn and R. J. Angelici, J. Catal., 2008, 260, 1–6 CrossRef CAS; (c) L. Aschwanden, B. Panella, P. Rossbach, B. Keller and A. Baiker, ChemCatChem, 2009, 1, 111–115 CrossRef CAS; (d) L. Aschwanden, T. Mallat, F. Krumeich and A. Baiker, J. Mol. Catal. A: Chem., 2009, 309, 57–62 CrossRef CAS; (e) A. Grirrane, A. Corma and H. Garcia, J. Catal., 2009, 264, 138–144 CrossRef CAS; (f) M.-H. So, Y. Liu, C. -M. Ho and C. -M. Che, Chem.–Asian J., 2009, 4, 1551–1561 CrossRef CAS PubMed; (g) H. Miyamura, M. Morita, T. Inasaki and S. Kobayashi, Bull. Chem. Soc. Jpn., 2011, 84, 588–599 CrossRef CAS; (h) S. Naya, K. Kimura and H. Tada, ACS Catal., 2013, 3, 10–13 CrossRef CAS.
- (a) R. D. Patil and S. Adimurthy, Adv. Synth. Catal., 2011, 353, 1695–1700 CrossRef CAS; (b) Z. Hu and F. M. Kerton, Org. Biomol. Chem., 2012, 10, 1618–1624 RSC; (c) M. Largeron and M.-B. Fleury, Angew. Chem., Int. Ed., 2012, 51, 5409–5412 CrossRef CAS PubMed; (d) R. D. Patil and S. Adimurthy, RSC Adv., 2012, 2, 5119–5122 RSC; (e) T. Sonobe, K. Oisaki and M. Kanai, Chem. Sci., 2012, 3, 3249–3255 RSC; (f) B. Huang, H. Tian, S. Lin, M. Xie, X. Yu and Q. Xu, Tetrahedron Lett., 2013, 54, 2861–2864 CrossRef CAS.
- (a) A. Prades, E. Peris and M. Albrecht, Organometallics, 2011, 30, 1162–1167 CrossRef CAS; (b) L. He, T. Chen, D. Gong, Z. Lai and K. Huang, Organometallics, 2012, 31, 5208–5211 CrossRef CAS.
- (a) K. Nakayama, M. Hamamoto, Y. Nishiyama and Y. Ishii, Chem. Lett., 1993, 1699–1702 CrossRef CAS; (b) S. Kodama, J. Yoshida, A. Nomoto, Y. Ueta, S. Yano, M. Ueshima and A. Ogawa, Tetrahedron Lett., 2010, 51, 2450–2452 CrossRef CAS; (c) G. Chu and C. Li, Org. Biomol. Chem., 2010, 8, 4716–4719 RSC.
- H. Yuan, W. Yoo, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2012, 134, 13970–13973 CrossRef CAS PubMed.
- (a) X. Lang, H. Ji, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 3934–3937 CrossRef CAS PubMed; (b) X. Lang, W. Ma, Y. Zhao, C. Chen, H. Ji and J. Zhao, Chem.–Eur. J., 2012, 18, 2624–2631 CrossRef CAS PubMed; (c) N. Li, X. Lang, W. Ma, H. Ji, C. Chen and J. Zhao, Chem. Commun., 2013, 49, 5034–5036 RSC.
- (a) N. Kornblum, J. W. Powers, G. J. Anderson, W. J. Jones, H. O. Larson, O. Levand and W. M. Weaver, J. Am. Chem. Soc., 1957, 79, 6562–6562 CrossRef CAS; (b) N. Kornblum, W. J. Jones and G. J. Anderson, J. Am. Chem. Soc., 1959, 81, 4113–4114 CrossRef CAS.
- (a) K. Omura and D. Swern, Tetrahedron, 1978, 34, 1651–1660 CrossRef CAS; (b) T. T. Tidwell, Synthesis, 1990, 857–870 CrossRef CAS.
- V. A. Schmidt and E. J. Alexanian, Angew. Chem., Int. Ed., 2010, 49, 4491–4494 CrossRef CAS PubMed.
- Y. Wang, F. Zhang and S. Chiba, Org. Lett., 2013, 15, 2842–2845 CrossRef CAS PubMed.
- B. E. Love and J. Ren, J. Org. Chem., 1993, 58, 5556–5557 CrossRef CAS.
- (a) L. Liu, S. Zhang, X. Fu and C.-H. Yan, Chem. Commun., 2011, 47, 10148–10150 RSC; (b) L. Liu, Z. Wang, X. Fu and C.-H. Yan, Org. Lett., 2012, 14, 5692–5695 CrossRef CAS PubMed; (c) H. Kim and H. Lee, Polymer, 2015, 72, 336–340 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra25434e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |