
Micelle-templated synthesis of Pt hollow nanospheres for catalytic hydrogen evolution†
Manickam Sasidharan*a,
Piyali Bhanjab,
Chenrayan Senthila and
Asim Bhaumikb
aSRM Research Institute, SRM University, Kattankulathur, Chennai, 603203, India. E-mail: sasidharan.m@res.srmuniv.ac.in
bDepartment of Material Science, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700 032, India. E-mail: msab@iacs.res.in
First published on 11th January 2016
Abstract
As an alternative to galvanic replacement reactions and hard-template strategies, we report an efficient, mild and simple synthesis strategy for fabrication of colloidal platinum (Pt) hollow nanospheres. An aqueous asymmetric triblock copolymer poly(styrene-b-vinyl-2-pyridine-b-ethylene oxide) [PS(20.1k)–PVP(14.2k)–PEO(26.0)] micelle with core–shell–corona architecture has been found to be an efficient soft scaffold for the synthesis of Pt hollow nanospheres using K2PtCl6 as a metal precursor and NaBH4 as a reducing agent. In the core–shell–corona type micelles, the core serves as a template for void volume creation, the shell domain acts reaction site for inorganic precursors, and the corona stabilizes the composite particles. The polymer/Pt composite particles were solvent-extracted by refluxing with dimethyl formamide (DMF) at 160 °C to remove polymeric materials and obtain hollow particles. Investigation of precursor concentrations suggested that the wall-structures become irregular and uneven as the molar ratio of PVP/Pt(IV) increases from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 to 1:25, whereas the use of polymers with large PS block length [PS(45k)–PVP(16k)–PEO(8.5)] results in the formation of spherical particles with slightly increased hollow void-space diameters. The polymeric micelles and Pt hollow nanospheres were thoroughly characterized by transmission electron microscope (TEM), X-ray diffraction (XRD), infra-red (FT IR), thermal (TG/DTA) and nitrogen sorption analyses. The catalytic activity of the Pt hollow nanospheres was investigated for hydrogen liberation from ammonia–borane (AB) by hydrolysis reaction at room temperature. The catalytic activity of the Pt hollow nanospheres reveals that they can serve as a promising heterogeneous catalyst towards hydrogen generation system using AB as solid hydrogen storage materials.
:12 to 1:25, whereas the use of polymers with large PS block length [PS(45k)–PVP(16k)–PEO(8.5)] results in the formation of spherical particles with slightly increased hollow void-space diameters. The polymeric micelles and Pt hollow nanospheres were thoroughly characterized by transmission electron microscope (TEM), X-ray diffraction (XRD), infra-red (FT IR), thermal (TG/DTA) and nitrogen sorption analyses. The catalytic activity of the Pt hollow nanospheres was investigated for hydrogen liberation from ammonia–borane (AB) by hydrolysis reaction at room temperature. The catalytic activity of the Pt hollow nanospheres reveals that they can serve as a promising heterogeneous catalyst towards hydrogen generation system using AB as solid hydrogen storage materials.
Introduction
Considerable attention has been paid to the synthesis of nanostructured platinum (Pt) with controlled size and shape.1–3 Much effort has also been focused on the morphological and structural features of Pt nanostructures to produce Pt catalysts with high surface area to volume ratios so that a large fraction of the metal atoms are exposed at the surface and accessible to reactant molecules, thereby available for catalysis.4,5 A number of synthetic methods have been reported in the past for preparation of Pt nanoparticles in organic solvents6–8 and in aqueous solutions.9,10 For instance, reduction of potassium tetrachloroplatinate in aqueous solution with hydrogen produced Pt nanoparticles of different shapes, like cubic, tetrahedral, and truncated octahedral, with diameters ranging from 2 to 11 nm.11 It is suggested that the formation of these nanostructures is thermodynamically favored owing to minimization of the surface energy of these shape-dependent nanoparticles and, perhaps, the free energy of the system as a whole plays an important role during the process of nucleation and growth. In addition, dendritic growth of small Pt nanoparticles12,13 and mesoporous monocrystalline Pt nanoparticles with uniform shape and size have also been investigated.14,15 Thus intense research has been devoted to the fabrication of dense Pt-metal nanoparticles with tailored properties in size, shape, and composition. Furthermore, the synthesis and characterization of nanoparticles were mainly carried out in the presence of capping agents with suitable functional groups to prevent them from agglomeration owing to van der Waals forces.16 Although the polymer-stabilized noble metal colloids have great potential applications, often they exhibit decreased catalytic activity.17,18 Thus, polymer-stabilized Pt nanoparticles show lower activity than the colloidal platinum in the sol state (nascent state), although their activity can be enhanced through removal by oxidation of the protective polymer through the calcination process. Thus, the protective polymer or organic stabilizing agent on the metal colloid is not preferable for the majority of catalytic applications. Therefore, preparation of nanoparticles without any capping agents to obtain “unprotected colloids” would be ideal for catalytic application as these particles would have a large number of low-coordinated surface atomic sites that are largely responsible for catalytic reactions.19Hollow metal micro/nanostructures are of great interest in many emerging areas of technology. In a large number of applications, such as catalysis, cosmetics, drug delivery, photonics, and rechargeable batteries, the chemical make-up and distribution of matter within the individual particles play important roles in determining the specific function. For instance, one can tune the void-space and wall-thickness in hollow particles to control drug-release kinetics, modulate the refractive index, increase the active area for catalytic applications, and improve the cyclic volume change during repeated charge/discharging processes in a rechargeable batteries. Furthermore, hollow metallic nanospheres exhibit catalytic activities different from their solid counterparts with the advantages of low density and the saving of materials. Traditional approaches to fabricate hollow nanospheres have focused on various sacrificial hard templates, including polystyrene spheres,20 silica spheres,21 stabilizing agents,22 sacrificial template,23 liquid droplets,24 and micro emulsion droplets.25 Bai et al. reported galvanic replacement reactions (also called sacrificial-template) for preparation of Pt hollow spherical particles of average diameter of 24 nm using cobalt (Co) nanoparticles as sacrificial templates and citrate as a capping agent.26 The wall-thickness of the reported Pt hollow nanospheres mainly consisted of ca. 2 nm Pt nanoparticles with uneven wall-thickness comprised of large pores and surface roughness. The hollow nanoparticles can fulfil our requirement in catalysis in a way that not only avoids the use of stabilizing agents but also thin-wall structures have a large number of exposed surface atoms for catalytic applications.
Recently, triblock copolymers with three different chemical entities (ABC type) with hydrophilic- and hydrophobic-parts have been extensively studied for the fabrication of inorganic hollow nanospheres. When dissolved in appropriate solvents followed by dialysis against water, these ABC type triblock copolymers form core–shell–corona (CSC) type micelles, which were efficiently used for construction of a variety of metal oxide, metal borate, silica, phosphosilicate and periodic mesoporous silica hollow nanospheres by self-assembly process in our group.27–32 The notable features of ABC type triblock copolymers are that when dissolved in an appropriate solvent, it forms CSC micelles having both hydrophilic and hydrophobic characters and self-assemble in such a way that the hydrophobic core part acts as a template for void formation, the central hydrophilic shell domain serves as reaction site for reaction of inorganic metal precursors, and the hydrophilic corona block stabilizes the composite micelles. Herein, we report a facile, efficient method for the synthesis of Pt hollow nanoparticles of average size 27 ± 2 nm templated by micelles of poly(styrene-b-vinyl-2-pyridine-b-ethylene oxide) (PS–PVP–PEO) using K2PtCl6 as the metal source and sodium borohydride (NaBH4) as the reducing agent under ambient reaction conditions. The obtained Pt hollow nanoparticles were thoroughly characterized by XRD, TEM, FTIR, TG/DTA, DLS, and nitrogen sorption analyses to confirm the morphology, crystallinity and phase purity.
Hydrogen is one of the most promising energy carriers and is expected to replace fossil fuels as the dominant energy source in the future owing to its carbon-free cycle. However, finding efficient hydrogen generation materials is one of the bottlenecks for using hydrogen energy. Recently, ammonia–borane (H3NBH3, AB) has attracted much attention as an efficient H2 storage material owing to its high hydrogen content (19.6 wt% H2) and low molecular weight (30.7 g mol−1).33,34 AB is highly stable at room temperature and also readily soluble in water. Unlike borohydrides, which require a base for stabilization, AB does not require any additional stabilizing agents and it releases hydrogen in the presence of a suitable catalyst.35,36 A number of catalysts have been investigated for effective release of H2 through hydrolysis of AB, including various metal salts like RuCl3, PdCl2, and CoCl2,37 noble metal nanoclusters and non-noble metals supported on Al2O3, carbon and silica.38,39 The efficacy of Pt hollow nanospheres in the hydrolysis of AB was determined by measuring the rate of hydrogen liberation using a jacketed round bottom flask with thermostat to control the temperature to 25 ± 0.2 °C.
Experimental section
Chemicals
Potassium hexachloroplatinate, (K2PtCl6, 99.9%), sodium borohydride (NaBH4, 99.0%), ammonia–borane (H3N–BH3, 99.0%), and N,N-dimethylformamide (DMF, 99.8%) were obtained from Sigma-Aldrich and used as received. PS–PVP–PEO triblock copolymers with PS, PVP, and PEO having varying chain lengths ca. PS(20.1k)–PVP(14.2k)–PEO(26k) and PS(45k)–PVP(16k)–PEO(8.5k) were obtained from Polymer Source Inc. The numbers in the parentheses are the average molecular weights of the block chains (20.1k, for example refers to 20100). Phosphotungstic acid hydrate (P2O5·24WO3·xH2O) and hydrochloric acid were obtained from SD fine chemicals. Water was purified with a Millipore Milli-Q water system.
Preparation of core–shell–corona micelles
A general procedure for the preparation of PS–PVP–PEO micelles involves dissolution of the desired amount (0.1 g) of PS(20.1k)–PVP(14.2k)–PEO(26k) in DMF containing 10 wt% water by stirring with a magnetic stirrer. The DMF was removed through dialysis against water to obtain micelles of PS–PVP–PEO. The resultant solution was transferred to a 100 mL volumetric standard flask and diluted with Millipore water to obtain a polymer micelle concentration of 1.0 g L−1. The micelle solution was adjusted to pH 4 with a dilute HCl solution, and was then used for fabrication of Pt hollow nanospheres. The micelle of (45k)–PVP(16k)–PEO(8.5k) was prepared according to the report by Zhang and Eisenberg for “crew-cut” micelles40 because the water soluble hydrophilic PEO chain is much shorter than the hydrophobic PS chains. Therefore, (45k)–PVP(16k)–PEO(8.5k) was dissolved in DMF at an initial concentration of 1 wt%. After complete dissolution of the polymer, water was added drop wise (1 wt% per minute) to the solution with vigorous stirring until the water content reached 5 wt%. The solution was then dialyzed as described above and the pH was also adjusted to 4 prior to the synthesis of Pt hollow nanospheres.Synthesis of Pt hollow nanospheres
A typical synthesis of Pt hollow nanospheres using PS–PVP–PEO micelles (1.0 g L−1) and NaBH4 reduction is as follows. For example, 10 mL of the above micelle solution was taken in a round bottom flask. The pH was adjusted to about 4 using dilute HCl and the micelle solution was vigorously stirred at room temperature. The micelle solution was initially purged with nitrogen gas to evacuate the air inside the vessel to remove the moisture inside the reaction vessel and then the desired amount of K2PtCl6 (PVP/Pt = 12) was added. Subsequently, a dilute solution of NaBH4 was added under N2 flow and the color of the solution changed from pale yellow to dark gray. The polymer/Pt composite particles were aged by stirring at room temperature for 48 h and separated by centrifugation (10000 rpm, 10 min). The solid was repeatedly washed with Milli-Q water and ethanol followed by drying at 50 °C. Similarly, other Pt hollow particles were prepared by changing the molar ratio of Pt/PVP like 12, 18, and 25 under identical experimental conditions. Finally, the polymer/Pt composite particles were solvent extracted by refluxing in DMF using a reflux condenser at 160 °C for 8 h. After drying with a high vacuum pump, the obtained Pt hollow nanospheres without any stabilizing agents were stored in an argon atmosphere in a glove box to prevent the oxidation of the Pt-surface owing to moisture.
Characterization of materials
The hydrodynamic diameter (Dh) of the polymeric micelles was measured with a Zetasizer Nano (Malvern Instruments). The correlation functions were analyzed by the cumulant method to determine the diffusion coefficient (D) of the micelles. The hydrodynamic diameter (Dh) was calculated from D using the Stokes–Einstein equation: Dh − kBT/3πηD, where kB is the Boltzmann constant, T the absolute temperature, and η the solvent viscosity. Powder X-ray diffraction (XRD) patterns were measured on a Rigaku RINT-2200 diffractometer with CuKα radiation (40 kV, 30 mA) from 10° to 70° 2θ in 0.01 steps at a scan speed of 2° min−1. The BET surface area was measured by N2 adsorption/desorption analysis at 77 K on a BELSORB 28SA analyzer after degassing of the sample at 150 °C for 3 h. The TEM images were recorded on a JEOL Electron Microscope operating at 80 kV and 200 kV. In the case of the micelle solutions, the TEM samples were prepared by casting a drop of micelle solution on a copper grid followed by staining with 1 wt% phosphotungstic acid, whereas for Pt nanospheres, a drop of an aqueous suspension of the sample was coated on a copper grid. The samples were finally dried at room temperature. Thermogravimetric and differential thermograms (TG/DTA) were obtained with a MAC Science TG-DTA 2100 under nitrogen. Fourier transform infrared (FTIR) spectra were recorded on a PerkinElmer spectrometer using the KBr pellet technique. UV-visible spectra of gold nanoparticles were recorded with a JASCO V-550 spectrometer using BaSO4 as the reference.Generation of hydrogen from ammonia–borane
The catalytic efficiency of Pt hollow nanospheres in the hydrolysis of ammonia–borane (AB) was determined by measuring the rate of hydrogen release at room temperature. In a typical reaction, a 100 mL round bottom flask was mounted on a thermostat with a temperature of 25 ± 0.2 °C by circulating water through its jacket from a constant temperature bath supplied by Heidolph, Germany. Then, a graduated glass U-tube (100 cm in height and 36 mm in diameter) partially filled with water was connected to the reaction flask through connectors and tubing without any gas leak. AB (190.8 mg, 6 mmol) was dissolved in 30 mL of water (Milli-Q), which corresponds to 18 mmol H2 at 25 °C, and this solution was carefully transferred to the reaction flask with a Teflon coated magnetic stirrer followed by addition of 1.91 mg of Pt hollow nanosphere (1 wt% with respect to AB) catalyst. The reaction was commenced immediately after the addition of the catalyst to the magnetically stirred AB by closing the flask and the volume of hydrogen gas evolved was measured by recording the displacement of the water level in the U-tube.Results and discussion
The stepwise fabrication of Pt hollow nanospheres using core–shell–corona micelles of PS(20.1k)–PVP(14.2k)–PEO(26k) at pH 4 is depicted in Scheme 1. Prior to synthesis, the polymeric micelles were characterized for their structure and morphology by DLS and TEM. The TEM sample preparation was carried out by casting a drop of the above micelles on a copper grid followed by staining with 1 wt% phosphotungstic acid, and finally the grid was dried at room temperature. Fig. 1 exhibits spherical micelles in which the PVP shell domain was stained with phosphotungstic acid. The average micelle particle size was found to be 40 ± 2 nm with the PS core having a diameter of 22 ± 1 nm, whereas the estimated PVP shell thickness was 18 ± 0.5 nm. However, the average hydrodynamic diameter (Dh) of the above micelles estimated by dynamic light scattering experiments was found to be 64 nm. The smaller size observed (40 ± 2 nm) in TEM is owing to exclusion of the corona part and accounts only for the core–shell parts. At pH 4, the PVP shell of the PS–PVP–PEO micelles, the PVP shell domain attains an extended/stretched configuration owing to repulsive forces among positively charged PVP units.41–43 These features of PS–PVP–PEO micelles are utilized in the current investigation to prepare Pt hollow nanospheres under mild conditions.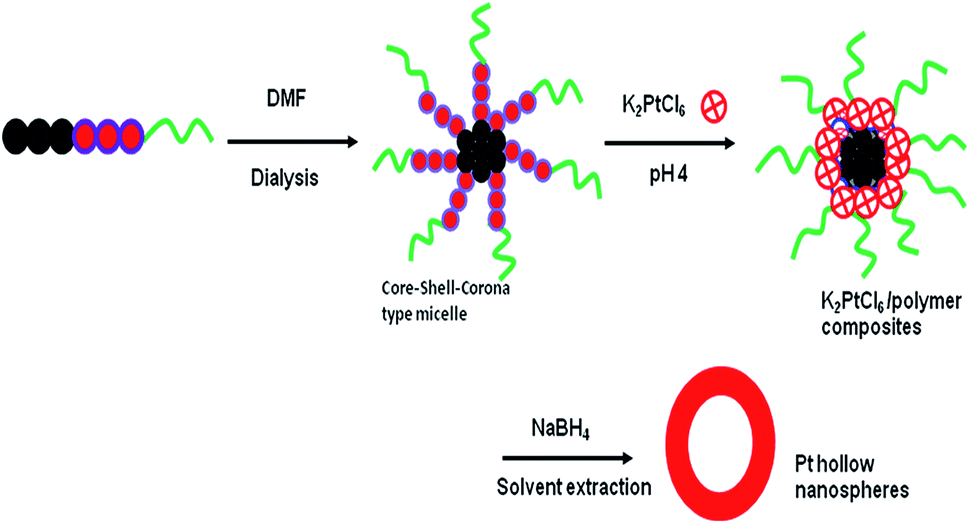 | ||
| Scheme 1 Schematic representation of the formation of hollow nanospheres. | ||
 | ||
| Fig. 1 TEM images of (A) PS–PVP–PEO micelles and (B) Pt/polymer composite particles. | ||
On addition of K2PtCl4 solution, the polyvinyl-2-pyridine (PVP) block with positive charges electro-statically interacts with PtCl62− to give organic/inorganic composite particles, as shown in Fig. 1B. The average size of the composite particles was 47 ± 2 nm, which is lower than the original micelle size estimated by DLS (64 nm). The PtCl62− species were reduced with NaBH4 (molar ratio of NaBH4/K2PtCl4 = 3) at room temperature and the color of the solution turned slowly to dark gray. The formation of Pt nanoparticles is clearly seen by drawing small aliquots of sample and analyzing by UV-visible spectroscopy after 30 minutes (ESI. Fig. S1†). Fig. S1A† shows surface plasmon resonance (SPR) bands at 522 nm corresponding to Pt-nanoparticles, whereas the band around 319 nm is ascribed to the presence of Pt(IV) ions in the reaction mixture.44 In addition, the FTIR spectrum of the composite particles (ESI. Fig. S2A†) shows a strong signal at 1720 cm−1 and a weak signal around 2913 cm−1, suggesting the presence of the N–H stretching mode of PVP units, whereas the band at 1405 cm−1 is attributed to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– bond stretching of the phenyl groups of the polymer backbone. Absorption involving C–H bonds is found at 1182 cm−1 (in-plane bending modes) and 821 cm−1 (C–H wagging modes).45 The TG/DTA analyses (ESI. Fig. S3†) indicated a complete decomposition of the polymeric template at about 300 °C; however, in the current strategy, the polymer was freed from the composite particles through solvent extraction by refluxing with DMF for 8 h repeated thrice to ensure complete removal. The FTIR spectrum (ESI, Fig. S2B†) shows complete removal of organic polymers from the absence of characteristic absorption bands. Furthermore, after solvent extraction, the UV-visible spectra showed exclusively a single band located at about 522 nm, suggesting that all Pt(IV) ions were reduced to Pt(0) (ESI, Fig. S1B†).
C– bond stretching of the phenyl groups of the polymer backbone. Absorption involving C–H bonds is found at 1182 cm−1 (in-plane bending modes) and 821 cm−1 (C–H wagging modes).45 The TG/DTA analyses (ESI. Fig. S3†) indicated a complete decomposition of the polymeric template at about 300 °C; however, in the current strategy, the polymer was freed from the composite particles through solvent extraction by refluxing with DMF for 8 h repeated thrice to ensure complete removal. The FTIR spectrum (ESI, Fig. S2B†) shows complete removal of organic polymers from the absence of characteristic absorption bands. Furthermore, after solvent extraction, the UV-visible spectra showed exclusively a single band located at about 522 nm, suggesting that all Pt(IV) ions were reduced to Pt(0) (ESI, Fig. S1B†).
The TEM micrographs in Fig. 2 show Pt hollow nanospheres in the absence of any stabilizing agents having different shell-thickness obtained by varying the molar ratio of K2PtCl6/PVP from 12, 18, to 25. The specific attributes of Pt hollow nanospheres, such as particle diameter, hollow void size, and wall-thickness, were estimated from the TEM image. For instance, Pt hollow particles synthesized with a molar ratio of K2PtCl6/PVP = 12 (Fig. 2A) showed an average particle size of 26 ± 2 nm with a wall thickness of about 5.5 ± 0.5 nm and the hollow void-space diameter was found to be 15 ± 0.5 nm. Almost all the hollow particles show a uniform spherical shape with a smooth shell wall. It can be seen from the TEM image that, similar to coalescing of other nanoscale materials owing to their very high surface area to volume ratio vis-à-vis surface free energy, the Pt hollow particles also exhibit aggregation inherent to particle size in the absence of any stabilizing agents. It is also worth mentioning that the wall-structure is completely formed without any breakage, unlike hollow particles obtained by galvanic replacement reactions or hard-template strategies. For instance, the hollow particles obtained by galvanic replacement reactions using a cobalt-nanoparticles template showed a high degree of non-uniformity in the wall-structure as well as void-volumes and also the surface was very rough.27 Even the hard-template approach using silica spherical particles also yielded hollow nanospheres with irregular shell-thickness, void-space, and particle-size.45 The significant development of this approach compared to previous strategies is that the shell-thickness increases to a certain extent by increasing the amount of K2PtCl6. Fig. 2B and C show TEM pictures of Pt hollow nanospheres obtained by varying the molar ratio of K2PtCl6/PVP to 18 and 25, respectively. Thus, as the metal precursor concentration increased from 18 to 25, the shell-thickness increased to 9.5 ± 0.5 nm (average particle size and void volumes were 29 ± 2 nm and 12 ± 1 nm, respectively) from 7.0 ± 0.5 nm. It is also commonly noticed that at K2PtCl6/PVP = 25, the hollow particles are rather not uniform and have a broad size distribution that ranges from 22 nm to 34 nm owing to deposition of a large amount of metal precursors in the shell domain of the micelles. Furthermore, two or more micelles may have combined to form large hollow particles, as seen from Fig. 2C. It is also expected that the variation in the chain length of the hydrophobic PS block of the template copolymer would be expected to increase the void space diameter.46
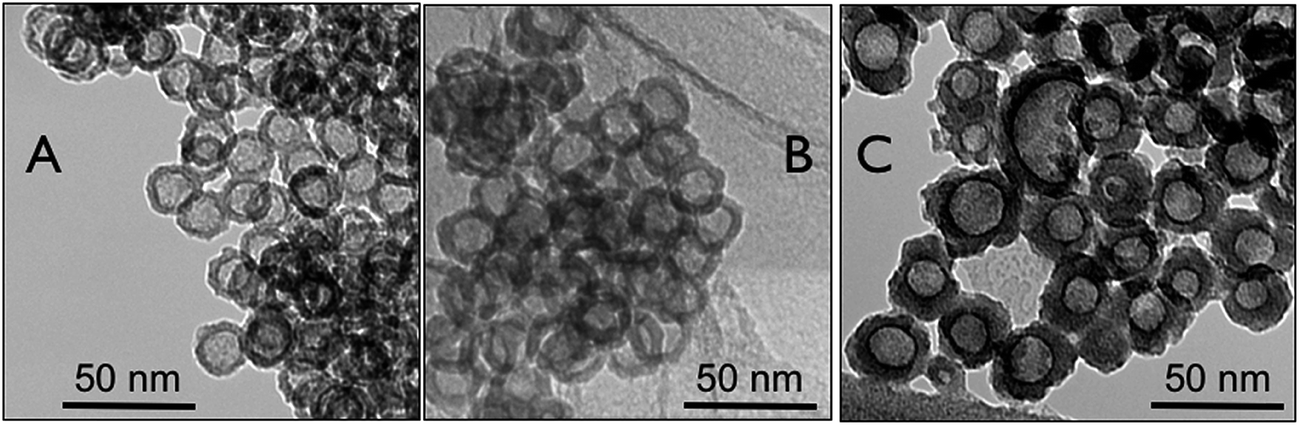 | ||
| Fig. 2 TEM images of Pt hollow nanospheres with varying precursors ratio; (A) K2PtCl6/PVP 12; (B) K2PtCl6/PVP 18; and (C) K2PtCl6/PVP 25. | ||
As the molecular weight of the PS block is increased from 20.1k to 45k, the hollow void space diameter is expected to enlarge because the PS block serves as the template for void space formation. Fig. 3A shows TEM micrographs with increased void volume synthesized using polymer PS(45k)–PVP(16k)–PEO(8.5k) with a higher PS molecular weight. The average particle size was found to be 29 ± 2 nm with a wall thickness of 5 ± 0.5 nm, whereas the estimated void-volume is 19 ± 0.5 nm. Therefore, from comparison of Fig. 2A and 3A, it is evident that the void space diameter of Pt-hollow particles can be increased over a few nanometers by changing the PS block length. Fig. 3B exhibits high-resolution TEM (HRTEM) images indicating lattice fringes originating from the Pt hollow nanoparticles and the lattice spacing of 0.235 nm corresponding to (111) plane for fcc Pt.13 The electron diffraction (ED) pattern of Pt nanoparticles also clearly shows the presence of (111), (200), and (220) diffractions (Fig. 3C), confirming phase pure Pt nanoparticles. The powder XRD patterns of Pt hollow nanospheres were recorded both prior to and after solvent extraction with DMF in the range of 30–70° 2θ (Fig. 4). Both samples exhibited diffraction pattern peaks corresponding to (111), (200), and (220) planes, similar to electron diffraction, confirming the crystallinity and phase purity of Pt hollow nanospheres. The BET surface area was found to be 36 m2 g−1 as estimated by nitrogen adsorption/desorption analyses (ESI, Fig. S4†) of Pt hollow nanoparticles under liquid-nitrogen at a temperature of 77 K. The above characterized Pt hollow nanospheres were investigated for the generation of hydrogen through the hydrolysis of ammonia–borane (AB) at 25 °C.
 | ||
| Fig. 3 (A) TEM images of Pt hollow nanospheres with slightly increased void volume. (B) High resolution TEM of Pt hollow nanospheres. (C) Electron diffraction pattern. | ||
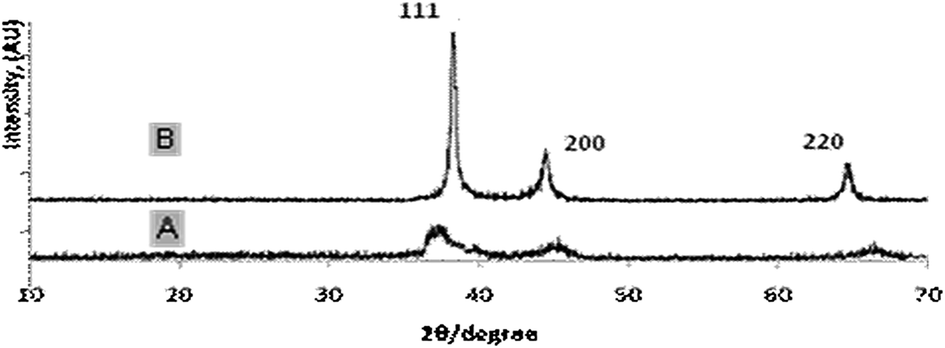 | ||
| Fig. 4 X-ray diffraction patterns of Pt hollow nanospheres: (A) Pt/polymer composite particles; and (B) solvent extracted Pt-hollow nanospheres. | ||
Nanoparticles of Pt with different shape and size were tested as catalysts in the hydrolysis of AB and the hydrogen liberation was found to depend on the composition and distribution of Pt within the nanostructures. The dependence of the catalytic activity of Pt nanoparticles was investigated with different Pt loading using Pt hollow nanospheres with wall-thickness of 5.5 ± 0.5 nm (obtained from Pt(IV)/PVP = 12) as standard catalysts. Fig. 5 shows the variation in the catalytic activity of Pt nanoparticles with Pt loading in the hydrolysis of AB under identical experimental conditions with an initial concentration of 6 mmol of AB. As the percentage of catalyst loading increases, the evolution of hydrogen was observed to be at a faster rate compared to lower loadings. The observed catalytic activity could be explained by the availability of a large number of nanoparticles with a huge number of active sites or binding sites at high Pt loadings. Furthermore, we also tested for any traces of ammonia formation during the hydrolysis of AB using an acid/base indicator and found no detectable amount of ammonia under the experimental conditions. In addition, to acquire meaningful catalytic data the hydrolysis of AB was carried out at a lower conversion rate using 1% Pt hollow nanospheres (controlling experiments at high Pt loading was also rather difficult owing to fast evolution of hydrogen) for further investigation. Fig. 6 exhibits the plots of the volume of generated hydrogen versus time over 1% Pt hollow nanospheres (wall-thickness of 5.5 ± 0.5 nm, Pt(IV)/PVP = 12) at temperatures ranging from 25 to 40 °C starting with an initial AB concentration of 6 mM. As expected, the volume of liberated hydrogen increases monotonically as the temperature increases from 25 to 40 °C and led to near complete conversion of AB in a short duration (less than 30 minutes). It is well known that the reaction rate generally increases with increasing reaction temperature. The AB concentration decreases with reaction time and volume of H2 generated increases nearly linearly with time. Under our experimental conditions, the reaction rate constant, k, is nearly constant for a given temperature, implying zero order kinetics for the AB hydrolysis reaction, as observed by other researchers.39 Thus, using the rate law −1/3d[NH3BH3]/dt = d[H2]/dt = k and rate equation k = exp−E/RT, where E is activation energy, R the gas constant, and T the reaction temperature,39 the Arrhenius plots were obtained from logk versus the reciprocal absolute temperature (ESI. Fig. S5†) and the estimated activation energy from the slope of straight line was found to be 22.7 kJ mol−1, which is comparable to the literature reports.38,39 To maintain the controlled release of hydrogen at room temperature, the subsequent studies were carried out at 25 °C using 1 wt% Pt hollow nanospheres.
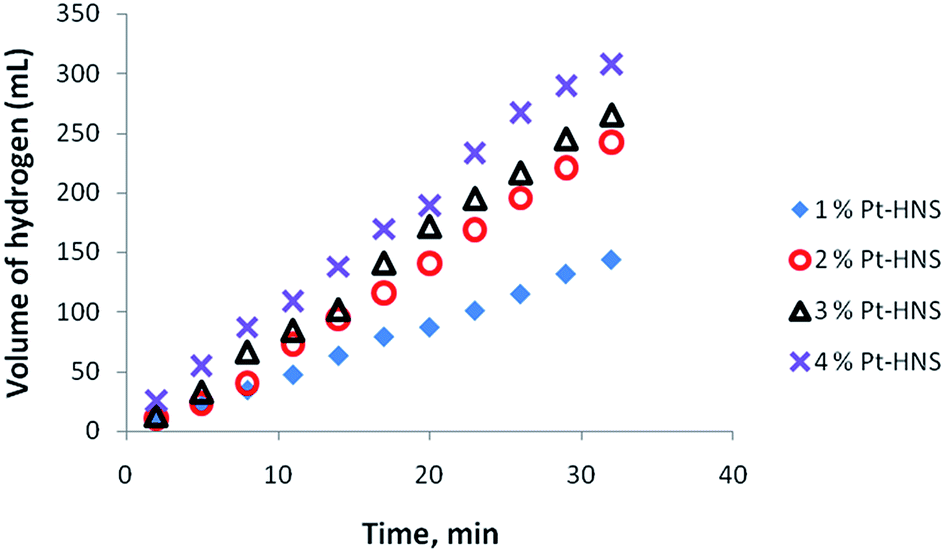 | ||
| Fig. 5 Effect of catalyst loading (Pt-hollow nanospheres) on the volume of generated hydrogen (mL) versus time for the hydrolysis of ammonia–borane. | ||
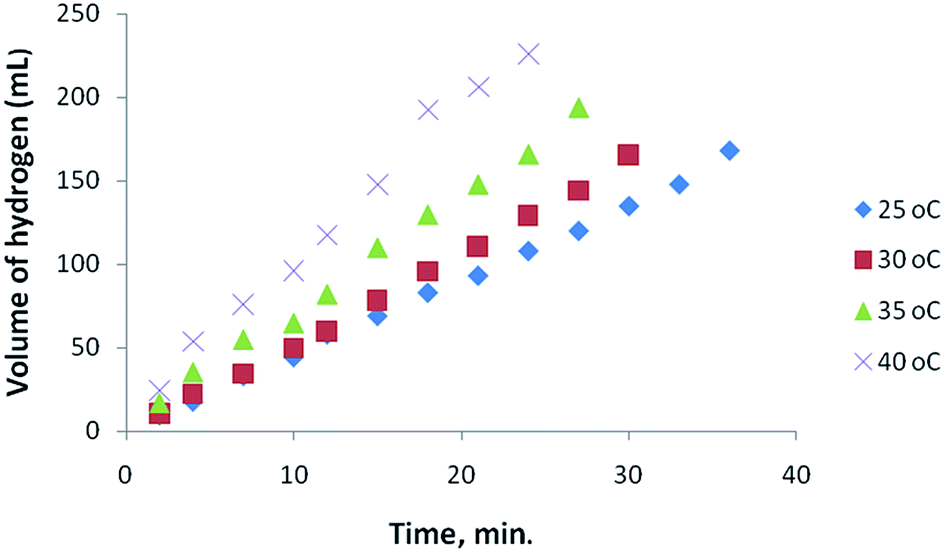 | ||
| Fig. 6 Effect of temperature (Pt-hollow nanospheres) on the volume of generated hydrogen (mL) versus time for the hydrolysis of ammonia–borane. | ||
Fig. 7 exhibits catalytic activities of Pt nanoparticles with different morphological features, such as hollow nanospheres, dense spheres, and ligand-stabilized nanoparticles of similar size and wall-thickness as the hollow nanospheres. Among the various particles tested for hydrolysis of AB, Pt hollow nanospheres with a wall-thickness of 5.5 nm showed the highest activity under similar experimental conditions; however, hollow nanoparticles with a wall thickness of 9.5 nm also realized nearly similar activity and no significant difference was observed. These observed results suggest that no appreciable difference in the catalytic activity was noticed when the shell-thickness of the hollow particles decreases below 10 nm. It is important to mention that Pt hollow nanospheres of size about 1–2 μm showed less activity than the bimetallic clusters47 and this can be accounted for by nano-size effects. As the particle size is reduced from the micrometer to the nanometer range, the surface area vis-à-vis binding sites of catalysts increases proportionally and hence the particles show enhanced catalytic activity compared to their corresponding micron sized particles. More interestingly, the ligand-stabilized Pt nanoparticles of size approximately 5–10 nm (ESI. Fig. S6†) show appreciably less activity, which could be owing to the following reason. In the case of functionalized or stabilized nanoparticles, the stabilizing agents always encapsulate or bind with the metal surface to reduce surface free energy vis-à-vis number of binding sites available for catalysis and hence it shows lower catalytic activity than the unmasked hollow nanospheres having shell thickness of 5–10 nm under investigation. We also studied dense spherical Pt particles of size 30–40 nm (ESI. Fig. S7†). These exhibited the lowest activity among the various catalysts investigated in this study and the decreased activity might be owing to the direct consequence of a lower number of binding sites available for catalysis. Obviously, the thin-wall structures (5–10 nm) of hollow nanospheres could efficiently promote catalytic reactions and effectively circumvent the adverse effects of masking agents in the liberation of hydrogen. After hydrolysis of AB, the catalyst was isolated by suction filtration and dried under nitrogen atmosphere. The isolated catalysts were reused again and tested for reusability. We reused the same Pt nanocatalyst successively for five times and negligible loss of catalytic activity was noticed (ESI. Fig. S8†). The minor decrease in catalytic conversion of AB in subsequent runs may be owing to the material loss during isolation or owing to passivation of the Pt hollow nanosphere surface by the increasing amount of metaborate (byproduct), which might decrease the accessibility of the active sites.48,49 Furthermore, it is worth pointing out that the Pt-hollow nanospherical particles mostly retained their spherical morphology, as shown in Fig. S9† of the ESI, and aggregation occurs to a lesser extent. The above study suggests that Pt hollow nanospheres can be used as effective heterogeneous catalysts for the liberation of hydrogen from AB under mild experimental conditions.
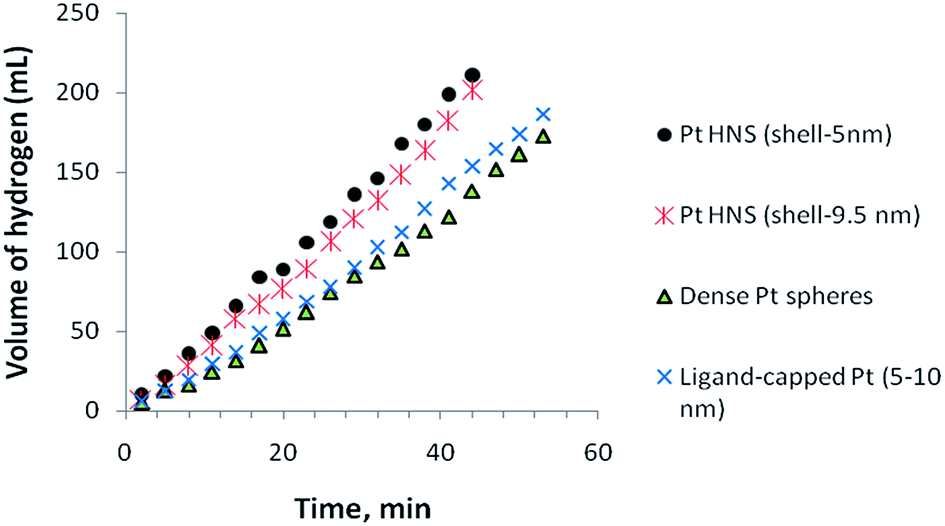 | ||
| Fig. 7 Effect of Pt-nanoparticles with different sizes (Pt-hollow nanospheres) on the volume of generated hydrogen (mL) versus time for the hydrolysis of ammonia–borane. | ||
Conclusions
ABC triblock copolymers of type PS–PVP–PEO with core–shell–corona architectures can be effectively used as soft templates for the fabrication of Pt hollow nanospheres of size less than 30 nm under mild experimental conditions. The synthesis of Pt hollow nanospheres with different K2PtCl6/PVP mole ratios confirmed that the wall-thickness can be increased over several nanometers. The use of triblock copolymers with different PS core block lengths, such as PS(20.1k)–PVP(14.2k)–PEO(26k) and PS(45k)–PVP(16k)–PEO(8.5k), led to the formation of hollow nanospheres with slightly increased void-space diameters. Investigation by FTIR spectroscopic study suggested that solvent extraction using DMF effectively removes polymeric templates from composite particles to provide Pt hollow nanospheres. High resolution TEM, electron diffraction pattern, and X-ray diffraction results confirmed the hollow spherical nature of Pt nanoparticles with very high crystallinity. The Pt hollow nanospheres serve as an efficient heterogeneous and reusable catalyst for the hydrolysis of ammonia–borane under mild conditions. The hollow nanoparticles with wall-thickness of less than 10 nm exhibited very good activity in the liberation of hydrogen from AB compared to either dense Pt spherical particles of similar size or ligand-stabilized Pt nanoparticles of size less than 10 nm, suggesting the potential application of Pt hollow nanospheres in hydrogen evolution reactions.Acknowledgements
M. S. thanks the Science & Engineering Research Board for their research support (No. SB/S1/PC-043/2013). P. B. thanks CSIR, New Delhi for a junior research fellowship.Notes and references
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- Y. Song, Y. Yang, C. J. Medforth, E. Pereira, A. K. Singh, H. Xu, Y. Jiang, C. J. Brinker, F. Swol and J. Shelnutt, J. Am. Chem. Soc., 2004, 126, 635–645 CrossRef CAS PubMed.
- H. Wang, H. Y. Jeong, M. Imura, L. Wang, L. Radhakrishnan, N. Fujita, T. Castle, O. Terasaki and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 14526–14529 CrossRef CAS PubMed.
- B. Lim, X. M. Lu, M. J. Jiang, P. H. C. Camargo, E. C. Cho, E. P. Lee and Y. N. Xia, Nano Lett., 2008, 8, 4043–4047 CrossRef CAS PubMed.
- J. Ren and R. D. Tilley, J. Am. Chem. Soc., 2007, 129, 3287–3291 CrossRef CAS PubMed.
- C. Wang, H. Daimon, Y. Lee, J. Kim and S. Sun, J. Am. Chem. Soc., 2007, 129, 6974–6975 CrossRef CAS PubMed.
- N. R. Jana and X. Peng, J. Am. Chem. Soc., 2003, 125, 14280–14281 CrossRef CAS PubMed.
- M. Zhao and R. M. Crooks, Adv. Mater., 1999, 3, 11–23 Search PubMed.
- Y. D. Liu, Z. Fang, L. Kuai and B. Y. Geng, Nanoscale, 2014, 6, 9791–9797 RSC.
- K. Niesz, M. Grass and G. A. Somorjai, Nano Lett., 2005, 5, 2238–2240 CrossRef CAS PubMed.
- J. M. Petroski, T. C. Green and M. A. El-Sayed, J. Phys. Chem. A, 2001, 105, 5542–5547 CrossRef CAS.
- M. A. Mahmoud, C. E. Tabor, M. A. El-Sayed, Y. Ding and Z. L. Wang, J. Am. Chem. Soc., 2008, 130, 4590–4591 CrossRef CAS PubMed.
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2009, 131, 9152–9153 CrossRef CAS PubMed.
- Y. Yamauchi, A. Takai, T. Nagura, S. Inoue and K. Kuroda, J. Am. Chem. Soc., 2008, 130, 5426–5427 CrossRef CAS PubMed.
- H. Wang, H. Y. Jeong, M. Imura, L. Wang, L. Radhakrishnan, N. Fujita, T. Castle, O. Teraski and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 14526–14529 CrossRef CAS PubMed.
- M. T. Reetz, W. Heibig and S. A. Quaiser, Chem. Mater., 1995, 7, 2227–2228 CrossRef CAS.
- M. Ohtaki, M. Komiyama, H. Hirai and N. Toshima, Macromolecules, 1991, 24, 5567–5572 CrossRef CAS.
- Q. Wang, H. Liu and H. Wang, J. Colloid Interface Sci., 1997, 190, 380–386 CrossRef CAS PubMed.
- H. S. Taylor, Proc. R. Soc. London, Ser. A, 1925, 108, 105 CrossRef CAS.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS PubMed.
- S. W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642–7643 CrossRef CAS PubMed.
- S. Moreton, K. Faulds, N. C. Shand, M. A. Bedics, M. R. Detty and D. Graham, Nanoscale, 2015, 7, 6075–6082 RSC.
- H. T. Schmidt and A. E. Odtafin, Adv. Mater., 2002, 14, 532–535 CrossRef CAS.
- T. Nakashima and N. Kimizuka, J. Am. Chem. Soc., 2003, 125, 6386–6387 CrossRef CAS PubMed.
- S. Schacht, Q. Huo, I. G. Voigt-Martin, G. D. Stucky and F. Schuth, Science, 1996, 273, 768–771 CAS.
- H.-P. Liang, H.-M. Zhang, J.-S. Hu, Y.-G. Guo, L.-J. Wan and C.-L. Bai, Angew. Chem., Int. Ed., 2004, 43, 1540–1543 CrossRef CAS PubMed.
- M. Sasidharan and K. Nakashima, Acc. Chem. Res., 2014, 47, 157–167 CrossRef CAS PubMed.
- M. Sasidharan, K. Nakashima, N. Gunawardhana, T. Yokoi, M. Inoue, S. Yusa, M. Yoshio and T. Tatsumi, Chem. Commun., 2011, 47, 6921–6923 RSC.
- M. Sasidharan, N. Gunawardhana, M. Inoue, S. Yusa, M. Yoshio and K. Nakashima, Chem. Commun., 2012, 48, 3200–3202 RSC.
- M. Sasidharan, H. Zenibana, M. Nandi, A. Bhaumik and K. Nakashima, Dalton Trans., 2013, 42, 13381–13389 RSC.
- M. Sasidharan, K. Nakashima, N. Gunawardhana, T. Yokoi, M. Ito, M. Inoue, S. Yusa, M. Yoshio and T. Tatsumi, Nanoscale, 2011, 3, 4768–4773 RSC.
- S. K. Das, M. K. Bhunia, D. Chakraborty, A. R. Khuda-Bukhsh and A. Bhaumik, Chem. Commun., 2012, 48, 2891–2893 RSC.
- B. Peng and J. Chen, Energy Environ. Sci., 2008, 1, 479–483 CAS.
- T. Umegaki, J. M. Yan, X. B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, Int. J. Hydrogen Energy, 2008, 34(5), 2303–2311 CrossRef.
- T. U. Umegaki, J. M. Yan, X. B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, Int. J. Hydrogen Energy, 2009, 34(9), 2303–2311 CrossRef CAS.
- P. V. Ramachandran and P. D. Gagare, Inorg. Chem., 2007, 46, 7810–7817 CrossRef CAS PubMed.
- Q. Xu and M. Chandra, J. Power Sources, 2006, 163, 364–370 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 163, 855–861 CrossRef.
- M. Chandra and Q. Xu, J. Power Sources, 2007, 168, 135–142 CrossRef CAS.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- J. F. Gohy, N. Willet, S. Varshney, J. X. Zhang and R. Jerome, Angew. Chem., Int. Ed., 2001, 40, 3214–3216 CrossRef CAS.
- L. Lei, J. F. Gohy, N. Willet, J. X. Zhang, S. Varshney and R. Jerome, Macromolecules, 2004, 37, 1089–1094 CrossRef CAS.
- M. Stepanek, J. Humpolickova, K. Prochazka, M. Hof, Z. Tuzar, M. Spirkova and T. Wolff, Collect. Czech. Chem. Commun., 2003, 68, 2120–2138 CrossRef CAS.
- T. Herricks, J. Chen and Y. Xia, Nano Lett., 2004, 4, 2367–2371 CrossRef CAS.
- H. Perez, J. P. Pradeau, P. A. Albouy and J. P. Omil, Chem. Mater., 1999, 11, 3460–3463 CrossRef CAS.
- M. Sasidharan, D. Liu, N. Gunawardhana, M. Yoshio and K. Nakashima, J. Mater. Chem., 2011, 21, 13881–13888 RSC.
- W. Qin, C. Yang, X. Ma and S. Lai, J. Alloys Compd., 2011, 509, 338–342 CrossRef CAS.
- T. J. Clark, G. R. Whittell and K. Manners, Inorg. Chem., 2007, 46, 7522–7527 CrossRef CAS PubMed.
- C. A. Jaska, T. Clark, S. B. Clendenning, D. Groeza, A. Turak and Z. H. Lu, J. Am. Chem. Soc., 2005, 127, 5116–5124 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-visible, FTIR spectra, TG/DTA, nitrogen adsorption/desorption isotherm, TEM pictures Pt nanoparticles of different sizes and catalyst reusability data. See DOI: 10.1039/c5ra26277a |
| This journal is © The Royal Society of Chemistry 2016 |