
Anti-choline esterase activity of ceramides from the Red Sea marine sponge Mycale euplectellioides†
Reda Abdelhameedab,
Mohamed Saleh Elgawishc,
Amira Mirad,
Amany K. Ibrahimb,
Safwat A. Ahmedb,
Kuniyoshi Shimizue and
Koji Yamada*a
aGraduate School of Biomedical Sciences, Nagasaki University, 1-14 Bunkyo-machi, Nagasaki 852-8521, Japan. E-mail: kyamada@nagasaki-u.ac.jp; Fax: +81-95-819-2462; Tel: +81-95-819-2462
bPharmacognosy Department, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt
cMedicinal Chemistry Department, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt
dPharmacognosy Department, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
eDivision of Systematic Forest and Forest Products Sciences, Department of Agroenvironmental Sciences, Faculty of Agriculture, Graduate School of Kyushu University, Fukouka 812-8581, Japan
First published on 9th February 2016
Abstract
The isolation and structure elucidation of new phytoceramides from a methanolic extract of the Red Sea sponge Mycale euplectellioides was exclusively studied. Structure elucidation was achieved using spectroscopic techniques, including 1D and 2D NMR and HRMS. The anti-choline esterase activity of the isolated ceramides was evaluated in vitro using a microplate-based Ellman’s assay. Bioassay guided isolation led to the isolation of a MEC-1 phytoceramide molecular species; further purification of MEC-1 afforded three pure phytoceramides: MEC-1-4, MEC-1-7 and MEC-1-8. Molecular modeling studies using glide docking showed tight binding of the ceramides to acetylcholine esterase (AChE). The ceramides showed a better docking score and glide Emodel value when compared to known AChE inhibitors. The ceramides interacted with an aromatic residue of the peripheral anionic site and penetrated deeply into the catalytic triad residues of the active site. Overall, the ceramides obtained using the approaches described here could be considered as promising lead compounds for the discovery and design of potent anti-choline esterase drug candidates, which would be used for Alzheimer’s eradication.
1. Introduction
With the increment in the elderly population around the world, neurodegenerative issues, including Alzheimer’s disease (AD), Parkinson’s disease, and Huntington’s disease continue to be devastating disorders for which compelling medications are required.1 AD, the most well-known type of dementia, is associated with memory dysfunction and cognition impairment. It is believed that 35.6 million individuals suffered from AD in 2010 and that the number will increase twofold at regular intervals, reaching more than 115 million individuals with the disease in 2050.2 The etiology of AD is still puzzling and remains to be elucidated, however, certain hallmarks, such as low levels of acetylcholine, β-amyloid (Aβ) deposits, τ-protein aggregation, oxidative stress, dyshomeostasis of biometals, and neuronal death, are common pathological factors in a patient suffering from AD.3Recently, some promising studies have shown that acetylcholinesterase inhibitors (AChEI) can mitigate neuropsychiatric side effects in AD patients and can improve cognitive capacity by increasing the acetylcholine levels inside the synaptic region.4 Currently, four drugs on the market are approved by the FDA for mild to moderate AD symptom treatment, of which three are AChEI (donepezil, rivastigmine and galantamine). Nevertheless, the approved AChEI have shown several side effects, for example, hepatotoxicity, insomnia, depression, fatigue, and gastro-intestinal upsets, and there is not yet conclusive proof demonstrating that the administration of AChEI or other groups could modify AD progression.5 Hence, the search is ongoing to discover safe and tolerable molecules of natural or synthetic origin for AD eradication.
Common natural products, particularly plant-based constituents, have been viewed as promising drug candidates for several diseases. They have significantly participated in medication disclosure and advancement for AD. For example, galantamine, physostigmine, and huperzine A have been isolated from Galanthus nivalis, Physostigma venenosum, and Huperzia serrata, respectively, and clinically used for AD symptomatic management.6 The diversity of marine organisms and microorganisms encourages and inspires scientists to look for other natural products which could be of great interest as sources of novel drug candidates. Marine secondary metabolites particularly those that can be utilized as medications have shown a noteworthy effect on the improvement of the pharmaceutical industry.7 Many excellent reviews have been published on compounds with potent pharmacological activity that are used to fight several devastating diseases experienced nowadays including cancer, HIV, osteoporosis, and AD.7–9 Among marine organisms, sponges of the genus Mycale are considered as one of the richest sources of bioactive metabolites.
Many bioactive compounds have been isolated from the genus Mycale, which belongs to different areas such as Kenya,10 New Zealand,11 India,12 USA,13 Japan,14 and Egypt,15 with different bioactivities. To the best of our knowledge, only one publication on the sponge Mycale tenuispiculata which shows weak AChE inhibitory activity is reported.16 Hence, the search is still ongoing with the Mycale genus, but with other species in an attempt to improve the AChE inhibitory activity. Throughout the years, alkaloids and terpenes have been regarded as the two largest groups to which most novel candidates isolated belong.7 Herein, ceramides have been isolated from Mycale euplectellioides and investigated for the first time as promising drug candidates for Alzheimer’s treatment.
Sphingolipids have emerged as an important class of biosynthetic compounds that modulate various cellular functions.17 Among the sphingolipids, ceramides (N-acyl-D-erythro-sphingosine) are key lipids, representing a central moiety in the biosynthesis of sphingolipids and glycosphingolipids. Ceramides have attracted the most attention due to their regulatory roles in diverse cellular events, including cellular metabolism, cell senescence, differentiation and apoptosis. Marine organisms are a prosperous source of ceramides with various structures and biological activities.18 Marine ceramides exhibit cytotoxicity, and antitumor, antifungal, and antibacterial activity.19,20 Recently, we have reported the antiepileptic activity of ceramides from the Red Sea sponge Negombata corticata.19 Moreover, CNS depression activity through GABA and serotonin receptor modulation has been reported under our supervision for ceramides from the Red Sea soft coral Sarcophyton auritum.21 It was reported that the chemical structure of the ceramides could extensively affect their biological function; the chain length and saturation at the sphingosin bond modulate the ceramide activity.22,23 Irie et al. declared that C6-dihydro-ceramide, a biologically inactive analogue, had no effects on cell survival or death, indicating that the presence of a double bond in the sphingosine base unit is essential for the activity.24
Continuing our efforts to discover the novel biological activities of marine bioactive constituents, we report on the isolation and structure elucidation of acetylcholine esterase inhibitor MEC-1 from the Red Sea marine sponge Mycale euplectellioides for the first time. Ceramides may provide valuable sources of lead compounds for AD management. Therefore, the AChE inhibitory activity of ceramides needs to be systemically demonstrated.
Computer-based modeling methodologies have been progressively used to investigate the pharmacological activity of many natural molecules and in the development of new medications. In contrast to traditional drug development methods, the expense of drug advancement would diminish using this new technique and the time required for the medication improvement cycle would be radically lessened.25 Thus, our objectives in this study are the isolation and structure elucidation of new ceramides from a marine sponge, in vitro investigation of the AChE inhibitory activity of the ceramides, and predicting the interactions of the ceramides in the active site of AChE through a molecular docking technique.
2. Materials and methods
2.1. General experimental information
1H NMR (400 MHz), 13C NMR (100 MHz), DEPT-135 and 2D-NMR spectra were recorded using the residual solvent signal as an internal standard with a Varian AS 400 (Varian Inc., Palo Alto, CA, USA). The UV spectra were recorded using a double beam Shimadzu UV-visible spectrophotometer (model UV-1601 PC, Kyoto City, Japan). IR spectra were recorded using a Nicolet FT-IR spectrophotometer (Nicolet Company, Nicolet, Canada) with a range of 400–4000 cm−1. High resolution mass spectra were recorded using a Bruker BioApex (Bruker Corporation). Pre-coated silica gel G-25 UV254 plates were used for thin layer chromatography (TLC) (20 cm × 20 cm) (E. Merck, Darmstadt, Germany). Silica gel Purasil 60A, 230–400 mesh was used for flash column chromatography (Whatman, Sanford, ME, USA).2.2. Animal organism collection and identification
The sponge Mycale euplectellioides was collected from Sharm El Sheikh at the Egyptian Red Sea, air-dried and stored at low temperature (−24 °C) until processed. Voucher specimens were deposited at the Zoological Museum of the University of Amsterdam under registration number ZMAPOR18964 and in the herbarium section of the Pharmacognosy Department, Faculty of Pharmacy, Suez Canal University, Ismailia, Egypt under registration number SAA-7.2.3. Extraction and isolation
All experimental protocols in the present study were approved by the Research Ethics Committee at the Faculty of Pharmacy, Suez Canal University, Ismailia, Egypt (approval no. PG-NR3-16-1). A sample (500 g, wet weight) of Mycale euplectellioides was defrosted and repeatedly extracted with methanol (3 × 2 L). The combined extracts were concentrated under vacuum to afford a crude extract (4 g). The crude extract was subjected to silica gel column chromatography using CHCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH:H2O (1:0:0–6.5:3.5:0.5), which provided seven fractions; M-1–M-7. The fraction eluted with 97% CHCl3:MeOH, M-2 (500 g), was chromatographed on a silica gel column using n-hexane:EtOAc (10:90–0:100) to give 12 sub-fractions (M-2-1–M-2-12). Fraction M-2-12 (262 mg) was further chromatographed on a sephadex LH-20 column using CHCl3:MeOH (1:1) followed by silica gel column chromatography using CHCl3:MeOH (98:2) isocratic elution to afford MEC-1 (17.7 mg, white amorphous powder). MEC-1 (6 mg) was finally purified using semi-preparative HPLC (Develosil C30-UG, 100% MeOH) to afford MEC-1-4 (1.2 mg), MEC-1-7 (0.4 mg), and MEC-1-8 (1.4 mg) as pure phytoceramides.
:MeOH:H2O (1:0:0–6.5:3.5:0.5), which provided seven fractions; M-1–M-7. The fraction eluted with 97% CHCl3:MeOH, M-2 (500 g), was chromatographed on a silica gel column using n-hexane:EtOAc (10:90–0:100) to give 12 sub-fractions (M-2-1–M-2-12). Fraction M-2-12 (262 mg) was further chromatographed on a sephadex LH-20 column using CHCl3:MeOH (1:1) followed by silica gel column chromatography using CHCl3:MeOH (98:2) isocratic elution to afford MEC-1 (17.7 mg, white amorphous powder). MEC-1 (6 mg) was finally purified using semi-preparative HPLC (Develosil C30-UG, 100% MeOH) to afford MEC-1-4 (1.2 mg), MEC-1-7 (0.4 mg), and MEC-1-8 (1.4 mg) as pure phytoceramides.
| Position | MEC-1 | |
|---|---|---|
| 1H | 13C | |
| a Spectra were acquired at 23 °C. Chemical shifts are given in δ (ppm) and are referenced to internal solvent signals for C5D5N at 7.19 (δH) and 123.5 (δC) ppm. | ||
| NH | 8.58 (1H, d, J = 8.9 Hz) | |
| 1a | 4.43 (1H, dd, J = 10.8, 5.2 Hz) | 62.0 (t) |
| 1b | 4.49 (1H, dd, J = 10.8, 4.6 Hz) | |
| 2 | 5.12 (1H, m) | 52.9 (d) |
| 3 | 4.35 (1H, m) | 76.7 (d) |
| 4 | 4.27 (1H, m) | 73.0 (d) |
| 1′ | 175.2 (s) | |
| 2′ | 4.61 (1H, m) | 72.4 (d) |
| –CH3 | 0.88 (m) | 14.2 (q) |
| 11.5 (q) | ||
| 19.3 (q) | ||
| 22.4 (q) | ||
:1) (0.2 mL) at 70 °C for 8 h in a sealed small-volume vial followed by evaporation under vacuum to dryness to afford the LCB acetates for further 1H-NMR, 13C-NMR and mass spectrometry analysis.2.4. Acetylcholine esterase (AChE) inhibition assay
AChE inhibition activity was measured using Ellman’s method.26 AChE hydrolyzes the substrate acetylthiocholine iodide (ACTI) into acetate and thiocholine. In a neutral or alkaline medium; thiocholine reacts with 5,5-dithiobis-2-nitrobenzoic acid (DTNB) to give yellow colored 2-nitro-5-thiobenzoate, which can be detected spectrophotometrically at 405 nm. Briefly, in a 96-well plate, 25 μL of 15 mM ACTI, 125 μL of 3 mM DTNB in buffer B (50 mM Tris–HCl, pH = 8, 0.1 M NaCl, 0.02 M MgCl2·6H2O), 50 μL of buffer A (50 mM Tris–HCl, pH 8, 0.1% BSA) and 25 μL of the test sample (dissolved in 25% DMSO) were mixed, and the absorbance was measured using a microplate reader (Biotek, Winooski, VT, USA) at 405 nm every 16 s for ten times. Then, 25 μL of (0.25 U mL−1 in buffer A) AChE (from Electrophorus electricus (electric eel)) was added and the absorbance was measured ten times every 16 s. A solution of 25% DMSO was used as a negative control. The absorbance was plotted against time and the enzyme activity was calculated from the slope of the line, and so was obtained and expressed as a percentage compared to an assay using a buffer without any inhibitor. To avoid any increase in the absorbance due to spontaneous hydrolysis of the substrate, the absorbance before addition of the enzyme was subtracted from the absorbance after adding the enzyme.2.5. Protein preparation for the docking study
For the present study, the X-ray crystal structure of AChE in a complex with donepezil (PDB ID: 1EVE) was obtained from the Protein Data Bank and further prepared using the protein preparation wizard, which is accessible in Glide, Schrodinger 10.1. After ensuring chemical accuracy, a preparation component adds hydrogen and neutralizes the side chains that are neither close to the binding cavity nor involved in the formation of salt bridges. The OPLS-2005 force field was used for this purpose and then the active site of the protein was defined. In the next step, water molecules were removed and H atoms were added to the crystal structure, most likely at the positions of hydroxyl and thiol hydrogen atoms. The protonation states and tautomers of the His residue and Chi ‘flip’ assignments for the Asn, Gln and His residues were selected using the protein assignment script provided by Schrodinger. Minimization was performed to relieve steric clashes using the OPLS2005 force field until the average root mean square deviation (RMSD) of the non-hydrogen atoms reached a maximum value of 0.3 Å.27–292.6. Ligand preparation
All the compounds were constructed using the fragment library of Maestro 10.1 and prepared by using LigPrep 2.1, which can produce a number of structures from each input structure with various ionization states, tautomers, stereochemistries and ring conformations. The OPLS-2005 force field was used for optimization, which produces a low energy conformer of the ligand.29,302.7. Molecular docking
To test the docking parameters, all the compounds were docked into the binding site of the AChE protein (PDB ID: 1EVE) using Grid-Based Ligand Docking With Energetics (Glide) software from Schrodinger. To soften the potential for non-polar parts of the receptor, we scaled the van der Waal radii of the receptor atoms by 0.8 with a partial atomic charge of 0.15. A grid box with coordinates X = 2.87, Y = 64.62 and Z = 67.93 was generated at the centroid of the active site. The ligands were docked with the active site using ‘extra precision’ glide docking (Glide XP) which docks ligands flexibly. Glide generates conformations internally and passes these through a series of filters. The specific methodology of XP docking has been explained elsewhere. The final best docked structure was chosen using a Glide score function. The lowest-energy docked complex was found in the majority of the similar docking conformations. Finally, the lowest-energy docked complex was selected for further study.27–293. Results and discussion
The methanolic extract obtained from Mycale euplectellioides showed acetylcholine esterase inhibitory activity using a TLC assay method. Bioassay guided fractionation led to the isolation of MEC-1. Further purification of MEC-1 using semi-preparative HPLC afforded three pure phytoceramides MEC-1-4, MEC-1-7, and MEC-1-8.3.1. Chemistry
MEC-1 (17.7 mg) was obtained as a white amorphous solid, and showed as a single spot on silica gel thin layer chromatography (TLC). MEC-1 exhibited strong hydroxy (3300 cm−1) and amide absorptions (1623, 1545 cm−1) in the IR spectrum.The 1H-NMR spectrum in C5D5N showed resonances of an amide proton doublet at δH 8.58 (1H, d, J = 8.9 Hz), the protons of a long methylene chain, centered at δH 1.25, and overlapped methyls at δH 0.85, indicating the presence of a sphingolipid skeleton. Characteristic resonances for the 2-amino-1,3,4,2′-tetrol region of the hydrocarbon chain were observed at δH 5.12 (1H, m, H-2), 4.61 (1H, m, H-2′), 4.49 (1H, dd, J = 10.8, 5.2 Hz, H-1b), 4.43 (1H, dd, J = 10.8, 4.6 Hz, H-1a), 4.35 (1H, m, H-3), and 4.27 (1H, m, H-4) in the 1H-NMR spectrum and at δC 52.9 (C-2), 72.4 (C-2′), 62.0 (C-1), 76.7 (C-3), and 73.0 (C-4) in the 13C-NMR spectrum, in addition to the amide carbonyl signal at δC 175.2 (C-1′). A series of molecular ion peaks due to [M + Na]+ were observed in the positive ion FAB-MS spectrum at m/z: 678, 692, 706, 720, and 734. Therefore, MEC-1 is presumed to be a molecular species consisting of phytosphingosine-type ceramides possessing 2-hydroxy fatty acid groups. Furthermore, MEC-1 is suggested to contain normal, iso, and ante-iso type terminal methyl groups,31 since the carbon signals for the terminal methyl groups were observed at δC 14.2 (normal type), 22.4 (iso type), 11.3 and 19.1 (ante-iso type) in the 13C-NMR spectrum (Fig. 1, Table 1). The core structure of MEC-1 was characterized by comparison of its 13C-NMR spectral data with that of known ceramides.19,32,33 The relative stereochemistry of the ceramide molecular species is suggested to be (2S, 3S, 4R, 2′R), since the aforementioned 1H-NMR (H-2, H-3, H-4, H-2′) and 13C-NMR signals (C-1, C-2, C-3, C-4, C-2′) are in good agreement with those of phytosphingosine-type ceramide molecular species possessing (2S, 3S, 4R, 2′R) configurations.34,35 Based on the results of MEC-1 methanolysis with methanolic hydrochloric acid followed by 1H-NMR, 13C-NMR, and FAB-MS analysis, the length and branching pattern of the resulting LCB and fatty acids could be determined. The n-hexane layer afforded a mixture of FAMEs, which were subjected to 1H-NMR, 13C-NMR, and EI-MS analysis. EI-MS analysis of the FAME mixture showed the presence of five components at m/z: 356, 370, 384, 398 and 412 [M+], which were designated as FAM-1, FAM-2, FAM-3, FAM-4, and FAM-5, indicating the presence of C-21, C-22, C-23, C-24 and C-25 fatty acid methyl esters, respectively.
 | ||
| Fig. 1 Structures of MEC-1. | ||
Furthermore, the FAME mixture was thought to possess normal and iso type terminal methyl groups, since the carbon signals for the terminal methyl groups in the 13C-NMR spectrum were observed at δC = 14.2 (normal) and δC = 22.4 (iso) On the other hand, the methanol layer contained the long chain bases, which were reacted with acetic anhydride to afford the corresponding acetate followed by analysis using 1H-NMR, 13C-NMR, and FAB-MS. The FAB-MS analysis of the LCB mixture indicated the presence of a C19 long chain base identified from the corresponding molecular ion peak at m/z 500 [M + H]+ and the 13C-NMR spectrum indicated that the LCBs exhibit normal, iso and ante-iso terminal methyl groups (Fig. 1, Table 1).
Based on the considerable interest in and importance of determining the molecular species composition of sphingolipids, the isolation and structure elucidation of the ceramide components in MEC-1 was conducted. By means of reversed phase HPLC, MEC-1 was separated to show eight peaks and they were recovered to give eight fractions in a range from MEC-1-1 to MEC-1-8. Three of these eight fractions, MEC-1-4, MEC-1-7 and MEC-1-8 displayed a single molecular ion peak [M + Na]+ in the positive ion FAB-MS spectrum. On the basis of the molecular mass of MEC-1-4 m/z 692 [M + Na+], MEC-1-7 m/z 706 [M + Na+], and MEC-1-8 m/z 720 [M + Na+], the structures of MEC-1-4, MEC-1-7 and MEC-1-8 were determined as shown in Fig. 2.
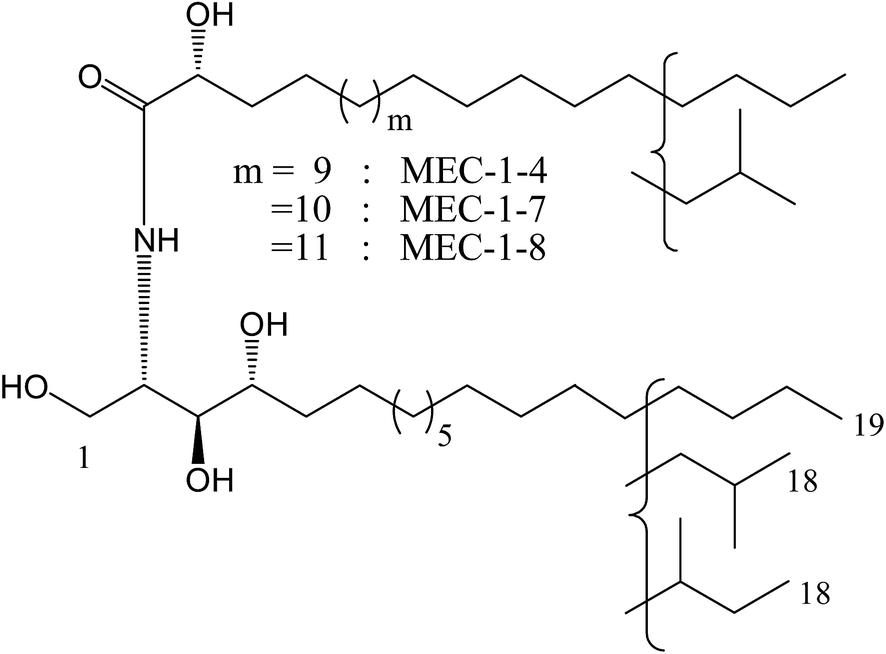 | ||
| Fig. 2 Structure of the isolated phytoceramides. | ||
3.2. Biological activity
| Compound | AChE inhibition, IC50 μM |
|---|---|
| MEC-1 | 20.9 |
| Galantamine | 1.7 |
3.3. Molecular docking
A powerful approach for predicting the coupling of a substrate with its receptor is docking simulation, which can be effectively executed using numerous applications. Nowadays, interest in the coupling of computational, chemical, and biological techniques has extensively grown in order to streamline drug design, discovery, and development.25 Using computer-based molecular docking simulations could save on the effort, time, and resources required by traditional drug development methodology. In this study, the potential inhibitory activity of the ceramides against AChE was determined using molecular simulation with glide docking.AChE is a serine protease, which catalyzes the hydrolysis of the neurotransmitter acetylcholine (ACh) in neuromuscular junctions and cholinergic brain synapses. Inhibiting the activity of cholinesterases increases the level of ACh in the brain, which affects the cognitive capacity positively.5 AChE has an overall ellipsoid shape containing a deep groove, usually called the gorge, which is about 20 Å deep. Although the hydrolysis process occurs at the base of the gorge, initial binding of ACh is thought to occur at its outer rim in a region called the peripheral site. At the bottom of the gorge, where the actual hydrolysis occurs, there are four main subsites, these being the esteratic site, the oxyanion hole, the anionic subsite and the acyl pocket. The esteratic site contains the catalytic machinery of the enzyme, which is dependent on a catalytic triad of Ser200–His440–Glu327. The catalytic triad enhances the nucleophilicity of the catalytic serine for attack of the substrate while the Glu stabilizes the histidinium cation of the transition state. The oxyanion hole consists of Gly118, Gly119 and Ala201, which stabilize the tetrahedral intermediate of ACh. The anionic subsite (hydrophobic subsite) is largely comprised of aromatic residues and contains Trp84, Phe330 and Glu199, which are believed to bind to quaternary ammonium ligands through pi-cation interactions. Trp84 is an important residue for binding ACh. Mutation of Trp84 to an aliphatic residue results in a severe decrease in reactivity toward ACh. The acyl pocket consists of Phe288 and Phe290, which are assumed to play a role in limiting the dimensions of the substrates that are able to enter the active site.6
Our docking study has shown that AChE can clearly accommodate ceramides within the aromatic gorge, giving them the opportunity to interact with peripheral anionic and quaternary ammonium binding sites (Fig. 3). The ceramide structure is more or less similar to some aliphatic bis-quaternary ammonium containing inhibitors like decamethonium. The simplicity of the skeleton of the ceramides gives them the ability to penetrate into the catalytic triad of AChE, compared to donepezil (Fig. 4). The orientation of Phe330 can control access to the bottom of the gorge and three major conformations, open, closed, and intermediate, were identified. The closed conformation was identified for an enzyme complex with huperzine A, and the intermediate was recognized for tacrine, while Phe330 adopted an open access position with gorge-spanning ligands such as donepezil and decamethonium.36 Due to the size and shape of the ceramides, a gorge spanning binding mode was selected for the molecular docking simulations. Therefore, a known X-ray structure of a AChE-E2020 (donepezil) complex was nominated as the template for our docking study.
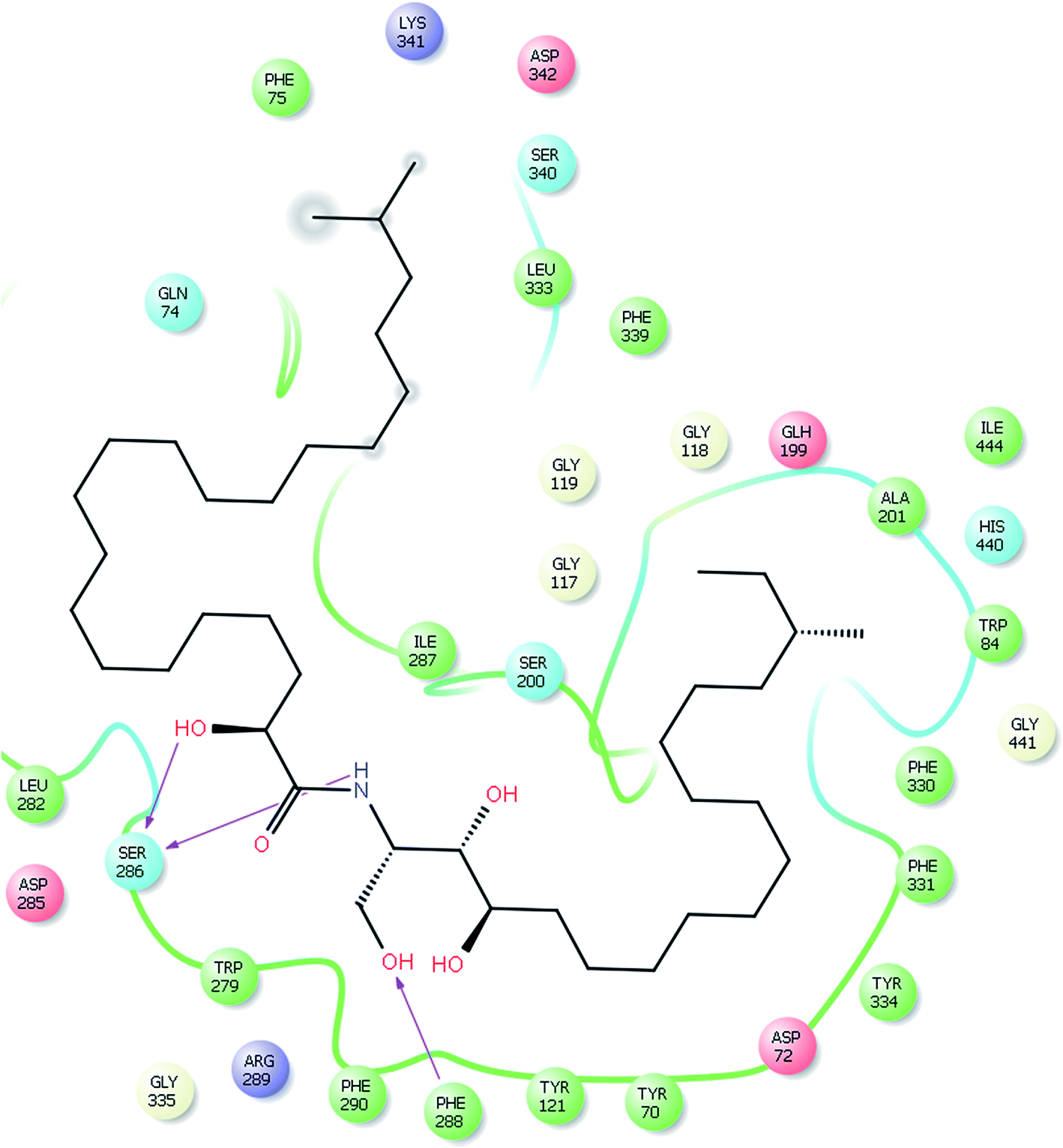 | ||
| Fig. 3 Glide XP docked structure of a ceramide within the active site of AChE, highlighting the hydrophobic interactions of the ceramide with the PAS and deep penetration into the esteratic site. | ||
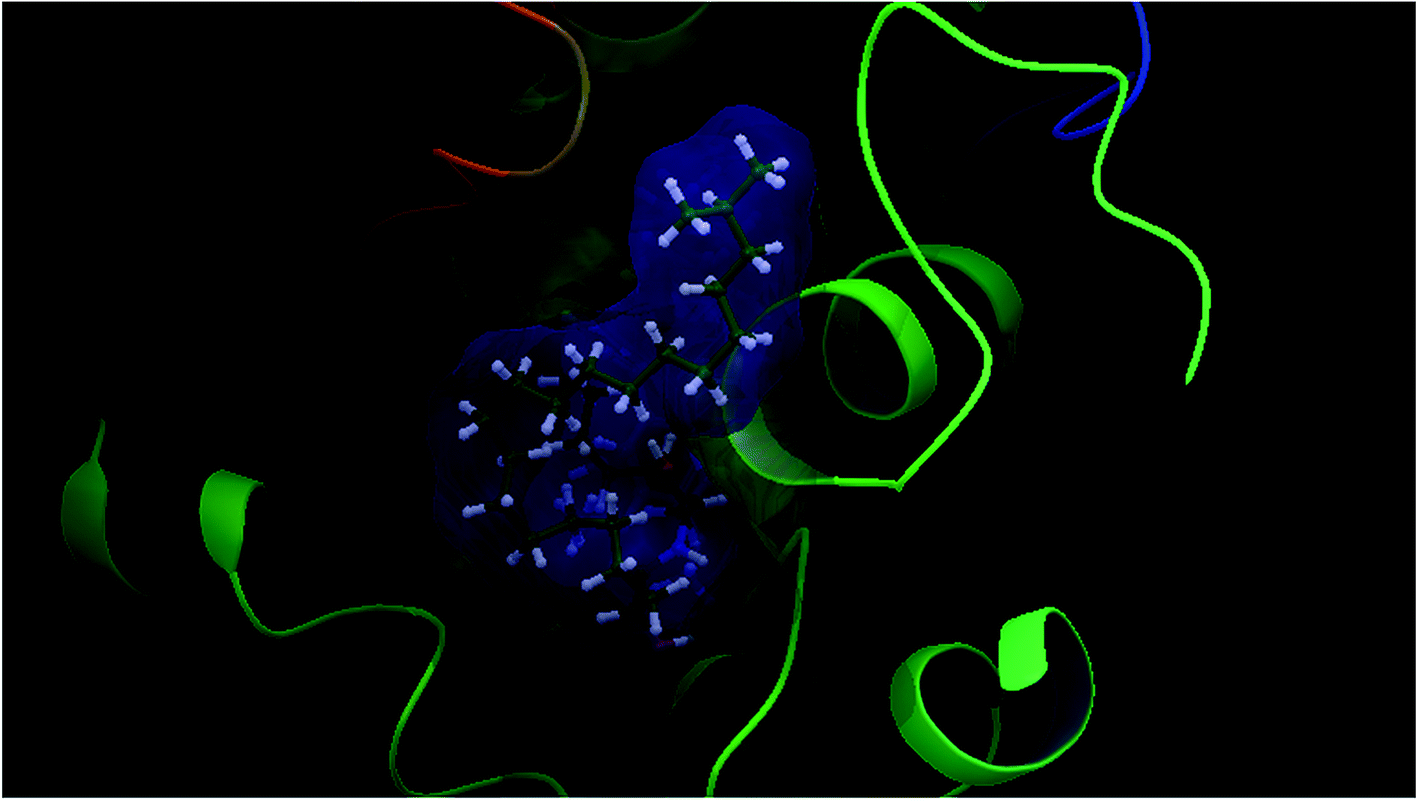 | ||
| Fig. 4 Superimposition of a ceramide (green colour) and donepezil (red colour) within AChE (PDBID: 1EVE), generated using glide docking. | ||
Investigation of the potential inhibitory activity of the ceramides towards AChE was carried out with TcAChE, obtained from the electric eel Torpedo californica. The structural similarity of TcAChE to mammalian AChE makes it the most popular enzyme used for in vitro and molecular docking studies and consequently it was used in our study.6 TcAChE-coupled donepezil was utilized for the protein preparation and grid generation, and then our target compounds were prepared via ligand preparation before glide docking. Although the normal, iso, and ante-iso forms of the MEC-1 polyceramides were difficult to separate at this stage, molecular docking studies of all of them were performed. The docking protocol was repeated several times and the best docking pose with the lowest energy was selected. The results of each docking protocol were identical to other repetitions of the protocol; meaning a high reproducibility of the glide docking methodology.
Ceramides interact with AChE via hydrogen bonding, hydrophobic contacts, and hydrophilic–hydrophobic interactions. The hydroxyl proton of the sphingosine moiety can form a hydrogen bond (2.51 Å) with the amide group of Phe288 and the hydroxyl proton of the fatty acid moiety and the amide proton of the sphingosine moiety form hydrogen bonds (1.95 Å and 2.2 Å, respectively) with the amide group of Ser286 (Fig. 5). These hydrogen bonds might participate in the stabilization of the ceramide–TcAChE complex. Ceramides can occupy the aromatic gorge of AChE, providing multiple-binding sites. They interact with peripheral anionic site (PAS) residues Tyr70, Tyr121, Trp279, and Tyr334 and oxyanion hole residue Phe330 via hydrophobic contacts. The binding to the PAS residues seems to play a vital role in the tight binding and energetic stabilization of the ceramides within AChE. The carboxylate groups of Asp72 and Gln74 also form hydrophilic–hydrophobic interactions with the methylene group of the fatty acid and sphingosine moieties. These interaction forces help to provide the tight contact of the ceramides with AChE. Extra precision glide docking of the ceramides with AChE showed a reasonable G score or docking score of −9.688 and a glide Emodel value of −70.083 kcal mol−1, compared to donepezil that gave a docking score of −10.404 and a glide Emodel value of −63.869 kcal mol−1. Under identical docking methodology, decamethonium showed a glide score of −9.349 and a glide Emodel value of −23.409 kcal mol−1. Moreover, the docking study results were compared with juliflorine, a potent natural PAS inhibitor of AChE which showed a docking score of −9.015 and a glide Emodel value of −71.588.37 Under our docking experiment conditions, the normal ceramides showed better binding compared to the iso and ante-iso ceramides (complete data are in preparation to be published later). Additionally, the length of the methylene chain of both the fatty acid and sphingosine moieties could affect the molecular docking score. This result is in agreement with previous publications,22–24 which claimed that the ceramides’ structure can strangely affect their biological activities. C6-Dehydrocermide, an unsaturated sphingosine base, could induce Aβ-aggregation, while dihydroceramides, like our studied molecules, lacked this role. Our docking study confirmed, by comparing the docking score of the saturated and unsaturated molecules, that the unsaturated molecules show a weak docking score (−5593). The low value of this docking score might be attributed to the 4,5-trans double bond which restricts the rotation of the long chain and makes the molecules more rigid for fitting into the receptor. Another important result of the docking study was that the ceramides showed weak fitting with butyrylcholinesterase (BChE) (data not shown). The major differences between AChE and BChE are the PAS and acyl pocket.6 The lacking of several aromatic amino acids in the PAS of BChE could affect the ceramide binding and give our studied compounds a high selectivity for AChE over BChE.
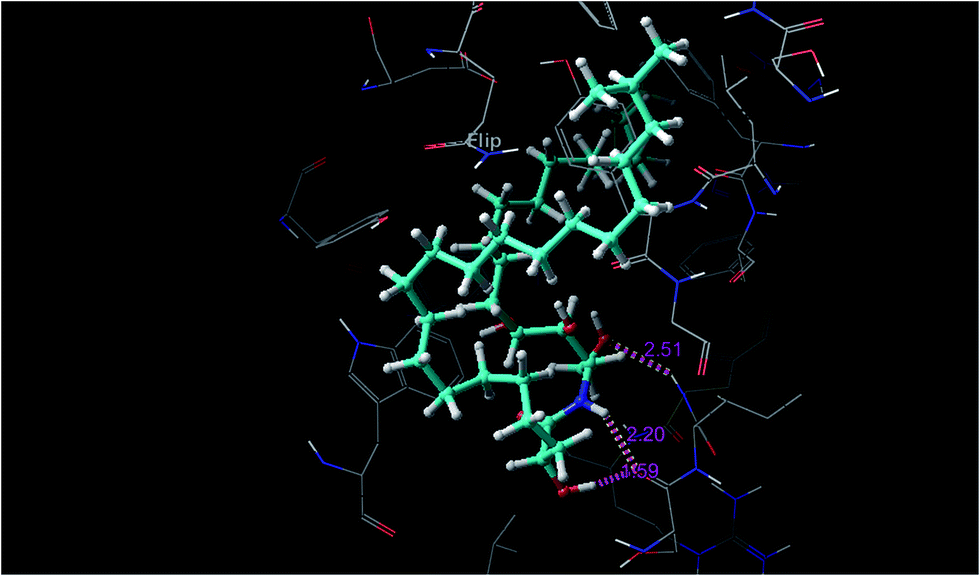 | ||
| Fig. 5 Hydrogen bond interactions between the ceramide and AChE (PDBID: 1EVE). | ||
A major observation is that all ceramides are potentially able to bind inside the catalytic triad gorge, however the whole molecule is not capable of interacting with amino acid residues of the enzyme (Fig. 3). This observation might be attributed to a size problem which causes reduced inhibitory activity compared to other AChEI. Our target compounds lack reactivity with Trp84 at the gorge bottom as in the case of donepezil. Moreover, the ceramides lack the quaternary ammonium group which plays a key role in the binding of AChEI like donepezil, tacrine, and huperzine A to the enzyme. Moreover, we utilized MEC-1 which contains a mixture of cermides; some of them showed good biological activity and others showed weak activity. Although the docking study showed a good docking score for the ceramides compared to known AChEIs, the in vitro assay showed moderate to weak activity. We attributed the moderate biological activity to the difficulty of pure ceramide separation. In future, our main goals are separation of each ceramide in the MEC-1 mixture and performing a complete biological and computational study.
From the docking results, we can deduce that the ceramides powerfully interact with amino acids residues of the PAS and penetrate inside the catalytic gorge, but do not interact with the triad of machinery amino acids. Thus, the ceramides are considered as non-competitive inhibitors of AChE. It has been reported that AChE may actively participate in the processing, maturation and deposition of β-amyloid peptides; this novel non-cholinergic function relates to its PAS.38,39 As our target molecules are binding to the PAS, they could be considered promising lead compounds for the development of potent anti-Alzheimer’s agents. After confirmation that the PAS at the rim of the cavity plays a vital role in the enzymatic activity and of the ability of the ceramides to interact with amino acids residues at the PAS, structural modification of a ceramide by the addition of a quaternary ammonium group for interaction with the catalytic triad and Trp84 at the base of the gorge is an important step in developing a potent dual acting anti-Alzheimer’s candidate.
4. Conclusion
Herein, the isolation and structure elucidation of phytoceramide acetylcholine esterase inhibitors from the genus Mycale was reported for the first time. In vitro biological screening was performed to detect the AChE inhibitory activity of the ceramide mixture MEC-1 using Ellman’s assay. It was found that MEC-1 showed moderate potency against AChE (IC50 = 20.9 ± 2.4 μM). Molecular docking studies using glide docking, Schrodinger 10.1 were conducted to demonstrate the binding mode of the ceramides inside the active gorge of AChE. The docking results showed that three binding forces, including hydrogen bonds, hydrophobic contacts and hydrophilic–hydrophobic interactions, participated in the tight binding of the ceramides to AChE. The ceramides can interact with the aromatic residues at the PAS and can penetrate deeply into the catalytic triad of the active site. Although the ceramides showed a better docking score and glide Emodel value compared to known AChEI, the in vitro biological screening showed moderate potency. This major observation may be attributed to the use of a ceramide mixture instead of pure compounds and thus more biological assays are required to solve this finding. The powerful interaction of ceramides with the PAS and their ability to fit into the active site makes them a promising dual acting anti-choline esterase inhibitor; hopefully suitable for use as a potent anti-Alzheimer’s agent. This study is considered as a part of our project to study and identify new biological activities of ceramides, rather than the familiar ones, especially in the area of CNS activity.Acknowledgements
We are grateful to Mr M. Inada and Dr N. Tsuda of the Scientific Support Section of Joint Research Center, Nagasaki University, for 1H-NMR, 13C-NMR and MS measurements. This work was supported in part by a Grant-in-Aid for Scientific Research No. 23590008 and No. 26460124 from the Japan Society for the Promotion of Science, which is gratefully acknowledged.References
- D. M. Walsh and D. Selkoe, Neuron, 2004, 44, 181–193 CrossRef CAS PubMed.
- http://www.alz.co.uk/research/les/WorldAlzheimerreport/WorldAlzheimerReport, 2009, Alzheimer’s disease international.
- E. Scarpini, P. Scheltens and H. Feldman, Lancet Neurol., 2003, 2, 539–547 CrossRef CAS PubMed.
- Y. Xu, J. P. Colletier, H. Jiang, I. Silman, J. L. Sussman and M. Weik, Protein Sci., 2008, 17, 601–605 CrossRef CAS PubMed.
- A. Corbett, et al., Nat. Rev. Drug Discovery, 2012, 11, 833–846 CrossRef CAS PubMed.
- P. J. Houghton, Y. Ren and M. Howes, Nat. Prod. Rep., 2006, 23, 181–199 RSC.
- J. W. Blunt, B. R. Copp, R. A Keyzers, M. H. Munro and M. R. Prinsep, Nat. Prod. Rep., 2015, 32, 116–211 RSC.
- D. Y. Choi and H. Choi, Arch. Pharmacal Res., 2015, 38, 139–170 CrossRef CAS PubMed.
- B. Pejin, M. Mojovic and A. G. Savic, Nat. Prod. Rep., 2014, 28, 2237–2244 CrossRef CAS PubMed.
- A. Rudi, Y. Benayahu and Y. Kashman, J. Nat. Prod., 2005, 68, 280–281 CrossRef CAS PubMed.
- A. Brackovic and J. E. Harvey, Chem. Commun., 2015, 514, 750–4756 Search PubMed.
- G. B. Reddy and N. Dhananjaya, Bioorg. Med. Chem., 2000, 8, 27–36 CrossRef CAS PubMed.
- D. Romo, N. S. Choi, S. Li, I. Buchler, Z. Shi and J. O. Liu, J. Am. Chem. Soc., 2004, 126, 10582–10588 CrossRef CAS PubMed.
- Y. Nakao, S. Yoshida, S. Matsunaga, N. Shindoh, Y. Terada, K. Nagai, J. K. Yamashita, A. Ganesan, R. W. van Soest and N. Fusetani, Angew. Chem., Int. Ed. Engl., 2006, 45, 7553–7557 CrossRef CAS PubMed.
- G. A. Mohamed, A. E. Abd-Elrazek, H. A. Hassanean, A. M. Alahdal, A. Almohammadi and D. T. Youssef, Nat. Prod. Res., 2014, 28, 1082–1090 CrossRef CAS PubMed.
- G. Beedessee, A. Ramanjooloo, R. Surnam-Boodhun, R. vanSoest and D. E. Marie, Chem. Biodiversity, 2013, 10, 442–451 CAS.
- R. C. Dickson, Annu. Rev. Biochem., 1998, 67, 27–48 CrossRef CAS PubMed.
- Y. A. Hannun and L. M. Obeid, Nat. Rev. Mol. Cell Biol., 2008, 9, 139–152 CrossRef CAS PubMed.
- S. A. Ahmed, S. I. Khalifa and M. T. Hamann, J. Nat. Prod., 2008, 71, 513–515 CrossRef CAS PubMed.
- E. E. Eltamany, A. K. Ibrahim, M. M. Radwan, M. A. ElSohly, H. A. Hassanean and S. A. Ahmed, Med. Chem. Res., 2015, 24, 3467–3473 CrossRef CAS.
- N. A. Eltahawy, A. K. Ibrahim, M. M. Radwan, S. Zayton, M. Gomaa, M. AElSohly, H. A. Hassanean and S. A. Ahmed, Bioorg. Med. Chem. Lett., 2015, 25, 5819–5824 CrossRef CAS PubMed.
- L. Puglielli, B. C. Ellis, A. J. Saunders and D. M. Kovacs, J. Biol. Chem., 2003, 278, 19777–19783 CrossRef CAS PubMed.
- A. Bielawska, H. M. Crane, D. Liotta, L. M. Obeid and Y. A. Hannun, J. Biol. Chem., 1993, 268, 26226–26232 CAS.
- F. Irie and Y. Hirabayashi, J. Neurosci. Res., 1998, 54, 475–485 CrossRef CAS PubMed.
- M. Dik-Lung, D. S. Chana and C. Leung, Chem. Sci., 2011, 2, 1656–1665 RSC.
- G. Ellman, D. Lourtney, V. Andres and G. A. Gmelin, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- S. K. Tripathi and S. K. Singh, Mol. BioSyst., 2014, 10, 2189–2201 RSC.
- LigPrep 2.5, Schrödinger, LLC, New York, 2011 Search PubMed.
- K. Yamada, R. Matsubara, M. Kaneko, T. Miyamoto and R. Higuchi, Chem. Pharm. Bull., 2001, 49, 447–452 CrossRef CAS PubMed.
- A. Loukaci, V. Bultel-Poncé, A. Longeon and M. Guyot, J. Nat. Prod., 2000, 63, 799–802 CrossRef CAS.
- N. Krishna, P. Muralidhar, M. Kumar, D. Rao and C. Rao, J. Nat. Prod., 2004, 67, 1423–1425 CrossRef CAS PubMed.
- X. Chen, Y. Wu and D. Chen, Tetrahedron Lett., 2002, 43, 3529–3532 CrossRef CAS.
- H. Azuma, R. Takao, H. Niiro, K. Shikata, S. Tamagaki, T. Tachibana and K. Ogino, J. Org. Chem., 2003, 68, 2790–2797 CrossRef CAS PubMed.
- C. Pilger, J. Mol. Graphics Modell., 2001, 19, 288–296 CrossRef CAS PubMed.
- M. I. Choudhary, S. A. Nawaz, Z. ul-Haq, M. K. Azim, M. N. Ghayur, M. A. Lodhi, S. Jalil, A. Khalid, A. Ahmed, B. M. Rode, A. Rahman, A. U. Gilani and V. U. Ahmad, Biochem. Biophys. Res. Commun., 2005, 332, 1171–1177 CrossRef CAS PubMed.
- G. V. de Ferrari, M. A. Canales, I. Shin, L. Weiner, M. I. Silman and N. C. Inestrosa, Biochemistry, 2001, 40, 10447–10457 CrossRef CAS PubMed.
- M. Bartolini, C. Bertucci, V. Cavrini and V. Andrisano, Biochem. Pharmacol., 2003, 65, 407–416 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR, 13C-NMR, FAB-MS, and EI-MS data for MEC-1 and MEC-1 after hydrolysis are available. See DOI: 10.1039/c5ra26424c |
| This journal is © The Royal Society of Chemistry 2016 |