
Thermochemical properties of rare-earth oxyhydrides from first principles phonon calculations†
Xin Liu,
Tor Svendsen Bjørheim* and
Reidar Haugsrud
FASE, Department of Chemistry, University of Oslo, Norway. E-mail: torsb@kjemi.uio.no
First published on 15th January 2016
Abstract
Rare-earth oxyhydrides – REHO (RE = La, Ce, Pr, Nd, Gd, Er) are among the oxyhydrides uncovered in recent years. Little is, however, known about their thermochemical properties as they are inherently prone to re-oxidation. In this contribution, we address the thermochemical properties of selected REHO from first principles phonon calculations to investigate possible trends in their formation from binary oxides and common reduction agents, and their thermal stability.
Hydrogen is known to take on all of its three oxidation states when incorporated in solids or liquids; metals dissolve minor amounts of H2(g) in the form of interstitial neutral H, while larger H concentrations result in metal hydride phases in which H− constitutes the anion lattice.1,2 A large number of ionic compounds, such as oxides, dissolve hydrogen in the form of positive protonic defects (H+) from for instance water vapour.3 However, trace amounts of hydride ions have been discovered in several proton conducting oxides under moderately reducing conditions.4,5 H− has been shown to be accommodated in certain transition metal oxides under highly reducing conditions (e.g., metal hydrides and/or high pressure H2), where it partially replaces the oxide ions and forms ionic bonds with the transition metal.6–13 Several studies have uncovered both stoichiometric oxyhydrides, as for instance SrCrHO2 (ref. 9) and the rare-earth oxyhydrides (REHO),6,13 and non-stoichiometric oxyhydrides, such as BaTiO3−xHx (ref. 7 and 10) and LaSrCoO3H0.7.8 H− has a filled 1s2 electronic configuration, and thus different frontier orbitals than O2−, leading to novel properties of these kinds of materials, such as magnetic8,9 and metallic properties.7,12 However, synthesis of oxyhydrides is challenging due to the strong reduction ability of hydride ions. Some oxyhydrides may be prepared by mixing transition metal oxides with CaH2(s),6–8,10–13 or by using high pressures of H2.9 Beside these two typical methods, for instance, Nd(OH)3 mixed with metallic Nd has also been shown to result in the rare-earth oxyhydride.6 In this thermodynamic study, we consider several synthesis routes for rare-earth oxyhydrides, exemplified by the reactions;
| RE2O3(s) + CaH2(s) = 2REHO(s) + CaO(s) | (1) |
RE2O3(s) + RE(s) + ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) H2(g) = 3REHO(s) H2(g) = 3REHO(s)
| (2) |
| RE(OH)3(s) + 2RE(s) = 3REHO(s) | (3) |
Further, similar to metal hydrides, oxyhydrides are inherently prone to re-oxidation upon heating in oxidizing atmospheres,6,11 forming either the corresponding oxides, or the hydroxides;
| 2REHO(s) + O2(g) = RE2O3(s) + H2O(g) | (4) |
| REHO(s) + H2(g) + ½O2(g) = RE(OH)3(s) | (5) |
The non-stoichiometric oxyhydrides, such as BaTiO3−xHx, exhibit the same – or closely related – crystal structures as the parent oxides, and the oxyhydrides may thus be described as highly deficient versions of the perfect oxides.7,10,12 The rare-earth oxyhydrides, however, take on crystal structures that are only similar to the parent oxides.6,11,13,14 Hence, while the physiochemical properties of the non-stoichiometric oxyhydrides, at least to some extent, can be assessed from the parent oxides, the properties of stoichiometric oxyhydrides may be rather different from those of their parent oxides. There are still few reports on preparation and physicochemical properties of stoichiometric rare-earth oxyhydrides.6,13 This is partly related to their poor stability with respect to oxidation, rendering experimental evaluations difficult. Therefore, computationally determined thermochemical properties can provide valuable information for potential experimental investigations.15–17 In this short communication, the thermochemical properties of the rare-earth oxyhydrides (REHO) are, for the first time, investigated from first principles phonon calculations in order to determine trends in their phase and thermal stability. The outcome of these calculations may serve as a basis to predict possible synthesis routes for novel oxyhydrides.
All calculation details are shown in the ESI.† The phonon calculations revealed no imaginary modes for either of the investigated rare-earth compounds, including the rare-earth metals (RE), rare-earth oxyhydrides (REHO), and the rare-earth oxides (RE2O3), suggesting that all structures are dynamically stable. Fig. 1A shows the relaxed crystal structure of LaHO, with rows of either O2− or H−, and alternating H− and O2− ions along the y-axis. All rare-earth oxyhydrides crystallize in a tetragonal structure with space group P4/nmm. Fig. 1B displays the evolution of the lattice parameter through the REHO series as emerging from the calculations. The cell parameters of REHO series follow the lanthanide contraction, i.e. they decrease with decreasing rare-earth radius18 (from LaHO to ErHO), also in agreement with the experimentally observed trend.6,11,13 Table 1 lists calculated interionic bond lengths, and the Bader charges19,20 of RE, H and O ions of the rare-earth oxyhydrides. The bond lengths (RE–O and RE–H) also decrease with decreasing rare-earth radius (from LaHO to ErHO), as expected from contraction of the unit cell through the RE series. The calculated La–H, La–O and O–H bond lengths of LaHO are 2.503, 2.448 and 2.895 Å, respectively, which are in good agreement with Malaman's experimental results,11 2.51, 2.48 and 2.85 Å, respectively. Bader charge analyses show that the rare-earth metal ions become more positive, while the hydride ions (H−) become more negative with decreasing rare-earth radius, especially for the heavier rare-earths (GdHO and ErHO). The Bader charge evolution suggests an increasing ionic character of the RE–H bond upon compression of the unit cell.
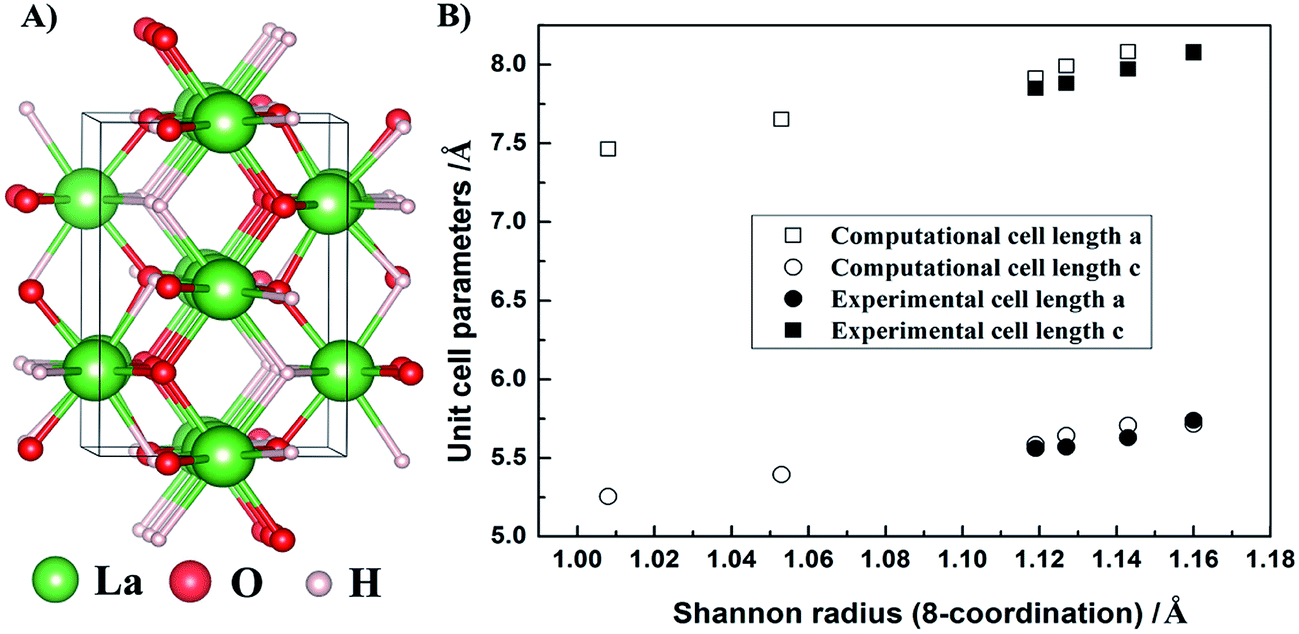 | ||
| Fig. 1 (A) Relaxed crystal structure of LaHO and (B) evolution of a/b (squares) and c (circles) cell parameters through the REHO series for RE = La – Er within the P4/nmm structure. Open symbols show computational results, while the solid show the experimental values.6,11,13 | ||
| Structure info (length Å, angle °) | Charge distribution | |||||||
|---|---|---|---|---|---|---|---|---|
| M–O | M–H | O–H | M–H–M | M–O–M | Metal | Oxygen | Hydride | |
| LaHO | 2.448 | 2.503 | 2.895 | 107.64 | 111.22 | 1.97 | −1.33 | −0.64 |
| CeHO | 2.456 | 2.490 | 2.890 | 108.49 | 110.70 | 2.03 | −1.37 | −0.66 |
| PrHO | 2.428 | 2.460 | 2.867 | 108.60 | 110.67 | 2.04 | −1.38 | −0.66 |
| NdHO | 2.405 | 2.435 | 2.839 | 108.62 | 110.67 | 2.04 | −1.38 | −0.66 |
| GdHO | 2.325 | 2.355 | 2.748 | 108.61 | 110.72 | 2.11 | −1.42 | −0.69 |
| ErHO | 2.267 | 2.298 | 2.628 | 108.58 | 110.77 | 2.14 | −1.44 | −0.70 |
Fig. 2 shows the evolution of the Gibbs free energies of reaction, ΔGreaction, according to eqn (1)–(3), respectively, through the REHO (RE = La, Ce, Pr, Nd, Gd and Er) series. The Gibbs free energies of formation of the oxyhydrides from CaH2(s) (Fig. 2A) are close to temperature independent, but display a notable compositional dependency. Of the included REHO, CeHO displays the lowest formation free energy (−18.88 kJ mol−1 at 300 K), while the formation free energies of NdHO, LaHO and PrHO are around −15 kJ mol−1. The slightly negative free energies of formation indicate that these compounds could be synthesized from CaH2, which is also supported by the experimental observation that NdHO emerges from a reactant mixture of CaH2 and Nd2O3.6 GdHO and ErHO, however, display significantly higher free energies of formation, indicating that these two oxyhydrides would be difficult to synthesize through this route.
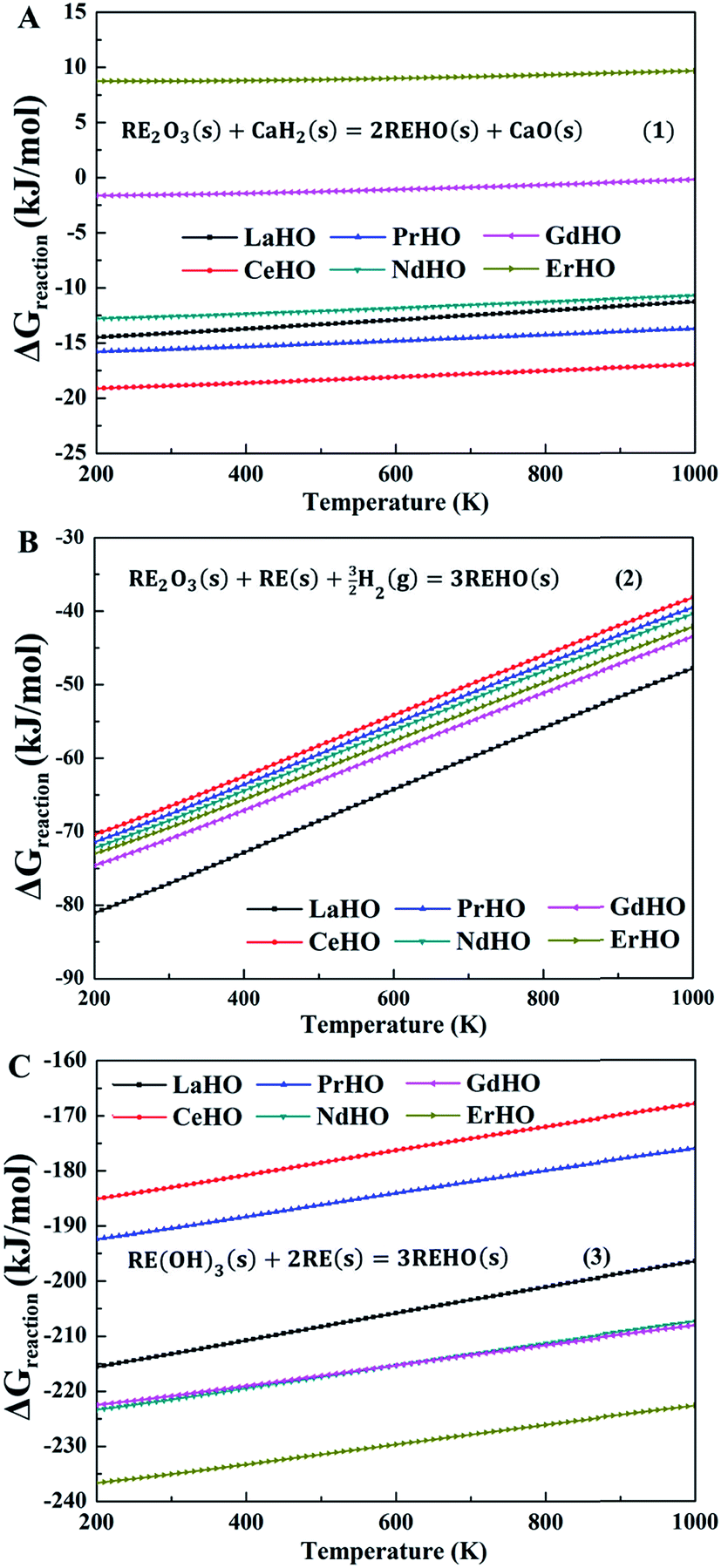 | ||
| Fig. 2 Gibbs free energy of reaction according to eqn (1)–(3) per mole of REHO (RE = La, Ce, Pr, Nd, Gd and Er) as a function of temperature. (A) CaH2 as the reducing agent. (B) RE metal and H2 gas (pH2 = 103 and pH2O = 10−5 bar) as reducing agents. (C) RE(OH)3 reduced by RE metal. | ||
The hydrides of the rare-earth metals in bulk form have successfully been prepared by heating the metal powder in H2,21,22 which, with combined partial oxidation, would form the REHO (eqn (2)). Fig. 2B displays the Gibbs formation free energies as a function of temperature, according to eqn (2). Interestingly, all REHO display negative formation free energies, which become even more negative at higher H2 pressures. The formation free energies also show a notable temperature dependency, which mainly originates from the entropy change due to H2(g). As such, this route is thermodynamically more favourable compared to that with CaH2 as reducing agent, indicating that the rare-earth oxyhydrides could be synthesized by heating a mixture of rare-earth metals and rare-earth oxides in H2(g). This synthesis route is also confirmed by Kageyama et al., who found that a high pressure and temperature method (5 GPa and 1000 °C), promoted formation of SrCrO2H and also accelerated metal hydride precursor decomposition.9 In addition, recent calculations on non-stoichiometric oxyhydrides in the authors' laboratory indicate that the external pressure could promote formation of hydride defects in oxides, because of a negative formation volume of the hydride defects.
Widerøe et al.6 reported that NdHO could be synthesized from a mixture of metal (Nd) and metal hydroxides (Nd(OH)3), i.e., eqn (3). The calculated thermodynamics of REHO formation according to eqn (3) is shown in Fig. 2C. The free energies display a stronger compositional dependency than those according to eqn (1) and (2). This strong dependency stems from the larger number of rare-earth compounds involved in the reaction compared to eqn (1) and (2). In addition, the free energies of formation of all included REHO are significantly lower than through the two other synthesis routes, and is therefore the most favourable synthesis route.
Fig. 3 shows the Gibbs free energies of decomposition of the REHO (RE = La, Ce, Pr, Nd, Gd and Er) series. We assume rather reducing conditions with pH2O = 10−5 and pH2 = 103 bar. pO2 is calculated by the water formation equilibrium constant, shown in the ESI, Fig. S1.† The calculated decomposition free energies of all rare-earth oxyhydrides are below −190 kJ mol−1, and thus favourable, even under these highly reducing conditions. This is in accordance with observations in references,6,9 showing that strongly reducing conditions are required to avoid oxidation of these oxyhydrides. There is another REHO decomposition route according to the reaction in eqn (5), shown in ESI, Fig. S3.† The increasing number of gas molecules involved on the reactant side of eqn (5) results in a stronger temperature dependency. Further, the poor stability of rare-earth hydroxides results in higher Gibbs decomposition free energies than for eqn (4).23 The higher decomposition free energies of eqn (5) compared to eqn (4) suggests that metal oxides would dominate as products upon decomposition of REHO above ambient temperatures.
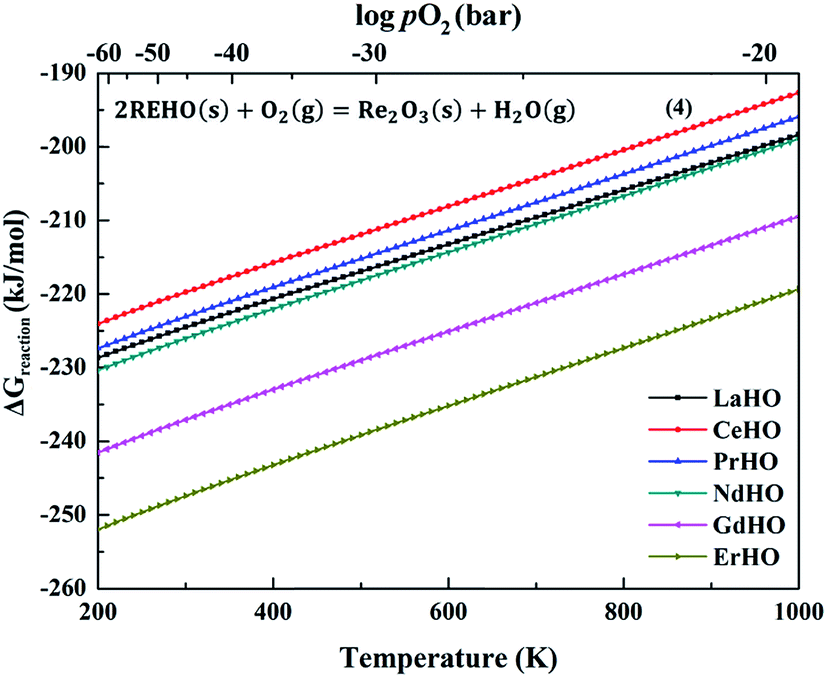 | ||
| Fig. 3 Gibbs free energy per mole REHO (RE = La, Ce, Pr, Nd, Gd and Er) as a function of temperature (pO2 is calculated by the equilibrium with pH2O = 10−5 and pH2 = 103 bar). | ||
In order to correlate trends in the various enthalpies to the stability of the different solid phases involved in the reactions, we have also evaluated the cohesive energies (Ecohesive and ΔCreaction) of the different materials and reactions,24 shown in Table 2. The lower cohesive energy, the more stable the material is. The cohesive energies through the RE2O3 series show stronger compositional dependence compared to REHO series, suggesting that the change in the stability of the metal oxides through the series dominates the compositional dependency of the reaction free energies. The reaction cohesive energies, ΔCreaction, are further calculated from the cohesive energy difference between the reactants and products. The calculation method is shown in ESI,† eqn (8). A higher reactant (e.g. RE2O3) cohesive energy will drive the reaction toward REHO formation. For instance, the high cohesive energy of Ce2O3 leads to the lowest formation free energy of CeHO in eqn (1).
| Oxyhydrides Ecohesive | Metal oxides Ecohesive | Eqn (1) | Eqn (2) | Eqn (3) | ||||
|---|---|---|---|---|---|---|---|---|
| ΔGreaction | ΔCreaction | ΔGreaction | ΔCreaction | ΔGreaction | ΔCreaction | |||
| La | −1567.97 | −3373.70 | −14.12 | 28.02 | −77.09 | 263.48 | −213.21 | 621.05 |
| Ce | −1540.54 | −3309.80 | −18.88 | 37.06 | −66.59 | 231.56 | −183.00 | 528.78 |
| Pr | −1551.12 | −3337.30 | −15.56 | 30.71 | −67.64 | 234.97 | −190.42 | 550.70 |
| Nd | −1558.21 | −3357.08 | −12.60 | 25.11 | −68.48 | 237.91 | −221.48 | 570.37 |
| Gd | −1573.83 | −3409.82 | −1.54 | 3.62 | −71.02 | 246.62 | −220.83 | 641.63 |
| Er | −1582.02 | −3446.40 | 8.76 | −16.58 | −69.44 | 243.00 | −235.05 | 685.03 |
The formation energies according to eqn (3) are significantly more exothermic than those of eqn (1) and (2), and show a stronger compositional dependence. Also the reaction cohesive energies of eqn (3) are much higher than those according to eqn (1) and (2), and generally increase with increasing atomic number, indicating that the reactants (RE metal and RE(OH)3) become more unstable. Experimental work shows that rare-earth hydroxides more easily dehydrate to the rare-earth oxide hydroxides (REO(OH)) with increasing RE atomic number.23 Therefore, the instability of RE(OH)3 results in the favourable formation energies of the heavier rare-earth oxyhydrides (ErHO and GdHO).
In summary, we have performed first principles DFT calculations of rare-earth oxyhydrides to explore their thermodynamic stability and predict possible synthesis routes. Our calculations reveal that the lattice parameters decrease through the REHO (RE from La to Er) series following the lanthanide contraction, in excellent agreement with the experimental data. Bader charge density analyses show that the rare-earth metal ions become more positive and the hydride ions (H−) become more negative with decreasing rare-earth radius, indicating increasing ionic character of RE–H bond upon compression of the unit cell.
Our results further suggest that: (1) CaH2 can be used to synthesize the lighter rare-earth oxyhydrides; (2) by heating a mixture of rare-earth metals and rare-earth oxides in H2(g), formation of oxyhydrides is thermodynamically favourable, and finally (3) rare-earth hydroxides are the most promising starting materials for fabrication of the heavier rare-earth oxyhydrides. Furthermore, the reaction cohesive energies are explored and shown to correlate strongly with the formation energies. Lastly, all rare-earth oxyhydrides are shown to be highly unstable even with trace amounts of O2. Their decomposition free energies are lower than −200 kJ mol−1, indicating that all these compounds are prone to be oxidized. In summary, the present calculations illustrate the application of density functional based methods to efficiently screen thermodynamic properties of unstable materials. In future work, we intend to adopt similar methods to address rational design of oxyhydrides and novel transport properties of hydride ions in oxyhydrides.
Acknowledgements
The authors gratefully acknowledge the financial support from FOXHOUND project and Norwegian metacenter for computational science (Notur) for providing computational resources under the project number NN4604k.References
- S. R. Ovshinsky, M. A. Fetcenko and J. Ross, Science, 2009, 260, 176–181 Search PubMed.
- K. Poeppelmeier, Science, 2002, 295, 1849 CrossRef CAS PubMed.
- T. Norby, Solid State Ionics, 1999, 125, 1–11 CrossRef CAS.
- S. Steinsvik, Y. Larring and T. Norby, Solid State Ionics, 2001, 143, 103–106 CrossRef CAS.
- F. W. Poulsen, Solid State Ionics, 2001, 145, 387–397 CrossRef CAS.
- M. Widerøe, H. Fjellvåg, T. Norby, F. Willy Poulsen and R. Willestofte Berg, J. Solid State Chem., 2011, 184, 1890–1894 CrossRef.
- T. Yajima, A. Kitada, Y. Kobayashi, T. Sakaguchi, G. Bouilly, S. Kasahara, T. Terashima, M. Takano and H. Kageyama, J. Am. Chem. Soc., 2012, 134, 8782–8785 CrossRef CAS PubMed.
- M. A. Hayward, E. J. Cussen, J. B. Claridge, M. Bieringer, M. J. Rosseinsky, C. J. Kiely, S. J. Blundell, I. M. Marshall and F. L. Pratt, Science, 2002, 295, 1882–1884 CrossRef CAS PubMed.
- C. Tassel, Y. Goto, Y. Kuno, J. Hester, M. Green, Y. Kobayashi and H. Kageyama, Angew. Chem., Int. Ed., 2014, 126, 10545–10548 CrossRef.
- Y. Kobayashi, O. J. Hernandez, T. Sakaguchi, T. Yajima, T. Roisnel, Y. Tsujimoto, M. Morita, Y. Noda, Y. Mogami, A. Kitada, M. Ohkura, S. Hosokawa, Z. Li, K. Hayashi, Y. Kusano, J. E. Kim, N. Tsuji, A. Fujiwara, Y. Matsushita, K. Yoshimura, K. Takegoshi, M. Inoue, M. Takano and H. Kageyama, Nat. Mater., 2012, 11, 507–511 CrossRef CAS PubMed.
- B. Malaman and J. F. Brice, J. Solid State Chem., 1984, 53, 44–54 CrossRef CAS.
- G. Bouilly, T. Yajima, T. Terashima, W. Yoshimune, K. Nakano, C. Tassel, Y. Kususe, K. Fujita, K. Tanaka, T. Yamamoto, Y. Kobayashi and H. Kageyama, Chem. Mater., 2015, 27, 6354–6359 CrossRef CAS.
- J. F. Brice and A. Moreau, Annales de Chimie Science des Matériaux, 1982, 7, 623–634 CAS.
- N. Hirosaki, S. Osgata and C. Kocer, J. Alloys Compd., 2003, 351, 31–34 CrossRef CAS.
- P. Hermet, K. Niedziolka and P. Jund, RSC Adv., 2013, 3, 22176–22184 RSC.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- Y. Liu, D. Duan, F. Tian, C. Wang, G. Wu, Y. Ma, H. Yu, D. Li and T. Cui, RSC Adv., 2015, 5, 103445–113450 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 254 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- M. Gupta and J. P. Burger, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 6074–6084 CrossRef CAS.
- D. J. Germano and R. A. Butera, J. Appl. Phys., 1979, 50, 2056–2058 CrossRef CAS.
- E. S. Stampler, W. C. Sheets, W. Prellier, T. J. Marks and K. R. Poeppelmeier, J. Mater. Chem., 2009, 19, 4375–4381 RSC.
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155406 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra26552e |
| This journal is © The Royal Society of Chemistry 2016 |