DOI:
10.1039/C5RA26553C
(Paper)
RSC Adv., 2016,
6, 9827-9834
Fast flow synthesis of highly reactive polyisobutylene co-initiated by an AlCl3/isopropyl ether complex
Received
21st December 2015
, Accepted 14th January 2016
First published on 18th January 2016
Introduction
Polyisobutylenes (PIBs) have broad applications based on their merits in low gas permeability and chemical stability.1–3 Because of the strong isomerization tendency of protonated PIB before β-H abstraction, conventional PIBs, industrially produced using H2O as initiator and AlCl3 as co-initiator, have a low content (<10 mol%) of exo-olefin end groups as well as low reactivity for further functionalization.4–9 On the contrary, highly reactive polyisobutylenes (HRPIBs) are denoted as PIBs with a high content of exo-olefin end groups (≥60 mol%, preferably ≥75 mol%) and specific molecular weight distribution (Mn = 500–5000).10–12 They draw much attention from both industry and academia due to their usage as highly reactive intermediates for synthesizing additives to lubricants, fuels and rubbers, such as maleic anhydride modified PIBs.13,14
Commercial HRPIBs can be produced by a single-step process via cationic polymerization of isobutylene (IB) in hexane using BF3 complexes with either alcohols and/or ethers as initiating systems at temperatures far below 0 °C.15–17 During last decades, several new methods have been reported to synthesize HRPIBs at elevated temperature to evade energy intensive process. For example, HRPIBs with Mn of 1400–7000 g mol−1 were prepared by using solvent-ligated complexes borate or aluminate [M(NCMe)6]2+ (M = Mn, Cu) at ambient temperatures, while long time required for high monomer conversion and high cost of initiating complex probably prevent their industrial acceptance.18–20 Recently, Kostjuk and Wu independently reported HRPIB synthesis at moderate temperatures in CH2Cl2 or CH2Cl2–hexane mixture (80/20 v/v) via a cost-effective initiating system consisting of AlCl3 with dialkyl ether, such as di-n-butyl ether (Bu2O) and diisopropyl ether (iPr2O).21–23 The adventitious water was regarded as initiator providing protons. In following, researchers investigated similar initiating systems consisting of various Lewis acids like FeCl3, TiCl4, GaCl3 and alkylaluminum dichloride (RAlCl2, R = Me, Et, iBu) and ethers like butyl methyl ether and ethyl ether, as to afford the adaptability of HRPIB synthesis to non-polar solvents like hexane and toluene or optimize ethers playing a key role in the selective β-H abstraction through electron donor effect.24–36 As using non-polar solvents with low solubility of water, some initiators were also tested like cumyl alcohol, tert-butyl chloride and 1-chlorine-2,4,4-trimethyl pentane (TMPCl). Unfortunately, they usually show poor performance compared with chlorinated solvents in terms of the polydispersity index (PDI) of product.37
In most of reports with Lewis acids concentration in the range of 10 mmol L−1 to 40 mmol L−1, considerable conversion could be reached within 2–3 minutes but 30 minutes or more were required for achieving a conversion platform or total conversion. Differently, we notice that CumOH/AlCl3OBu2 initiating system in CH2Cl2 could afford 60% conversion at 3 min as high as that at 30 min and near 3 times of monomer conversion could be obtained with H2O/FeCl3/iPr2O initiating system in CH2Cl2 under low concentration of FeCl3 at 5 mmol L−1 compared with H2O/AlCl3/Bu2O initiating system. We envision fast synthesis of HRPIB within tens of seconds or less may be achievable under low concentration of AlCl3 with acidity stronger than FeCl3, as long the polymerization kinetics can benefit from the decrease of ether concentration accompanied with Lewis acid. The main challenge in decreasing the concentration of AlCl3/ether complex and controlling an intensive HRPIB synthesis is how to realize effective mixing quickly enough matching with fast reaction process. From this point of view, the microflow system, with the merits of perfect mixing and heat transfer performances and narrow residence time distribution, may be an idea platform.38,39 Many polymerizations have been successfully carried out in the microflow reactor with improved product or process.40–42
In this work, with the AlCl3 concentration in the range from 4 mmol L−1 to 10 mmol L−1 and enough iPr2O addition, we firstly successfully synthesized HRPIB (PDI < 1.9) with over 60% monomer conversion at 20 °C after a 12 s stay in a microflow system. The evolutions of HRPIBs were measured at various molar ratios of iPr2O to AlCl3 to reveal the effects of free iPr2O on active species initiating or deactivating. And then, we extended the temperature range for HRPIBs synthesis from −20 °C to 50 °C and found the depression effect of free iPr2O on the temperature-sensitivity of Mn. Through adjusting the molar ratio of iPr2O to AlCl3 to assure smooth and distinct reaction within tens of seconds, we tailored Mn proportionally by varying monomer concentration with guarantee of good mixing conditions. Finally, we proposed a mechanism with respect to the generation and transformation of complex of AlCl3 with iPr2O for understanding and designing an easily controllable and fast synthesis for HRPIB with Mn in a broad range from 500 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000+.
000+.
Experimental
Materials
Dichloromethane (CH2Cl2, 99.9+%, anhydrous), isopropyl ether (iPr2O, 99.0+%), and aluminum chloride (AlCl3, 99+%, anhydrous) were purchased from J&K Scientific (China). Isobutylene (IB, 99.9+%, anhydrous) and ethanol (analytical reagent) were obtained from Korea Noble Gas (Korea) and Sinopharm Chemical (China), respectively. CH2Cl2 was dried over Molecular Sieves 5A overnight to decrease the content of water to about 7 ppm (determined by Coulometric Karl fischer moisture meter (METTLER TOLEDO, Switzerland)), and then kept in a sealed bottle before use. Isopropyl ether was distilled to remove stabilizer butylated hydroxytoluene (BHT), and then dried over Molecular Sieves 5A overnight. Isopropyl ether and AlCl3 were preserved in a glovebox (MIKROUNA, China).
Preparation of catalytic complex solution
The catalytic complex solution, composed of AlCl3, isopropyl ether (iPr2O), and CH2Cl2 as solvent, was prepared in a glovebox, shortly before experiments. In details, AlCl3 and iPr2O were quantitatively added into a dry bottle in sequence. And then, CH2Cl2 was poured into the bottle to obtain transparent solution. The content of AlCl3 in the solution was determined by UV-Vis spectrophotometer (UV-2450, SHIMADZU). It should be noted that most of the adventitious water, as initiator in the system, comes from the catalytic complex solution.
Polymerization of IB
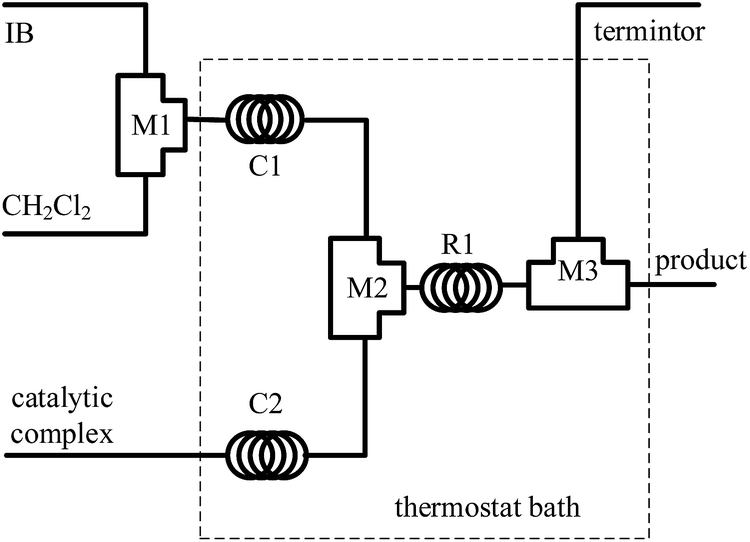
The polymerization of IB was carried out in a microflow system composed of three T-shaped micromixers (M1 for the mixing of IB and diluent, CH2Cl2), M2 for the mixing of IB solution and catalytic complex solution, and M3 for the injection of terminator agent, ethanol), two precooling (or preheating) coiled tubes (C1 and C2, inner diameter 900 μm), and a microtube reactor (R, inner diameter 900 μm), as shown in Fig. 1. The polymerization of IB proceeds throughout R1, and the reaction time could be changed by the flow rate and the length of R1. Three syringe pumps were used to deliver IB, CH2Cl2, and catalytic complex solution, respectively. IB transferred as vapour from cylinder was liquefied at −30 °C into the syringe and perfused under a pressure of about 3 bars. The additional CH2Cl2 was introduced to adjust IB concentration in the reaction system.
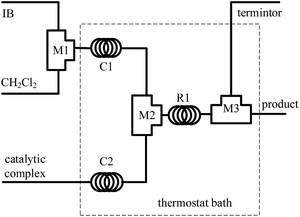 |
| | Fig. 1 Schematic diagram of flow synthesis setup. M1, M2 and M3 are tees as micromixers; C1 and C2 are curved tubes for achieving the pre-set temperature; R1 is microtube as reactor. | |
Characterization
Molecular weight and dispersity of the polymers were measured by a Waters gel permeation chromatography (GPC) composed of a Waters 2707 auto sampler, a 1515 Isocratic HPLC pump, a 2414 refractive index detector and three GPC columns (the molecular weight could be detected from 500–4 × 106) placed in a 38 °C thermostat. Tetrahydrofuran (THF) was used as the eluent at a flow rate of 1.0 mL min−1. The instrument was calibrated with narrow molecular weight distribution polystyrene standards. The results were processed by the Breeze 2 software from Waters. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a JNM-ECA 600 MHz spectrometer using CDCl3 as solvents.
Results and discussion
Feasibility of fast HRPIB synthesis
Exploiting relatively low iPr2O and AlCl3 concentrations (10 mmol L−1 or less), we firstly conducted the polymerization of IB at 20 °C with the residence time of 12 s to test the feasibility of fast HRPIB synthesis. The results listed in Table 1 can be classified into two groups. The first group, including entries 1 to 3, is featured that the iPr2O concentration is clearly lower than the AlCl3 concentration. In this group, the conventional PIBs containing mainly endo-, tri- or tetra-substituted double bonds were obtained, the polydispersity index (PDI) is around 5.0 or higher, and the conversion of monomers is kept at a high level (>80%). It should be noticed that the concentration of AlCl3, much higher than the concentration of adventitious water, is always far lower than the concentration of PIB, indicating the occurrence of intensive chain transfer process. The second group, including entries 4 and 5, is featured that the iPr2O concentration is close to or higher than the AlCl3 concentration. The PIBs with a high exo-olefin group content (>70%) and low PDI (<2.0) were obtained in this group, indicating a success in HPRIBs synthesis. With the increase of the iPr2O concentration, the conversion of monomers is still considerable but decreases to some extent. By comparison, the second group shows high and stable number-average molecular weight (Mn), but much weak chain transfer processes. Obviously, besides of regulating the end group distribution, the addition of iPr2O could decrease the conversion of monomers by reducing the chain propagation rate (related to PDI value) and the chain transfer rate.
Table 1 Polymerization of IB co-initiated by AlCl3 × iPr2O at various iPr2O and AlCl3 concentrations in CH2Cl2a
| Entry |
iPr2O (mmol L−1) |
AlCl3 (mmol L−1) |
Convnb (%) |
Mn (g mol−1) |
PDI |
[PIB]c (mmol L−1) |
End groups distribution (mol%) |
| Exo |
Tri + endo |
Tetra |
| T = 20 °C; t = 12 s; [IB] = 0.75 mol L−1. Gravimetric conversion. [PIB] = [IP] × 56 × convn/Mn. |
| 1 |
1.4 |
4.1 |
81 |
880 |
5.8 |
38.7 |
9 |
50 |
41 |
| 2 |
3.6 |
6.0 |
83 |
890 |
6.0 |
39.1 |
10 |
50 |
40 |
| 3 |
8.6 |
10.0 |
85 |
1240 |
4.6 |
28.8 |
12 |
43 |
45 |
| 4 |
7.1 |
7.5 |
60 |
2180 |
1.9 |
11.5 |
76 |
12 |
12 |
| 5 |
10.7 |
8.1 |
50 |
2040 |
1.8 |
10.3 |
72 |
17 |
11 |
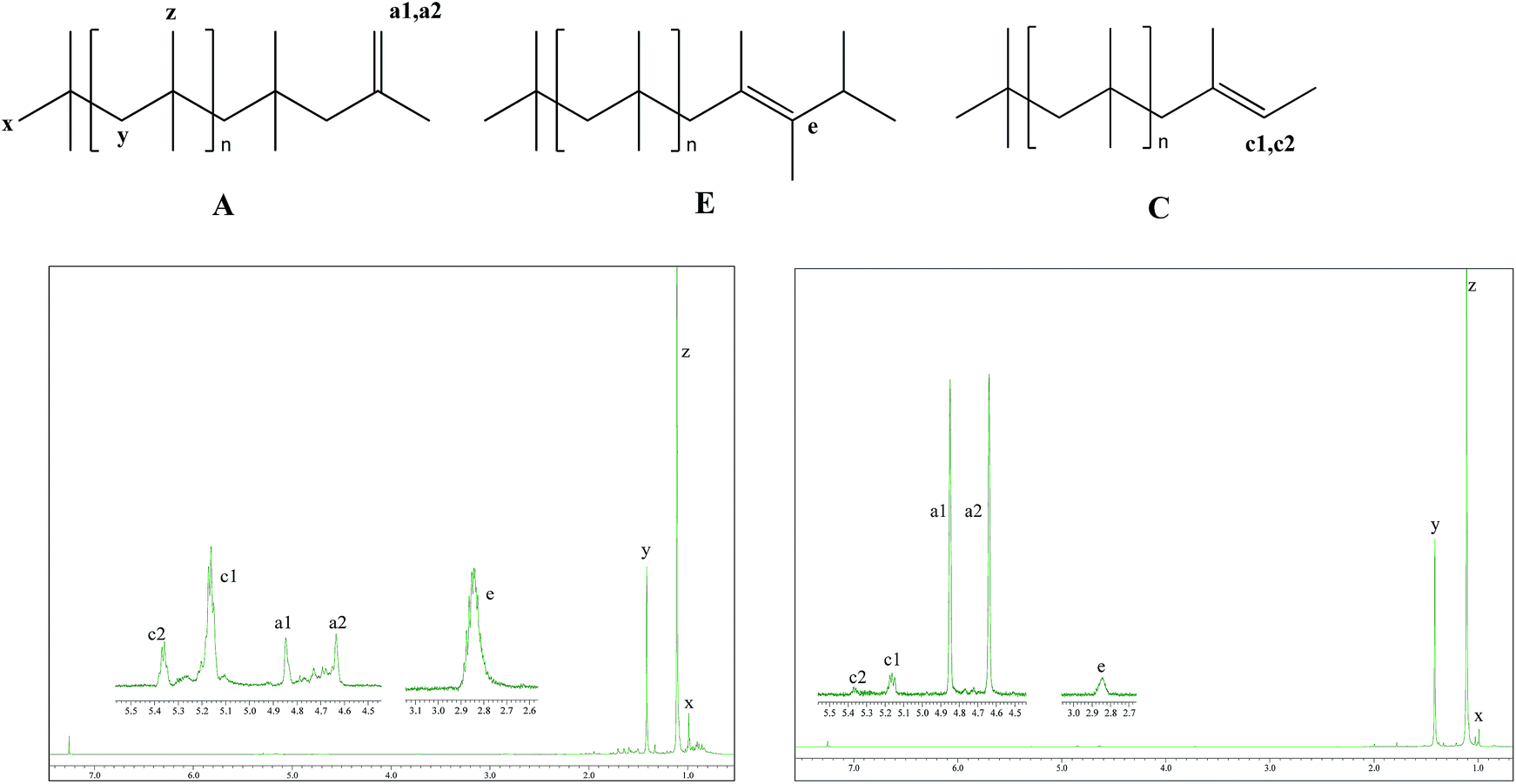
Fig. 2 presents the 1H NMR spectra of entry 1 and entry 4. Main resonance signals observed are located at δ = 1.1 (z), 1.41 (y), 0.99 (x), 4.85 (a1), 4.64 (a2), 5.17 (c1), 5.37 (c2), and 2.83 (e).43 Therein, the single peaks, z and y are assigned to –CH3 and –CH2 protons of the structural units along the main chain of PIB, respectively; the single peak, x is assigned to the protons in head groups of –C(CH3)3 in PIB chains; the single peaks, a1 and a2 in the expansion of olefin region, are assigned to the protons of exo-olefin end groups, –CH2–C(CH3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2; the quartet peaks, c1 and c2 are attributed to –C(CH3)CH(CH3) end groups in PIB chains; the single peak, e is assigned to the protons in –CH(CH3)2 end groups in PIB chains. Accordingly, most PIB chains generated in the experiments have structures A, C, or E, as presented in Fig. 2. Compared with entry 1, structure A takes the majority instead of structures C and E in entry 4. Besides, the absence of the signals at 1.68 and 1.96 ppm in both of entries indicates that the PIB chains were free of tert-Cl terminal groups and the termination via chlorine transfer from counter anion did not take place during polymerization.44
CH2; the quartet peaks, c1 and c2 are attributed to –C(CH3)CH(CH3) end groups in PIB chains; the single peak, e is assigned to the protons in –CH(CH3)2 end groups in PIB chains. Accordingly, most PIB chains generated in the experiments have structures A, C, or E, as presented in Fig. 2. Compared with entry 1, structure A takes the majority instead of structures C and E in entry 4. Besides, the absence of the signals at 1.68 and 1.96 ppm in both of entries indicates that the PIB chains were free of tert-Cl terminal groups and the termination via chlorine transfer from counter anion did not take place during polymerization.44
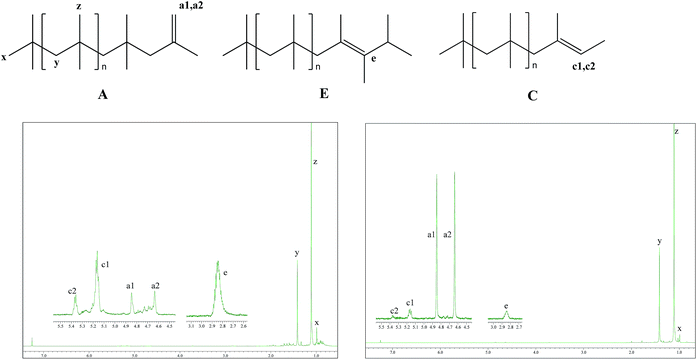 |
| | Fig. 2 1H NMR spectra of selected PIBs from Table 1. The left and the right correspond to entry 1 and entry 4, respectively. | |
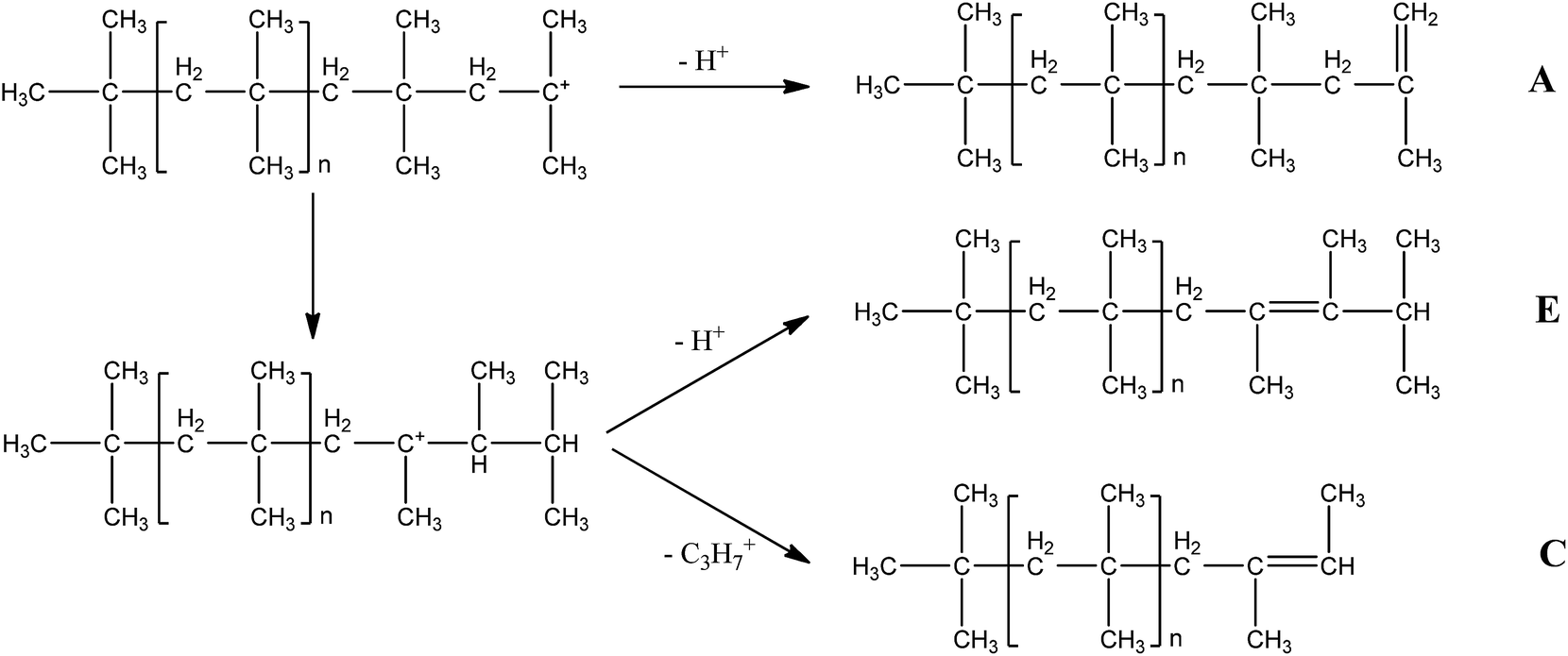
Fig. 3 shows possible shifts and β-proton abstraction processes causing the formation of various structures. The structures C and E are formed by isomerization from the normal growing tertiary carbocation via carbenium ion rearrangements by hydride and/or methide shifts and followed proton or isopropyl elimination. For the synthesis of conventional PIBs, extensive hydride and/or methide shifts take place before β-proton abstraction to create large amount of byproducts containing internal unsaturated bonds. On the contrary, the sufficient addition of iPr2O seems to inhibit these shifts or enhance the β-proton abstraction to obtain HRPIBs.9
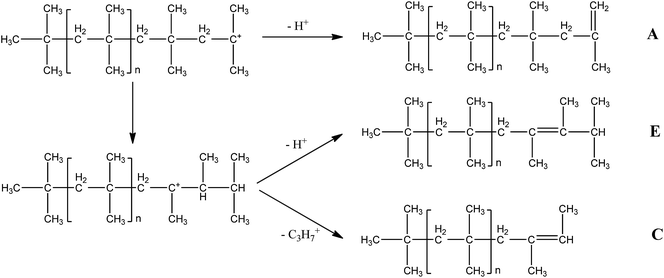 |
| | Fig. 3 Possible shifts and β-proton abstraction processes in the synthesis of HRPIBs. | |
Evolution of IB polymerization in flow system
In the microflow system, the reaction time can be easily adjusted by changing the length of reaction tube individually. Monitoring the variance of monomer conversion and product's molecular weight distribution with time can help understand the proceeding of the polymerization. Considering that the molar ratio of iPr2O to AlCl3 (iPr2O:AlCl3) shows distinct influence on the monomer conversion after specific duration in Table 1, two groups of experiments with different values of iPr2O:AlCl3 were conducted for comparison. Table 2 lists the results. For the first group (entries 1 to 4, iPr2O:AlCl3 = 1.1), we almost obtained consistent monomer conversion and HRPIB products, but the excess of PIB over AlCl3 (much higher than water) indicates chain transfer evidently. A reasonable explanation is that most of PIB chains have been terminated within 1 s, but the characteristic time for chain transfer is far less than 1 s. For the second group (entries 5 to 11, iPr2O:AlCl3 = 1.4), the monomer conversion, as well as the PIB concentration, increased gradually with time ranging from 0.5 s to 18 s; Mn and PDI changed little. Compared with the first group, both the chain propagation and the chain termination were retarded much in the second group at high iPr2O:AlCl3, and more times of chain transfer carried out before chain termination to achieve relatively high PIB concentration. A polymerization process dominated by fast but not instantaneous chain reactions is easy to control in a microflow system. Therefore, a proper excess of iPr2O over AlCl3 (such as iPr2O:AlCl3 = 1.4) is preferred to making a compromise between the controllability and efficiency of IB polymerization. Besides, high iPr2O:AlCl3 seems to afford high ratio of exo-olefin-terminated PIBs with low molecular weight, which can be interpreted by the ether-assisted chain transfer mechanism.45,46
Table 2 Time dependence of polymerization of IB co-initiated by AlCl3 × iPr2O in CH2Cl2a
| Entry |
T (s) |
Convnd (%) |
Mn (g mol−1) |
PDI |
[PIB]e (mmol L−1) |
End groups distribution (mol%) |
| Exo |
Tri + endo |
Tetra |
| T = 20 °C; [IB] = 0.75 mol L−1. [AlCl3] = 6.1 mmol L−1; iPr2O:AlCl3 = 1.1. [AlCl3] = 7.5 mmol L−1; iPr2O:AlCl3 = 1.4. Gravimetric conversion. [PIB] = [IP] × 56 × convn/Mn. |
| 1b |
1 |
56 |
2250 |
1.9 |
10.4 |
75 |
14 |
11 |
| 2b |
2 |
65 |
2230 |
2.0 |
12.2 |
75 |
13 |
12 |
| 3b |
4 |
59 |
1960 |
1.8 |
12.6 |
80 |
11 |
9 |
| 4b |
6 |
53 |
2110 |
1.9 |
10.5 |
83 |
10 |
7 |
| 5c |
0.5 |
16 |
1290 |
1.6 |
5.19 |
90 |
5 |
5 |
| 6c |
1 |
17 |
1240 |
1.6 |
5.74 |
90 |
8 |
2 |
| 7c |
2 |
24 |
1320 |
1.7 |
7.61 |
88 |
7 |
5 |
| 8c |
4 |
32 |
1270 |
1.6 |
10.6 |
90 |
6 |
4 |
| 9c |
6 |
42 |
1380 |
1.7 |
12.7 |
88 |
7 |
5 |
| 10c |
12 |
61 |
1290 |
1.7 |
19.8 |
89 |
6 |
5 |
| 11c |
18 |
63 |
1280 |
1.7 |
20.6 |
89 |
6 |
5 |
Effects of temperature
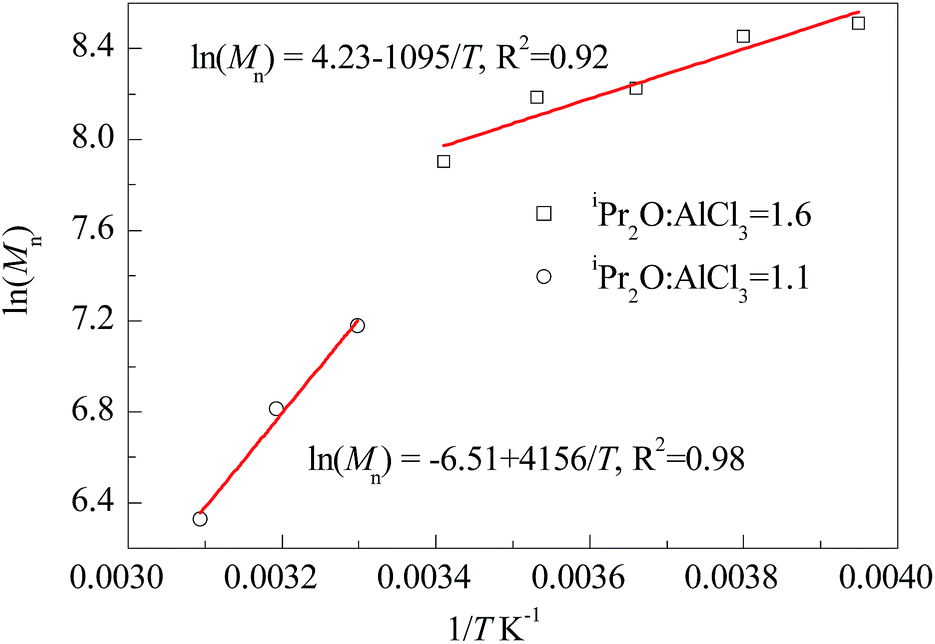
It is well known that the stability of carbenium ions is highly sensitive to temperature, and temperature is usually a critical and complicated influence factor on the carbonic polymerization. Meanwhile, for the AlCl3/iPr2O co-initiating system, the strength of such influences may be related to the amount of iPr2O addition. We carried out the polymerization of IB at various temperatures ranging from −20 °C to 50 °C and listed the results in Table 3. At a glance, HRPIBs could be successfully synthesized throughout the wide range of temperature; high temperature corresponds to low Mn and low content of exo-olefin terminated chains; the adjustment of Mn from 500 to 5000 is available. In details, for the entries 1 to 5 with iPr2O:AlCl3 = 1.6, even larger than 1.4, it can be speculated that there still exists considerable unterminated chains after a duration of 12 s. At −20 °C or −10 °C, nearly 100% monomers could convert into HRPIBs with relatively high Mn, which implies the chain propagation is still a fast step. Further increasing the temperature, the conversion decreases, but the PIB concentration increases gradually. It indicates chain transfer may be accelerated with the temperature increasing. For the entries 6 to 7 with iPr2O:AlCl3 = 1.1, a 12 s stay is enough for totally chain terminating. The PIB concentration is dependent on the chain transfer ability before chain termination. Obviously, the increasing of temperature could afford intensive chain transfer, which can compensate the effect of Mn decreasing to sustain the conversion. In general, the high temperature is favour for chain transfer, and the excessive iPr2O could weaken this temperature dependency. Besides, the inverse effect of polymerization temperature on molecular weights can be described by Arrhenius equation, i.e. lnMn = lnA − ΔE/RT. The overall activation energy (ΔE) could be calculated from the slope of the linear Arrhenius plot of lnMn vs. 1/T for specific co-initiating system and temperature intervals. Fig. 4 shows that the ΔE is strongly dependent on the molar ratio of iPr2O to AlCl3, corresponding to −9.1 kJ mol−1 at iPr2O:AlCl3 = 1.6 and −34.6 kJ mol−1 at iPr2O:AlCl3 = 1.1, respectively. The former is higher than what reported by Kostjuk47 (−19.8 kJ mol−1) and Wu (−13.9 kJ mol−1)22 for similar initiating systems. Considering the large presence of free ether in our system, it indicates the free ether could remarkably affect the chain transfer (β-proton abstraction) by weak interaction. Moreover, low temperature sensitivity is preferred to assuring an expected range of molecular weight at elevated temperature, while high temperature sensitivity afford convenience for molecular weight regulation by changing temperature.
Table 3 Polymerization of IB co-initiated by AlCl3 × iPr2O in CH2Cl2 at various temperatures in CH2Cl2a
| Entry |
T (°C) |
Convnd (%) |
Mn (g mol−1) |
PDI |
[PIB]e (mmol L−1) |
End groups distribution (mol%) |
| Exo |
Tri + endo |
Tetra |
| t = 12 s; [IB] = 0.75 mol L−1. [AlCl3] = 4.2 mmol L−1; iPr2O:AlCl3 = 1.6. [AlCl3] = 6.1 mmol L−1; iPr2O:AlCl3 = 1.1. Gravimetric conversion. [PIB] = [IP] × 56 × convn/Mn. |
| 1b |
−20 |
100 |
4980 |
2.4 |
8.41 |
95 |
2 |
3 |
| 2b |
−10 |
100 |
4690 |
2.2 |
8.93 |
93 |
3 |
4 |
| 3b |
0 |
88 |
3740 |
2.1 |
9.85 |
88 |
6 |
6 |
| 4b |
10 |
86 |
3580 |
2.1 |
10.1 |
76 |
13 |
11 |
| 5b |
20 |
73 |
2710 |
1.9 |
11.3 |
71 |
14 |
15 |
| 6c |
30 |
52 |
1310 |
1.7 |
16.6 |
85 |
8 |
7 |
| 7c |
40 |
67 |
910 |
1.8 |
30.8 |
77 |
12 |
11 |
| 8c |
50 |
61 |
560 |
1.7 |
45.6 |
74 |
12 |
14 |
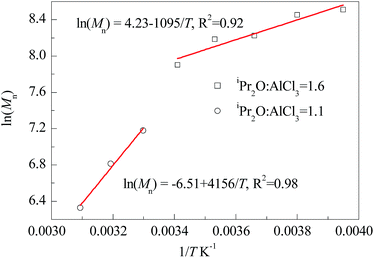 |
| | Fig. 4 Arrhenius plots of ln(Mn) versus 1/T for polymerization of IB co-initiated by AlCl3 × iPr2O. | |
Effects of monomer concentration
The productivity of polymerization process is limited by the monomer concentration, and the latter affects the supply of monomers for chain propagation as well. We attempted to increase the monomer concentration to improve the productivity under controlled molecular weight distribution. Table 4 shows some results. As seen, with the increasing of the monomer concentration, the Mn almost increases proportionally, regardless of temperature and the ratio of iPr2O to AlCl3. Correspondingly, other parameters, including conversion, the PIB concentration, and end groups distribution, change little. It seems that the monomer concentration has little effect on the rate of chain transfer, but determines the rate of chain propagation. Therefore, the molecular weight of HRPIBs may be quantitatively regulated by changing the monomer concentration individually. Meanwhile, considering the temperature difference between entries 1 to 3 and entries 4 to 6, the effects of temperature are mainly reflected on the variance of end group distribution as exploiting relatively high ratio of iPr2O to AlCl3 (such as 1.4 or more). As for achieving high content of exo-olefin terminated chains, low temperature is preferred. However, a proper selection of the ratio of iPr2O to AlCl3 (such as 1.4 for the second group in Table 2) seems available to synthesize PIBs with around 90 mol% of exo-olefin end groups and controllable molecular weight distribution at room temperature.
Table 4 Polymerization of IB co-initiated by AlCl3 × iPr2O at various IB concentration in CH2Cl2a
| Entry |
[IB] (mol L−1) |
Convnd (%) |
Mn (g mol−1) |
PDI |
[PIB]e (mmol L−1) |
End groups distribution (mol%) |
| Exo |
Tri + endo |
Tetra |
| t = 12 s. T = 20 °C; [AlCl3] = 4.2 mmol L−1; iPr2O:AlCl3 = 1.6. T = −20 °C; [AlCl3] = 4.5 mmol L−1; iPr2O:AlCl3 = 1.4. [AlCl3] = 6.1 mmol L−1. Gravimetric conversion. [PIB] = [IP] × 56 × convn/Mn. |
| 1b |
0.75 |
73 |
2710 |
1.9 |
11.3 |
71 |
15 |
14 |
| 2b |
1.50 |
52 |
4650 |
1.8 |
9.4 |
72 |
11 |
17 |
| 3b |
2.24 |
60 |
7550 |
1.9 |
10.0 |
66 |
16 |
18 |
| 4c |
0.75 |
56 |
3480 |
2.1 |
6.8 |
94 |
3 |
3 |
| 5c |
1.50 |
54 |
6790 |
1.9 |
6.7 |
95 |
2 |
3 |
| 6c |
2.24 |
52 |
10640 |
1.9 |
6.1 |
96 |
3 |
1 |
Roles of iPr2O in IB polymerization process control
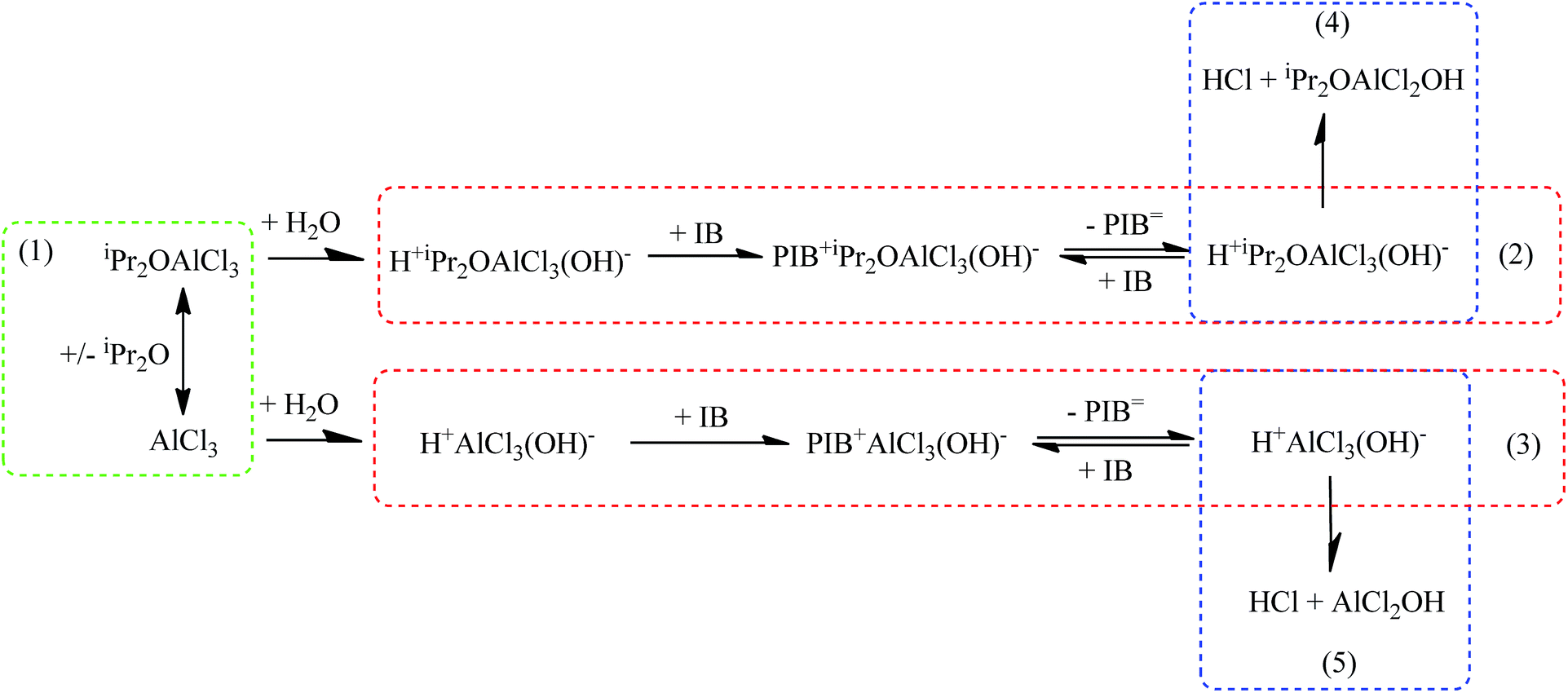
As mentioned above, the addition of iPr2O has multiple effects on the polymerization process of IB due to its complex reaction mechanism, as schematically illustrated in Fig. 5. In the catalytic complex solution, the iPr2O can combine with AlCl3 to generate the catalytic complex, iPr2OAlCl3 (eqn (1)). Both iPr2OAlCl3 and AlCl3 can combine with adventitious water to generate active species initiating chain propagations (eqn (2) and (3)). The molecular weight of chains is dependent on the ratio of chain propagation probability to chain transfer probability, and increasing the monomer concentration is favour for chain propagation (forward reaction in eqn (2)) and molecular weight increment. Compared with the polymer chains initiated by H+AlCl3(OH)−, the polymer chains initiated by H+iPr2OAlCl3(OH)− have low propagation rate and high chain transfer proneness due to the insertion of iPr2O in active species. The fast ether-assisted chain transfer reduces the probability of isomerization resulted from hydride or methide shifts in propagating chains. Intensive isomerization corresponding to a little deficiency of iPr2O implies that most of chains are initiated by H+AlCl3(OH)− with initiation efficiency higher than H+iPr2OAlCl3(OH)−. So an excess of iPr2O to guarantee full conversion from AlCl3 to the catalytic complex is necessary to synthesize HRPIBs. The collapse of active species according to eqn (4) and (5) may take place and result in chain termination, which can be inhibited by the existence of free iPr2O around counterions. However, abundant free iPr2O may retard propagation seriously, decreasing the conversion and allowing more isomerization. A selection of the mole ratio of iPr2O to AlCl3 should be based on the overall consideration of decreasing free AlCl3 as well as avoiding abundant free iPr2O. In general, controlling the iPr2O addition at low amount is prerequisite for achieving fast HRPIBs synthesis.
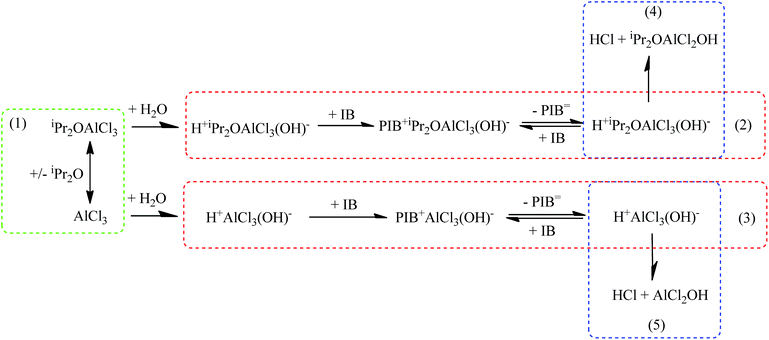 |
| | Fig. 5 Suggested mechanism of polymerization of IB co-initiated by AlCl3 × iPr2O. | |
Considering the monomer conversion follows the supply of monomers towards the propagating chain and the chain termination is independent of monomer supply, enhancing the mixing between monomer solution and catalytic complex solution can help increase the molecular weight or conversion. The comparisons of experiments conducted under various mixing conditions (Table 5) present the importance of high efficiency mixing for fast HRPIBs synthesis. A microflow system is easy to control the mixing performance, which usually approaches a platform when the flow capacity is over a threshold.
Table 5 Effects of mixing conditions on polymerization of IB co-initiated by AlCl3 × iPr2O in CH2Cl2a
| Entry |
F(IB + CH2Cl2 + catalytic complex)b (mL min−1) |
Convnc (%) |
Mn (g mol−1) |
PDI |
[PIB]d (mmol L−1) |
End groups distribution (mol%) |
| Exo |
Tri + endo |
Tetra |
| t = 12 s; T = −20 °C; [AlCl3] = 4.5 mmol L−1; iPr2O:AlCl3 = 1.4. F(IB):F(CH2Cl2):F(initiator) = 1:7:8. Gravimetric conversion. [PIB] = [IP] × 56 × convn/Mn. |
| 1 |
3.2 |
30 |
2120 |
1.8 |
5.9 |
95 |
2 |
3 |
| 2 |
8 |
52 |
3580 |
2.4 |
6.1 |
93 |
4 |
3 |
| 3 |
16 |
56 |
3480 |
2.1 |
6.8 |
94 |
3 |
3 |
Conclusions
Through IB polymerization co-initiated by AlCl3/iPr2O at low concentrations (4 mmol L−1 to 10 mmol L−1) in a microflow system, we achieved 50% to 100% conversion towards HRPIBs with 65% to 96% exo-olefin terminated chains after 12 s or less duration within a wide temperature window from −20 °C to 50 °C. The molecular weight distribution was narrow with a PDI around 1.9, and the average molecular weight was adjustable from 500 to 15000 by changing temperature or monomer concentration. The excess of iPr2O over AlCl3 is necessary to decrease the existence of free AlCl3 and intensive isomerization co-initiated by free AlCl3. The considerable existence of free iPr2O can inhibit the chain termination and allow the polymerization to present chain transfer dominated kinetics. The increasing of free iPr2O can weaken the temperature sensitivity of molecular weight and avoid the failure in molecular weight control at elevated temperature. The retarding effects of free iPr2O on the chain propagation will decrease the apparent reaction rate and increase the probability of isomerization. A key access to fast synthesis of HRPIBs with 90% exo-olefin terminated chains is to make a compromise between multiple effects of the excessive iPr2O by selecting a proper ratio of iPr2O to AlCl3, such as 1.4. This work provides useful information in aspect of fundamentals and methodology to produce high quality HRPIBs expeditiously and cost-effectively.
Acknowledgements
The authors are gratefully thankful for the support of the National Natural Science Foundation of China (21176136, 21422603) and National Science and Technology Support Program of China (2011BAC06B01) on this work.
References
- J. P. Kennedy and B. Ivan, Designed Polymers by Carbocationic Macromolecular Engineering: Theory and Practice, Hanser, Munich, Germany, 1991, pp. 173–177 Search PubMed.
- J. P. Kennedy and E. Marechal, Carbocationic Polymerization, John Wiley & Sons, New York, 1982, pp. 475–488 Search PubMed.
- S. F. Rach and F. E. Kühn, Sustainability, 2009, 1, 35–42 CrossRef CAS.
- J. D. Burrington, J. R. Johnson and J. K. Pudelski, Top. Catal., 2003, 23, 175–181 CrossRef CAS.
- I. Puskas, E. M. Banas and G. Nerheim, J. Polym. Sci., Polym. Symp., 1976, 56, 191–201 CrossRef CAS.
- J. J. Harrison, D. C. Young and C. L. Mayne, J. Org. Chem., 1997, 62, 693–699 CrossRef CAS PubMed.
- W. Günther, K. Maenz and D. Stadermann, Angew. Makromol. Chem., 1996, 234, 71–90 CrossRef.
- E. N. Kresge, R. H. Schatz and H. C. Wang, Encyclopedia of Polymer Science and Engineering, Wiley, New York, 1985, pp. 423–450 Search PubMed.
- P. Dimitrov, J. Emert, J. Hua, S. Keki and R. Faust, Macromolecules, 2011, 44, 1831–1840 CrossRef CAS.
- P. Boerzel, K. Bronstert and F. Hovemann, BASF AG, DE 2702604, 1978.
- H. P. Rath, US 5,910,550, BASF AG, 1997, Chem. Abstr., 1997, 128, 23292 Search PubMed.
- J. M. Kerr, J. Mcmahon and J. M. Scotland, EP 0671419, BP Chemicals Ltd., 1995, Chem. Abstr., 1995, 123, 314902 Search PubMed.
- H. Mach and P. Rath, Lubr. Sci., 1999, 11, 175–185 CrossRef CAS.
- J. J. Harrison, C. M. Mijares and J. Hudson, Macromolecules, 2002, 35, 2494–2500 CrossRef CAS.
- H. P. Rath, BASF AG, US 5,286,823, 1994.
- H. P. Rath, A. Lange and H. Mach, BASF AG, US 7,071,275, 2006.
- H. P. Rath, BASF AG, WO 99/64482, 1999.
- M. Vierle, Y. Zhang, E. Herdtweck, M. bohnenpoll, O. Nuyken and F. E. Kuhn, Angew. Chem., Int. Ed., 2003, 42, 1307–1310 CrossRef CAS PubMed.
- Y. Li, M. Cokoja and F. E. Kuehn, Coord. Chem. Rev., 2011, 13–14, 1541–1557 CrossRef.
- S. V. Kostjuk, RSC Adv., 2015, 5, 13125–13144 RSC.
- I. V. Vasilenko, A. N. Frolov and S. V. Kostjuk, Macromolecules, 2010, 43, 5503–5507 CrossRef CAS.
- Q. Liu, Y. X. Wu, Y. Zhang, P. f. Yang and R. W. Xu, Polymer, 2010, 51, 5960–5969 CrossRef CAS.
- S. V. Kostjuk, H. Y. Yeong and V. Brigitte, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 471–486 CrossRef CAS.
- R. F. Storey, B. J. Chisholm and L. B. Brister, Macromolecules, 1995, 28, 4055–4061 CrossRef CAS.
- R. Kumar, P. Dimitrov, K. J. Bartelson, J. Emert and R. Faust, Macromolecules, 2012, 45, 8598–8603 CrossRef CAS.
- J. P. Russum, C. W. Jones and F. J. Schork, Macromol. Rapid Commun., 2004, 25, 1064–1068 CrossRef CAS.
- R. Kumar, B. Zheng, K. W. Huang, J. Emert and R. Faust, Macromolecules, 2014, 47, 1959–1965 CrossRef CAS.
- B. Yang and R. F. Storey, Polym. Chem., 2015, 6, 3764–3774 RSC.
- X. Yang, A. Guo, H. Xu and Y. X. Wu, J. Appl. Polym. Sci., 2015, 132, 42232 Search PubMed.
- N. Hand, R. T. Mathers, K. Damodaran and S. P. Lewis, Ind. Eng. Chem. Res., 2014, 53, 2718–2725 CrossRef CAS.
- I. V. Vasilenko, D. I. Shiman and S. V. Kostjuk, Polym. Chem., 2014, 5, 3855–3866 RSC.
- R. Kumar, J. Emert and R. Faust, Polym. Bull., 2015, 72, 49–60 CrossRef CAS.
- R. Kumar, P. De, B. Zheng, K. W. Huang, J. Emert and R. Faust, Polym. Chem., 2014, 6, 322–329 RSC.
- K. Bartelson, R. Kumar, P. De, J. Emert and R. Faust, Polymer, 2013, 54, 4858–4863 CrossRef CAS.
- S. Banerjee, J. Emert, P. Wright, T. Skourlis, R. Severt and R. Faust, Polym. Chem., 2015, 6, 4902–4910 RSC.
- S. Banerjee, B. H. Jha, P. De, J. Emert and R. Faust, Macromolecules, 2015, 48, 5474–5480 CrossRef CAS.
- P. Dimitrov, J. Emert and R. Faust, Macromolecules, 2012, 45, 3318–3325 CrossRef CAS.
- N. K. Madhvanand and K. M. Lioubov, Ind. Eng. Chem. Res., 2009, 48, 6465–6485 CrossRef.
- J. L. Steinbacher and D. T. McQuade, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6505–6533 CrossRef CAS.
- A. Miyazawa, T. Kase and T. J. Shibuya, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1841–1844 CrossRef CAS.
- A. Nagaki, K. Kawamura and J. Yoshida, J. Am. Chem. Soc., 2004, 126, 14702–14703 CrossRef CAS PubMed.
- T. Iwasaki, A. Nagaki and J. Yoshida, Chem. Commun., 2007, 1263–1265 RSC.
- I. Puskas, E. M. BanasX, G. J. Nerheim and G. J. Ray, J. Polym. Sci., Polym. Symp., 1976, 56, 191–201 CrossRef CAS.
- K. L. Simison, C. D. Stokes, J. J. Harrison and R. F. Storey, Macromolecules, 2006, 39, 2481–2487 CrossRef CAS.
- D. I. Shiman, V. V. Irina and S. V. Kostjuk, Polymer, 2013, 54, 2235–2242 CrossRef CAS.
- S. V. Kostjuk, I. V. Vasilenko, D. I. Shiman, A. N. Frolov and L. V. Gaponik, Macromol. Symp., 2015, 349, 94–103 CrossRef CAS.
- I. V. Vasilenko, D. I. Shiman and S. V. Kostjuk, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 750–758 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.