
N-(7-Chloroquinolinyl-4-aminoalkyl)arylsulfonamides as antimalarial agents: rationale for the activity with reference to inhibition of hemozoin formation†
Saroj Vermaa,
Shashi Pandeya,
Pooja Agarwalb,
Pravesh Vermac,
Shreekant Deshpandea,
Jitendra Kumar Saxenac,
Kumkum Srivastavab,
Prem M. S. Chauhana and
Yenamandra S. Prabhakar*a
aMedicinal and Process Chemistry Division, Academy of Scientific and Innovative Research, CSIR-Central Drug Research Institute, Jankipuram Extension, Lucknow-226 031, India. E-mail: yenpra@yahoo.com; Fax: +91-522-2771941; Tel: +91-522-2772450
bParasitology Division, Academy of Scientific and Innovative Research, CSIR-Central Drug Research Institute, Jankipuram Extension, Lucknow-226 031, India
cBiochemistry Division, Academy of Scientific and Innovative Research, CSIR-Central Drug Research Institute, Jankipuram Extension, Lucknow-226 031, India
First published on 26th February 2016
Abstract
New N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides were synthesized and tested in vitro against P. falciparum 3D7 and K1 strains and hemozoin formation. Compounds 16 and 17 showed promising antimalarial activity (IC50 3D7: 0.05 and 0.01 μM; K1: 0.41 and 0.36 μM) and exceedingly well inhibited hemozoin formation (IC50, 3.23 and 2.40 μM). Considering hemozoin inhibition was due to the affinity of the compounds towards heme, its μ-oxo dimer and/or heme detoxification protein (HDP), they were used in docking experiments for probing intermolecular interactions with the compounds. The docked structures indicated that quinoline, 4-amino and sulfonamide moieties interacted with one or more key constituents of the targets. In heme and μ-oxo dimer, the key constituents for interactions were ferriprotoporphyrin and propionic acid side chains. In the case of HDP, the key constituents were His175 and Glu126, and rationalized the antimalarial activity and functional use of the sulfonamide moiety. Thus, these compounds opened an avenue for exploration of hemozoin inhibition in malaria chemotherapy.
1. Introduction
Malaria is one of the most widespread parasitic infections in the world affecting nearly 300 million people annually.1 In the symptomatic stage, the parasite makes its presence visible in the host’s erythrocytes and thrives on haemoglobin.2 In this stage while nourishing on hemoglobin, the parasite discards the nonproteinaceous constituent heme, a toxic substance to its milieu, by transforming it into a non-toxic insoluble crystalline polymer form called hemozoin or β-hematin.3 In the infected erythrocytes, the biochemical factors controlling the conversion of heme to hemozoin are not fully understood.3 However, in normal situations, depending upon pH and ionic strength of the biological milieu, heme exists in monomer, head-to-tail dimer, μ-oxo-dimeric and/or in non-crystalline aggregate form.3 All the same, the scenario suggests that inhibition of heme to hemozoin formation (Fig. 1) is a worthwhile handle for exploring the chemotherapeutic agents of malaria.3,4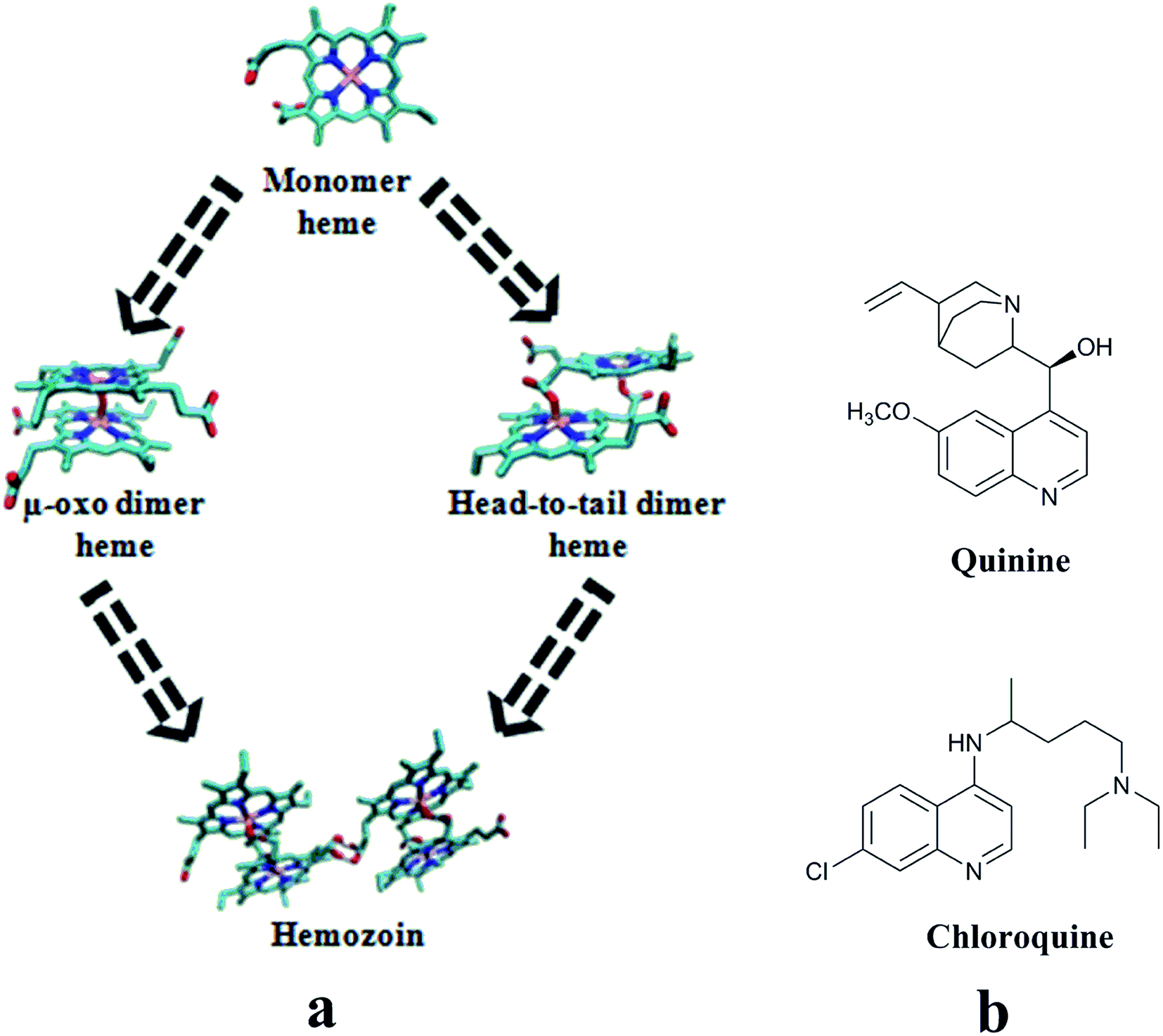 | ||
| Fig. 1 (a) Schematic representation of heme to hemozoin formation path; (b) compounds well-known for heme binding and interfering with the hemozoin formation. | ||
Among the antimalarial agents, chloroquine (CQ) is well-known for forming a complex with heme in its monomer and μ-oxo dimer forms.5–8 Also, it is one of the most commonly used drugs for the clinical management of malaria.9 Amid the possibilities, the most appealing basis for the drug action of CQ (and its analogues) include the accumulation of the compound in the food vacuole of the parasite and its role in blocking/inhibition of heme to hemozoin formation in the milieu.2,4,9 In light of this, various groups investigated the physiological conditions of the milieu for accumulation of the drug in the food vacuole and different proteins/enzymes, phospholipids and neutral lipids of the parasite involved in hemozoin formation.10–19 The findings of Asher and co-workers suggested that certain physicochemical conditions i.e., high pH, physiological temperature, high salt concentration are necessary for the formation of pi-stacked aggregates of μ-oxo dimers of heme.10 Furthermore, the experiments of Dorn and co-workers have led to suggest that the heme to hemozoin formation is more a physicochemical controlled process than an enzymatic one.11 Slater et al.,12 Bendrat et al.13 and Egan et al.14 have investigated the role of phospholipids and heme-polymerase in hemozoin formation. Studies by Slater et al. with heme polymerase from P. falciparum trophozoites extracts showed that the enzyme converts heme to hemozoin in acidic medium, and quinoline derivatives (chloroquine and quinine) target this enzyme.12 Jani et al.15 have studied the involvement of heme detoxification protein (HDP) in hemozoin formation. Reports also exist for the involvement of histidine-rich proteins (Pf HRP I, II and III) alone as well as together with lipids in triggering hemozoin formation.16,17 The studies of Chugh et al. showed that falcipain 2 and HDP together process the conversion of heme to hemozoin. Interestingly, HDP has been seen to play a major role in hemozoin formation with 1000-fold more efficiency than lipids. In this they suggested CQ interferes in the process.18 Furthermore, Nakatani and co-workers highlighted the importance of histidine residues of HDP in anchoring the heme in the hemozoin formation.19 These studies clearly showed that CQ interferes in the heme to hemozoin formation in more than one way. It is quite likely that different CQ prototypes might be responding with different affinities in the explored mechanisms leading to varied drug response end points. This encouraged us to search for new chemical entities for the inhibition of hemozoin formation.
It is common practice in medicinal chemistry to hold-on to a known or established scaffold and functionally modify it with different features to result in new chemical entities for exploration. Compounds entrenched with the sulfonamide moiety are known to be endowed with a wide spectrum of pharmacological activities.20–22 In view of this, several researchers have appended this feature to the chloroquinoline scaffold to derive new antimalarial agents. Ryckebusch et al. reported N-(3-(4-((7-chloroquinolin-4-ylamino)methyl)piperazin-1-yl)propyl)sulfonamide derivatives with noteworthy antimalarial activity.23 A different set of 4-amino-7-chloroquinolyl sulfonamides with varying 4-amino side chains as well as sulfonamide substituents showing significant antimalarial activity was reported by Ekoue-Kovi and co-workers.24 Salahuddin et al. synthesized some 7-chloro-4-(piperazin-1-yl)quinoline sulfonamide analogs and evaluated them against β-hematin formation and P. falciparum dihydropteroate synthase.25 Keeping in line with our explorations for diverse antimalarial agents,26,27 we synthesized new and much simpler N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides and tested them against CQ-sensitive/-resistant P. falciparum strains and for the inhibition of hemozoin formation. Simultaneously, the possible molecular basis for the mode of action of newly synthesized derivatives was explored in silico using the molecular modeling and docking studies with the monomer and μ-oxo dimer of heme. Furthermore the in silico study is extended to the role of HDP in hemozoin formation vis-à-vis the newly synthesized compounds. In the absence of an X-ray crystal structure of HDP, its homology model was developed and further refined using molecular dynamics simulations to use it as a biological target to understand the possible binding modes/residues in the inhibition of hemozoin formation. The possible binding, if any, of N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides with HDP was also investigated.
2. Materials and methods
2.1. General synthetic procedure of N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides
The target compounds, N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides (4–19) were prepared in a two-step reaction using the earlier reported synthetic procedure (Scheme 1).23 In the first step N-1-(7-chloroquinolin-4-yl)alkanamine (2/3) was prepared from 4,7-dichloroquinoline (1) and appropriate alkyldiamine in a nucleophilic aromatic substitution reaction. In a round bottom flask, a mixture of 4,7-dichloroquinoline (1 equiv.) and 1,2-diaminoethane (or 1,3-diaminopropane) (5 equiv.) was slowly heated with stirring for 1 h from room temperature to 80 °C. The heating of the mixture was continued with constant stirring at raised temperature (120–130 °C) for 6–8 h to complete the reaction. The progress of reaction was monitored through TLC (10% MeOH/CHCl3). On completion of reaction, the mixture was cooled to room temperature and poured into ice-cold water to precipitate the product. It was filtered off and washed with ethyl acetate to furnish the desired compound (2/3) in quantitative yield (70–85%).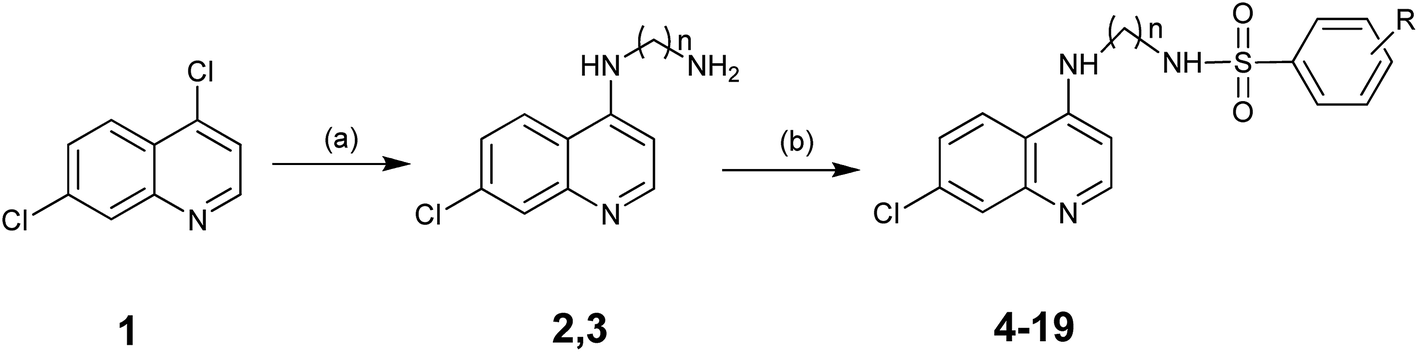 | ||
| Scheme 1 Reagents and conditions: (a) ethane-1,2-diamine/propane-1,3-diamine, 120–130 °C, neat reaction; (b) R-aryl sulfonyl chloride, DIEA, DCM, room temperature. | ||
In the second step, 3 equiv. of N,N-diisopropylethylamine (DIEA) and 1.1 equiv. of desired aryl sulfonyl chloride were added with constant stirring to compound 2 (or 3) (1 equiv.) dissolved in dichloromethane (DCM). The stirring of the mixture was continued for 6–8 h, while monitoring the completion through TLC (10% MeOH/CHCl3), to afford N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides (4–19). On completion of reaction, the mixture was washed with brine, and dried with anhydrous sodium sulfate followed by evaporation of dichloromethane under reduced pressure to obtain crude N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides (4–19) (yield 60–75%). The crude compounds were further purified through column chromatography. The structures of the final compounds were characterized by analyzing their ESI-MS, 1H NMR, 13C NMR and HRMS spectra.
2.2. Biological screening
The culture medium (50 μL) was dispensed in a 96 well plate followed by 50 μL of highest concentration (in subsequent screens with two-fold serial dilutions) of test compounds (in duplicate wells) which were added in row B. Following this, 50 μL of 2.0% parasitized cell suspension containing 0.8% parasitaemia (asynchronous culture containing more than 80% ring stages) was added to each well except 4 wells in row ‘A’ which received matching volume of non-parasitized erythrocyte suspension. The plates were incubated at 37 °C in CO2 incubator in an atmosphere of air mixed with 5% CO2 for 72 h. Following this, 100 μL of lysis buffer containing 2× concentration of SYBR Green-I (Invitrogen) was added to each well, and the plates were incubated for 60 min at 37 °C. The plates were examined at 485 ± 20 nm (excitation) and 530 ± 20 nm (emission) for relative fluorescence intensities (RFUs) per well using the fluorescence plate reader (FLX800, BIOTEK). The IC50 values were determined through Logit regression analysis of dose response curves using a pre-programmed Excel spreadsheet.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 30 °C. The pellet was suspended in 100 mM Tris–HCl buffer (pH 7.4) containing 2.5% sodium dodecyl sulfate (SDS). The pellet obtained after centrifugation was washed three times with triple distilled water (TDW) to remove free heme attached to β-hematin. Following this the pellet was solubilized in 50 μL of 2 M NaOH and the volume was made up to 1.0 mL with TDW and the absorbance was measured at 400 nm. The 50% inhibitory concentration (IC50) values of the compounds were determined from dose response curves using non-linear regression analysis.
000 rpm for 10 min at 30 °C. The pellet was suspended in 100 mM Tris–HCl buffer (pH 7.4) containing 2.5% sodium dodecyl sulfate (SDS). The pellet obtained after centrifugation was washed three times with triple distilled water (TDW) to remove free heme attached to β-hematin. Following this the pellet was solubilized in 50 μL of 2 M NaOH and the volume was made up to 1.0 mL with TDW and the absorbance was measured at 400 nm. The 50% inhibitory concentration (IC50) values of the compounds were determined from dose response curves using non-linear regression analysis.2.3. In silico studies
000 minimization steps) followed by equilibration for enough length of time (5000000 steps, equivalent to 10 ns). The stability of the protein during the course of simulation was assessed using the root mean square deviation (RMSD) between the initial and successive MD trajectories of the protein generated in the study. After MD simulations, the protein quality was reassessed by PROCHECK, ERRAT plot and ProSA, etc.3. Results and discussion
3.1. Synthesis and biological evaluation
Incorporating the privileged features of both CQ and sulfonamide into a single molecular system, new N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides (4–19) (Table 1) were synthesized as potential antimalarial agents. While CQ has inherent antimalarial activity, the sulfonamide moiety is thought to help through enhancing the interactions of these compounds with the intended targets. All the synthesized compounds (Table 1) were tested for the antimalarial activity against 3D7 (CQ-sensitive) and K1 (CQ-resistant) strains of P. falciparum, inhibition of β-hematin formation and cytotoxicity in the Vero cell line. Several of these analogues showed moderate to good antimalarial activity against CQ-sensitive strains. Some of the compounds also showed moderate activity against CQ-resistant strain of P. falciparum. Among the analogues, compound 17 showed antimalarial activity (IC50: 3D7, 0.01 μM; K1, 0.36 μM) close to CQ (IC50: 3D7, 0.01 μM; K1, 0.26 μM). Apart from this, compounds 12, 16 and 19 showed IC50 values 0.04, 0.05 and 0.07 μM, respectively, against the sensitive strain. In case of the resistant strain, these compounds showed IC50 values between 0.38 and 0.65 μM. For the remaining compounds the IC50 values against the sensitive strain are of the order of 1.00 μM or more with hardly any activity against the resistant strain (Table 1). In β-hematin formation inhibition assay all compounds acted in a dose dependent manner (Fig. 2). Interestingly they inhibited β-hematin formation better than that of CQ (IC50, 11.41 μM) (Table 1) with compound 17 showing the best activity (IC50, 2.40 μM). In spite of these compounds responding better than CQ in the β-hematin inhibition assay, most of them, except 16 and 17, showed far lower antimalarial activity when compared to that of CQ. The antimalarial activity observed in CQ and derivatives is known be a cumulative effect from different modes of action.3,12,18 From the β-hematin inhibition assay it appears that the present compounds have fared well here, but have fallen short in some other counts which are equally important for the end response. While this demands further investigations, all the same, the compounds open a new avenue due to their role in the inhibition of β-hematin. In the Vero cell line, none of the compounds showed cytotoxicity within the prescribed test concentration limits (ten-times the IC50 values) (Table 1).| Compd. | Substitution | IC50a/μM | CC50b/μM | β-Hematin inhibitory activity IC50/μM | Docking score | ||||
|---|---|---|---|---|---|---|---|---|---|
| R | n | 3D7 | K1 | Monomer | μ-Oxo dimer | HDP | |||
| a 3D7, chloroquine sensitive strain; K1, chloroquine resistant strain.b Vero cell line. | |||||||||
| 4 | H | 3 | 0.10 | >1.00 | 21.76 | 5.11 | 2.81 | 4.90 | 4.74 |
| 5 | H | 2 | 0.36 | >1.00 | 76.52 | 4.70 | 2.69 | 4.22 | 3.72 |
| 6 | p-F | 3 | 0.11 | >1.00 | 20.54 | 4.62 | 1.92 | 5.08 | 5.14 |
| 7 | p-F | 2 | 0.21 | 0.90 | 23.14 | 4.61 | 2.71 | 5.27 | 3.67 |
| 8 | p-Br | 3 | >1.00 | >1.00 | 25.53 | 6.82 | 4.76 | 5.19 | 5.02 |
| 9 | p-Br | 2 | >1.00 | >1.00 | 126.41 | 4.22 | 2.89 | 5.57 | 4.69 |
| 10 | p-NO2 | 3 | 0.40 | >1.00 | 70.53 | 7.60 | 2.08 | 5.33 | 4.13 |
| 11 | p-NO2 | 2 | >1.00 | >1.00 | 333.33 | 6.61 | 2.02 | 6.02 | 3.60 |
| 12 | p-CH3 | 3 | 0.04 | 0.38 | 4.29 | 5.21 | 2.62 | 4.81 | 4.21 |
| 13 | p-CH3 | 2 | 0.50 | >1.00 | 66.67 | 10.48 | 2.25 | 4.55 | 4.53 |
| 14 | p-t-Bu | 3 | 0.27 | 0.99 | 25.77 | 4.95 | 3.29 | 4.12 | 5.36 |
| 15 | p-t-Bu | 2 | >1.00 | >1.00 | 71.44 | 3.92 | 2.39 | 4.75 | 4.46 |
| 16 | 2,4,6-i-Pr3 | 3 | 0.05 | 0.41 | 6.92 | 3.23 | 2.62 | 6.18 | 5.20 |
| 17 | 2,4,6-i-Pr3 | 2 | 0.01 | 0.36 | 8.58 | 2.40 | 3.00 | 4.22 | 5.94 |
| 18 | p-Ph | 3 | 0.31 | >1.00 | 26.88 | 5.35 | 4.66 | 5.05 | 3.18 |
| 19 | p-Ph | 2 | 0.07 | 0.65 | 82.47 | 5.39 | 2.42 | 6.28 | 3.42 |
| CQ | — | — | 0.01 | 0.26 | 120.00 | 11.41 | 2.79 | 5.27 | 4.03 |
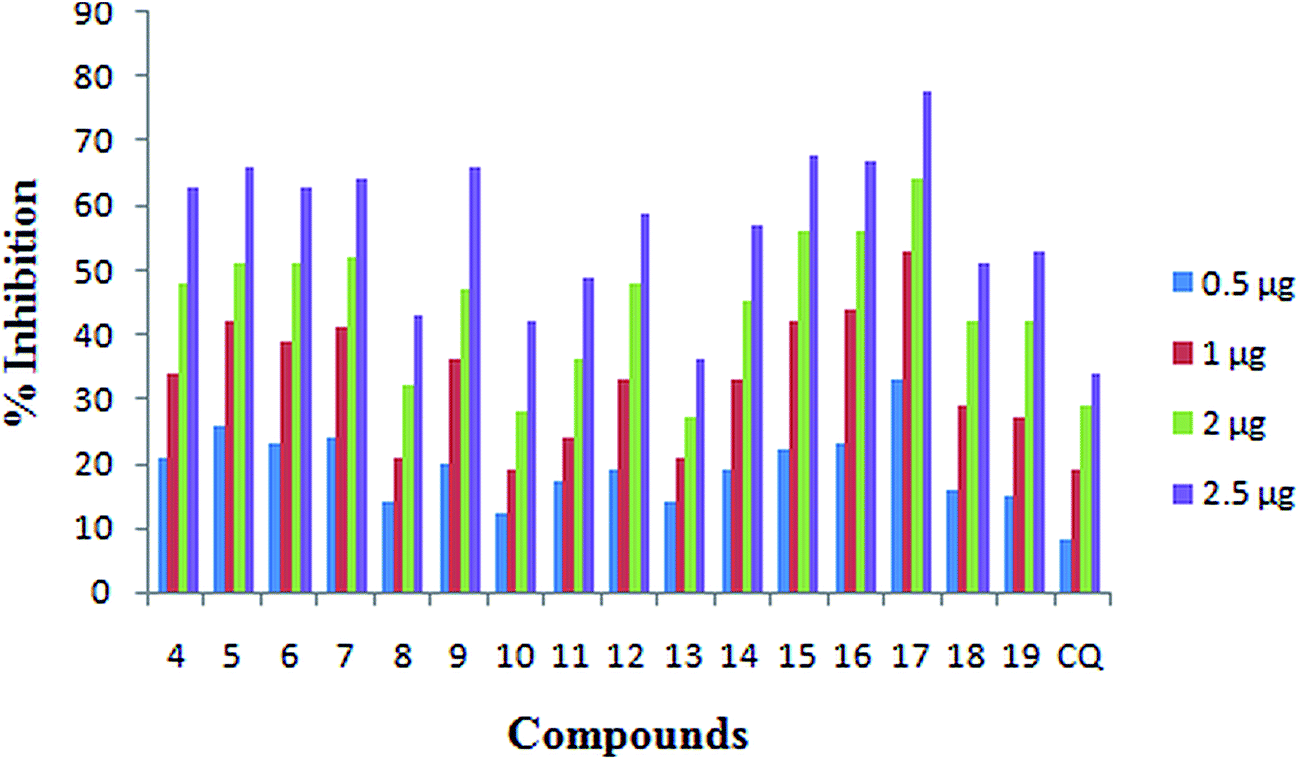 | ||
| Fig. 2 Dose response graph of N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides for β-hematin inhibitory activity. | ||
3.2. Structure–activity relationship
In CQ (and CQ analogues) the presence of a distal nitrogen and its disposition from the 4-amino moiety are crucial for the antimalarial activity. Our earlier studies also indicate that the distal nitrogen (or a functional moiety capable of playing its role) and substitutions beyond it hold scope for modulating the antimalarial activity.46 In the present compounds (Table 1) the C2-/C3-linkers extending from the 4-amino moiety acted like arms to place the sulfonamide moiety in a region corresponding to the distal nitrogen of CQ. Compounds with both C2 and C3 linkers showed average to good activity against the CQ sensitive strain (Table 1). However, in the case of the CQ resistant strain, only some compounds showed activity (Table 1). In view of the limited number of compounds with well-defined activity profile, the structure–activity inferences of the physicochemical properties were deduced by comparing the substituent characteristics vis-à-vis the observed activities.47The activity profiles of the compounds (Table 1) suggest that both the length of spacer between 4-amino and distal group, as well as the characteristics (steric/hydrophobic properties) of the substitution beyond the distal group, are important for the activity. A comparison of compounds 6 and 7 with those of 4 and 5 indicate that the fluoro group of the former compounds is comparable to the hydrogen (of unsubstituted phenyl) in terms of the steric character (e.g., molar refractivity of H is 1.03 and that of F is 0.92). Also, the σ value (Hammett constant) of fluoro is very small (σ = 0.06). The comparable activity profiles of these four compounds hint that the substitutions on the aryl moiety (attached to the distal group) may confer steric/hydrophobic aspects with little or minimal electronic characteristics for activity. Except for compound 10, the antimalarial activities of all other analogues are in agreement with the aforesaid observation. Furthermore, the length of spacer and steric bulk of the substitution beyond the distal group are in concomitantly inverse relationship with respect to the activity (e.g., compounds 4 vs. 5 and 16 vs. 17). We noticed the existence of this kind of relationship in CQ derived antimalarial agents reported in the literature as well.48
Furthermore, the activity profile of the compounds (Table 1) indicated that substitution of the aromatic moiety attached to the distal group with bulky hydrophobic groups is favorable for antimalarial activity against CQ-sensitive and resistant strains as well as for inhibition of β-hematin formation (e.g., compounds 4 vs. 16 and 5 vs. 17). The loss of activity observed in compounds 14 and 15 with a tert-butyl substitution on para-position of aryl may be attributed to position/region specific steric restriction resulting from the additional methyl in comparison to an isopropyl (e.g., compounds 16 and 17) at that location. The compounds 18 and 19 may be viewed as an outcome of replacing tert-butyl of compounds 14 and 15 by a phenyl group. Here, phenyl may be offering a different environment as compared to tert-butyl; this may explain the differences observed in the activities of these compounds. The structural features which are found to be influencing the antimalarial activity are also reflected in the β-hematin formation inhibition activity. For these compounds, the antimalarial activity against 3D7 and inhibition of β-hematin formation are correlated (n = 12, r2 = 0.585).
3.3. In silico studies
The model was further refined by subjecting it to equilibration through molecular dynamics (MD) simulations under physiological conditions for 10 ns. It reached the equilibrium state in approximately 2.5 ns and thereafter continued to remain in that state till the end of dynamics. The plots of Ramchandran and Errat, and the free energies of folding of the models from the trajectories sampled in the equilibrated region (after 2.5 ns to 10 ns) were comparable or even better than that of the initial one. All the parameters associated with the diagnosis of model indicate that in the equilibrium region, the model has retained its shape without much change in its structure (Fig. 3).
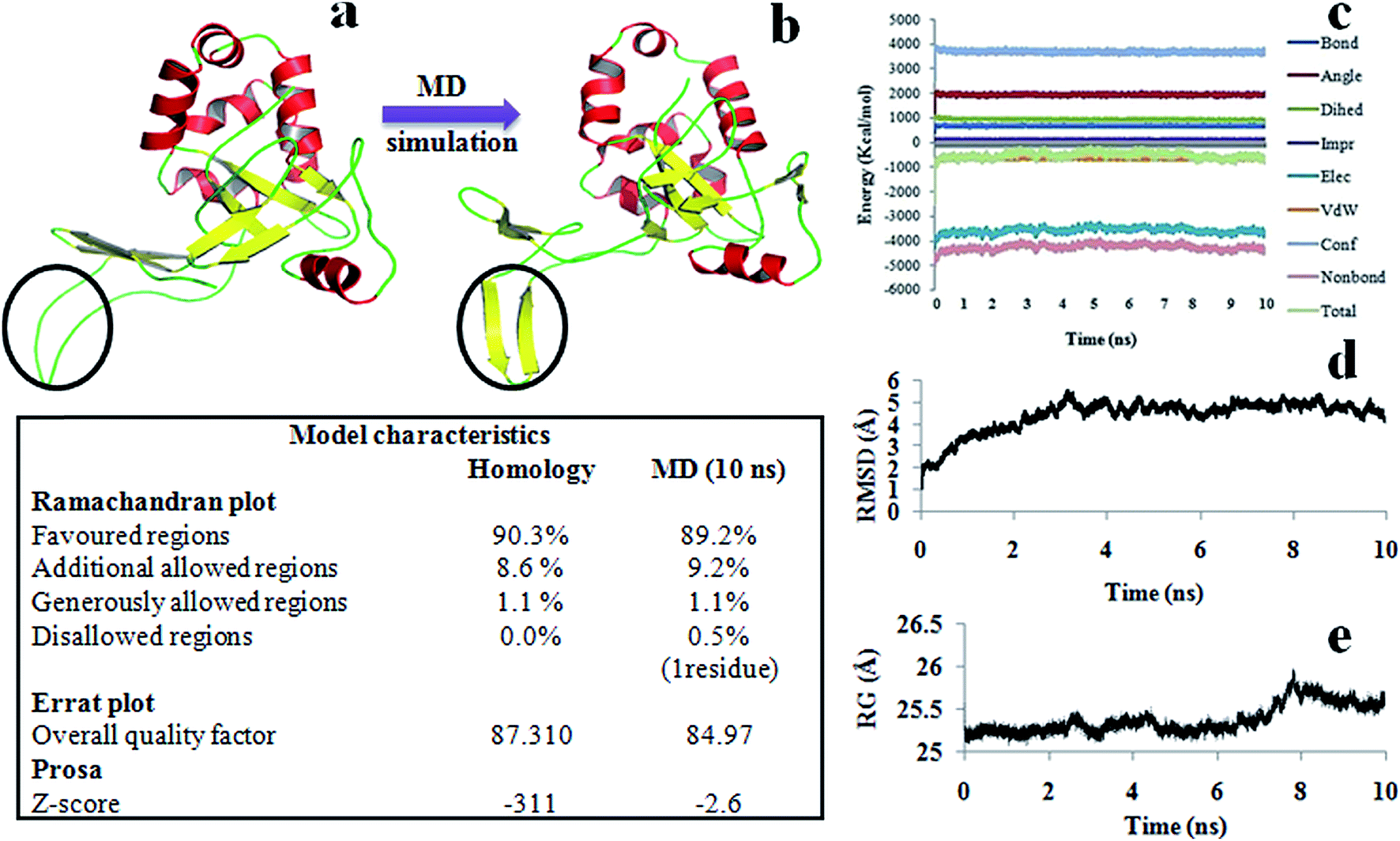 | ||
| Fig. 3 (a) 3D structure of Pf-HDP generated from homology modeling; (b) homology model of Pf-HDP at the end of 10 ns molecular dynamics simulations in NAMD; (c–e) different components of molecular energy profiles of Pf-HDP trajectories, RMSD, and radius of gyration, respectively. The data in the box shows some formal characteristics of the Pf-HDP model as developed from homology modeling procedure and after subjecting it to 10 ns MD simulations. During the dynamics the residues (Asp30, Pro31, Glu32, Asn35, Arg36, Lys37) present in the circled region transformed from loop to beta sheet and continued to maintain this shape until the end of dynamics. | ||
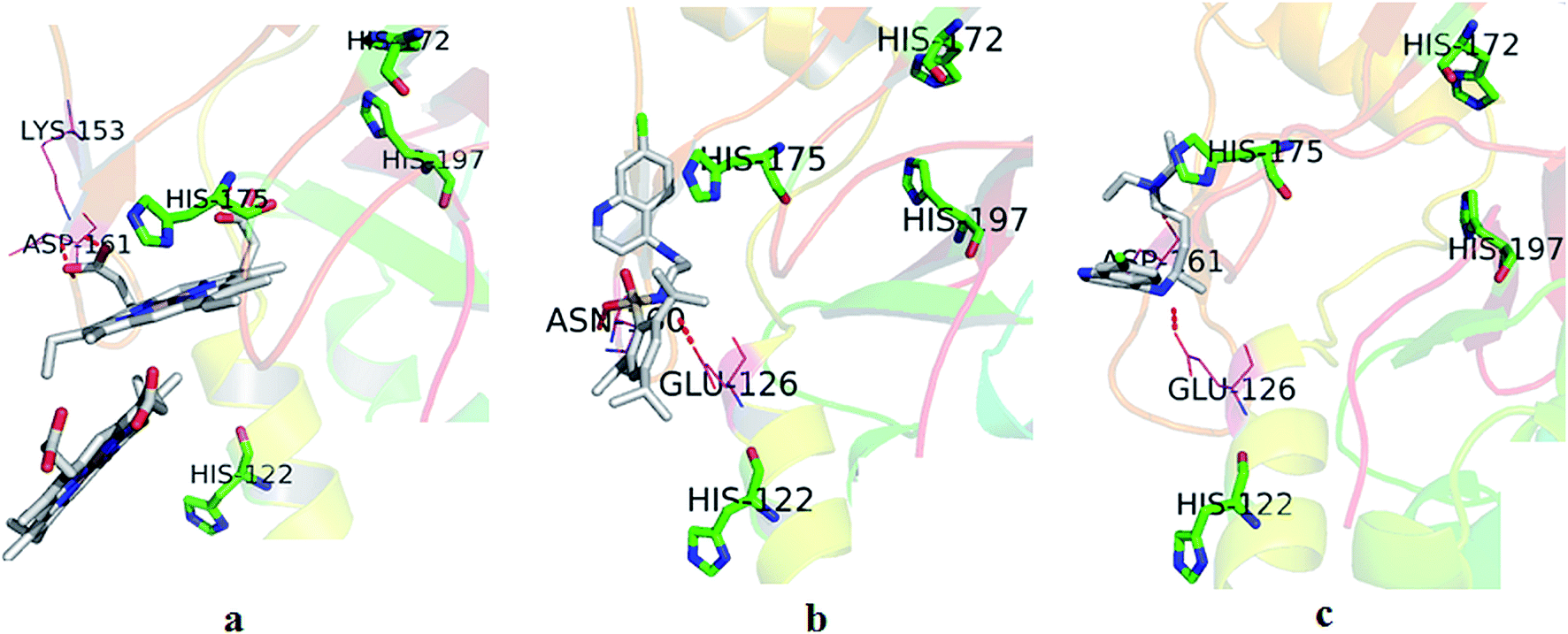 | ||
| Fig. 4 Surflex dock derived heme binding pocket of HDP together with docked poses of (a) heme(s), (b) compound 17 and (c) CQ. | ||
3.3.3.1. HDP. As the HDP of the parasite plays an important role in the transformation of digested heme into hemozoin and in view of some of the recent mechanistic studies with CQ related analogues favouring the inhibition of hemozoin formation as an important event for the antimalarial activity,3,18 we considered it appropriate to dock the synthesized compounds to HDP to examine the likelihood of their binding to the protein. Here, the synthesized compounds were docked into the identified binding pocket of HDP composed of histidines 122, 172, 175 and 197 (along with many other residues) which resulted in scores between 3.18 and 5.94 with the best score attributed to compound 17 (Table 1). The best docked pose of compound 17 in the heme binding of HDP pocket is shown in Fig. 4b. In most of the compounds, except for 7 and 11, the quinoline ring and SO2NH group, respectively, showed arene–arene interactions with the His175 imidazole moiety and H-bond interaction(s) with Glu126 of HDP. Apart from these, in compound 17 an H-bond interaction is seen between the SO2NH moiety and Asn160. In the docked pose of compound 7, its SO2NH moiety showed H-bonds with Lys153 and Asp161. In the case of compound 11, its SO2NH moiety showed an H-bond with Ser153 only. In the HDP–CQ docking experiment, CQ has occupied a position proximate to His175 (Fig. 4c). Furthermore, in the docked pose, the 4-amino of CQ and distal nitrogen showed H-bond interactions with Glu126 and Asp161, respectively. Here it is important to note that compounds which have the tendency to interact with one or more designated histidines residues of HDP can block the entry of heme, and thereby hemozoin formation. The docking interactions and resulting scores of the compounds have very well reflected this phenomenon.
3.3.3.2. Heme monomer. In the experiments with heme the synthesized compounds have resulted in docking scores ranging from 1.92 to 4.76 (Table 1). The highest activity analogue, compound 17, shows a docking score of 3.0. In the docked pose, its quinoline ring is oriented almost parallel to the ferriprotoporphyrin of heme (Fig. 5a). In this state, the quinoline nitrogen showed polar interaction with the Fe of ferriprotoporphyrin. Furthermore, a hydrogen bond is seen between the NH of sulfonamide and the carboxyl group of one of the propionic acid side chains of heme. In the heme–CQ docked pose, the CQ quinoline ring is oriented in a plane parallel to ferriprotoporphyrin and an H-bond interaction is seen between the CQ distal nitrogen and the carboxyl group of the propionic acid side chain of heme (Fig. 5b). Compound 8, though inactive, showed the highest score (4.76) in docking with heme. Interestingly, its docked pose led to three significant interactions with heme. Of these, one is due to a polar interaction (between Cl of quinoline and Fe) and the other two are due to H-bonds (4-amino of quinoline and carboxyl group of one propionic acid side chain; NH of sulfonamide and carboxyl group of the other propionic acid side chain). While these interactions may suggest the scope and ways to improve the activity, in the present compound some of the physicochemical features may have become detrimental for the activity. Among the analogues, compound 6 showed lowest docking score (1.92) with the heme. An examination of its docked pose indicated that it interacted with the heme in a sideways manner and showed no H-bonds or significant polar interactions. The docking scores of all compounds show correspondence with the intermolecular interactions resulting from their docked poses.
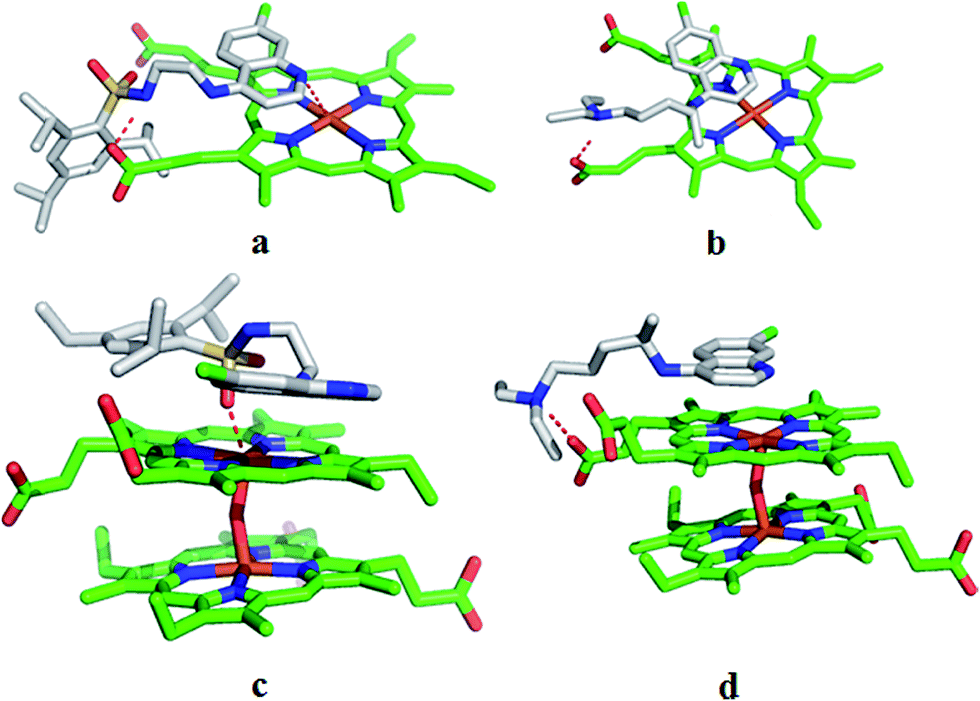 | ||
| Fig. 5 Surflex dock derived docked poses of compound 17 and CQ with monomer (a and b) and μ-oxo dimer (c and d) of heme. | ||
3.3.3.3. μ-Oxo dimer of heme. In the docking experiments with μ-oxo dimer of heme, the compounds showed scores from 4.12 to 6.28 with compound 17 having a score of 4.22 (Table 1, Fig. 5c). The earlier proposed mechanism for CQ complexing with monomer heme suggested the involvement of a dative bond between quinoline nitrogen and Fe.3 In the case of μ-oxo dimer of heme and CQ, the proposed mechanism suggested the involvement of π–π interactions between the two for the stabilization of the complex,3,8 which is noticed in the present study (Fig. 5d). In docking with μ-oxo dimer, all compounds (Table 1) oriented parallel to one face of ferriprotoporphyrin. Moreover in all compounds, except 5 and 7, the oxygen of sulfonamide is involved in the polar interaction with Fe. In the docked poses of compounds 5 and 7, while they oriented parallel to ferriprotoporphyrin, the quinoline nitrogen showed polar interaction with the Fe. Furthermore, in these compounds the NH of sulfonamide group showed an H-bond with the carboxyl group of the propionic acid side chain. The higher docking scores seen in some of the compounds may be attributed to the additional interactions with the target; for example in compound 16 the higher score (6.18) may be due to an additional interaction (H-bond) observed between 4-amino of quinoline and a carboxyl group of one of the propionic acid side chains of the μ-oxo dimer.
In docking experiments several of the compounds showed better scores than that of CQ. In these compounds the presence of a sulfonamide moiety and additional aromatic ring along with other features may have facilitated this. The docking scores of the compounds from the HDP, heme and μ-oxo dimer of heme are not parallel to each other, which is not an unexpected phenomenon. The changes in the docking energetics (due to changing poses) from one system to the other may be primarily responsible for this. However, interestingly, for the compounds with 3D7 and/or K1 activities, excepting 5, their hemozoin inhibition activity and the cumulative dock scores from HDP, heme and μ-oxo dimer of heme are in agreement with each other. On excluding compound 5, the correlation between hemozoin inhibition activity (log(1/IC50)) and the cumulative dock scores of the compounds is 0.56 (n = 11, r2 = 0.56). We cannot clearly explain the deviation seen in the case of compound 5. Nevertheless, in brief, the dock-scores of these compounds are largely in conformity with the observed antimalarial activity profiles (Table 1).
4. Conclusions
In conclusion, two compounds (16 and 17) from the newly synthesized N-(7-chloroquinolinyl-4-aminoalkyl)arylsulfonamides showed good in vitro antimalarial activity against 3D7 and K1 strains of P. falciparum and remarkably inhibited hemozoin formation. Also in the active analogues, the antimalarial activity and inhibition of hemozoin formation are reasonably well correlated. Considering the hemozoin formation inhibition of the compounds were due to their affinity for heme, its μ-oxo dimer and/or heme detoxification protein (HDP), these biological targets were modeled and investigated for probable intermolecular interactions with the compounds using the docking studies. The analysis of docking interactions indicated that in the active analogues the quinoline, 4-amino and sulfonamide (distal group) moieties of the compounds have provided one or more interactions with the key constituents of the biological targets. In heme and in its μ-oxo dimer the key constituents were found to be ferriprotoporphyrin (mostly showing interactions with quinoline and/or 4-amino moieties) and propionic acid side chains (mostly showing interactions with distal group). In the case of HDP, the key constituents were His175 (mostly showing interactions with quinoline moiety) and Glu126 (mostly showing interactions with distal group). Apart from these, residues Asn160, Lys153 and/or Asp161 also showed interactions with some of the compounds. These findings are largely in conformity with the observed antimalarial activity profiles of the compounds and justified the introduction of the sulfonamide moiety and variants in place of distal nitrogen and beyond.As malaria is often associated with economic hardship and poverty, containing the cost of explorations and products is highly desirable feature. In view of this, the novelty of these compounds lies in their simple synthetic procedure for a wider exploration. Moreover, the inclusion of sulfonamide moiety in these compounds considerably improved the inhibition of hemozoin formation over CQ. The low end point of antimalarial response of these compounds may amenable to further optimization. Thus the study has opened scope for the compounds to explore them for antimalarial activity through the inhibition of hemozoin formation.
Acknowledgements
The authors thank Dr Renu Tripathi for the helpful discussions. SV and PA thankfully acknowledge financial support, in the form of Senior Research Fellowships, of the Department of Science and Technology (DST-INSPIRE) New Delhi and the Council of Scientific and Industrial Research, New Delhi, respectively. The SAIF, CSIR-CDRI is thankfully acknowledged for the analytical and spectral data. CDRI Communication No. 9188.References
- World malaria report, 2014, World Health Organization, Geneva, 2014, vol. 789, pp. 1–227, http://www.who.int/malaria/publications/world_790malaria_report_2014/wmr-2014-no-profiles.pdf Search PubMed.
- R. Thome, S. C. Lopes, F. T. Costa and L. Verinaud, Immunol. Lett., 2013, 153, 50–57 CrossRef CAS PubMed.
- A. P. Gorka, A. de Dios and P. D. Roepe, J. Med. Chem., 2013, 56, 5231–5246 CrossRef CAS PubMed.
- S. Kumar, M. Guha, V. Choubey, P. Maity and U. Bandyopadhyay, Life Sci., 2007, 80, 813–828 CrossRef CAS PubMed.
- A. Leed, K. DuBay, L. M. Ursos, D. Sears, A. C. De Dios and P. D. Roepe, Biochemistry, 2002, 41, 10245–10255 CrossRef CAS PubMed.
- A. C. de Dios, R. Tycko, L. M. B. Ursos and P. D. Roepe, J. Phys. Chem. A, 2003, 107, 5821–5825 CrossRef CAS.
- K. F. Schwedhelm, M. Horstmann, J. H. Faber, Y. Reichert, M. Buchner, G. Bringmann and C. Faber, Open Spectrosc. J., 2008, 2, 10–18 CrossRef CAS.
- A. C. Aguiar, M. Santos Rde, F. J. Figueiredo, W. A. Cortopassi, A. S. Pimentel, T. C. França, M. R. Meneghetti and A. U. Krettli, PLoS One, 2012, 7, e37259 CAS.
- D. A. Fidock, P. J. Rosenthal, S. L. Croft, R. Brun and S. Nwaka, Nat. Rev. Drug Discovery, 2004, 3, 509–520 CrossRef CAS PubMed.
- C. Asher, K. A. de Villiers and T. J. Egan, Inorg. Chem., 2009, 48, 7994–8003 CrossRef CAS PubMed.
- A. Dorn, R. Stoffel, H. Matile, A. Bubendorf and R. G. Ridley, Nature, 1995, 374, 269–271 CrossRef CAS PubMed.
- A. F. Slater and A. Cerami, Nature, 1992, 355, 167–169 CrossRef CAS PubMed.
- K. Bendrat, B. J. Berger and A. Cerami, Nature, 1995, 378, 138–139 CrossRef CAS PubMed.
- T. J. Egan, Mol. Biochem. Parasitol., 2008, 157, 127–136 CrossRef CAS PubMed.
- D. Jani, R. Nagarkatti, W. Beatty, R. Angel, C. Slebodnick, J. Andersen, S. Kumar and D. Rathore, PLoS Pathog., 2008, 4, e1000053 Search PubMed.
- D. J. Jr Sullivan, I. Y. Gluzman and D. E. Goldberg, Science, 1996, 271, 219–222 Search PubMed.
- A. V. Pandey, V. K. Babbarwal, J. N. Okoyeh, R. M. Joshi, S. K. Puri, R. L. Singh and V. S. Chauhan, Biochem. Biophys. Res. Commun., 2003, 308, 736–743 CrossRef CAS PubMed.
- M. Chugh, V. Sundararaman, S. Kumar, V. S. Reddy, W. A. Siddiqui, K. D. Stuart and P. Malhotra, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5392–5397 CrossRef CAS PubMed.
- K. Nakatani, H. Ishikawa, S. Aono and Y. Mizutani, Sci. Rep., 2014, 20(4), 1–7 Search PubMed.
- D. Vicente and E. Pérez-Trallero, Enferm. Infecc. Microbiol. Clin., 2010, 28, 122–130 CrossRef PubMed.
- T. H. Maren, Annu. Rev. Pharmacol., 1976, 16, 309–327 CrossRef CAS PubMed.
- S. Mharakurwa, T. Kumwenda, M. A. Mkulama, M. Musapa, S. Chishimba, C. J. Shiff, D. J. Sullivan, P. E. Thuma, K. Liu and P. Agre, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18796–18801 CrossRef CAS PubMed.
- A. Ryckebusch, R. Deprez-Poulain, M. A. Debreu-Fontaine, R. Vandaele, E. Mouray, P. Grellier and C. Sergheraert, Bioorg. Med. Chem. Lett., 2002, 12, 2595–2598 CrossRef CAS PubMed.
- K. Ekoue-Kovi, K. Yearick, D. P. Iwaniuk, J. K. Natarajan, J. Alumasa, A. C. de Dios, P. D. Roepe and C. Wolf, Bioorg. Med. Chem., 2009, 17, 270–283 CrossRef CAS PubMed.
- A. Salahuddin, A. Inam, R. L. van Zyl, D. C. Heslop, C. T. Chen, F. Avecilla, S. M. Agarwal and A. Azam, Bioorg. Med. Chem., 2013, 21, 3080–3089 CrossRef CAS PubMed.
- B. K. Sharma, S. Verma and Y. S. Prabhakar, Curr. Comput.-Aided Drug Des., 2013, 9, 317–335 CrossRef CAS PubMed.
- S. Verma, U. Debnath, P. Agarwal, K. Srivastava and Y. S. Prabhakar, J. Chem. Inf. Model., 2015, 55, 1708–1719 CrossRef CAS PubMed.
- S. Singh, R. K. Srivastava, M. Srivastava, S. K. Puri and K. Srivastava, Exp. Parasitol., 2011, 127, 318–321 CrossRef CAS PubMed.
- T. J. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- A. V. Pandey, N. Singh, B. L. Tekwani, S. K. Puri and V. S. Chauhan, J. Pharm. Biomed. Anal., 1999, 20, 203–207 CrossRef CAS PubMed.
- M. J. Gardner, N. Hall, E. Fung, O. White, M. Berriman, R. W. Hyman, J. M. Carlton, A. Pain, K. E. Nelson, S. Bowman, I. T. Paulsen, K. James, J. A. Eisen, K. Rutherford, S. L. Salzberg, A. Craig, S. Kyes, M. S. Chan, V. Nene, S. J. Shallom, B. Suh, J. Peterson, S. Angiuoli, M. Pertea, J. Allen, J. Selengut, D. Haft, M. W. Mather, A. B. Vaidya, D. M. Martin, A. H. Fairlamb, M. J. Fraunholz, D. S. Roos, S. A. Ralph, G. I. McFadden, L. M. Cummings, G. M. Subramanian, C. Mungall, J. C. Venter, D. J. Carucci, S. L. Hoffman, C. Newbold, R. W. Davis, C. M. Fraser and B. Barrell, Nature, 2002, 419, 498–511 CrossRef CAS PubMed.
- S. Verma and Y. S. Prabhakar, Curr. Med. Chem., 2015, 22, 1603–1630 CrossRef CAS PubMed.
- D. T. Jones, J. Mol. Biol., 1999, 287, 797–815 CrossRef CAS PubMed.
- M. Yoneyama, T. Tomizawa, N. Tochio, S. Koshiba, M. Inoue, T. Kigawa and S. Yokoyama, Solution structure of the FAS1 domain of human transforming growth factor-beta induced protein IG-H3, http://www.rcsb.org/pdb/explore/explore.do?structureId=1X3B.
- N. J. Clout, D. Tisi and E. Hohenester, Structure, 2003, 11, 197–203 CrossRef CAS PubMed.
- R. G. Moody and M. P. Williamson, FEBS Open Bio, 2013, 3, 71–77 CrossRef CAS PubMed.
- B. Webb and A. Sali, Curr. Protoc. Bioinf., 2014, 47, 5.6.1–5.6.32 Search PubMed.
- N. Guex, M. C. Peitsch and T. Schwede, Electrophoresis, 2009, 30, S162–S173 CrossRef PubMed.
- R. A. Laskowsky, M. W. MacArthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef.
- M. Wiederstein and M. J. Sippl, Nucleic Acids Res., 2007, 35, W407–W410 CrossRef PubMed.
- L. Willard, A. Ranjan, H. Zhang, H. Monzavi, R. F. Boyko, B. D. Sykes and D. S. Wishart, Nucleic Acids Res., 2003, 31, 3316–3319 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- SYBYL, version 7.3, Tripos Associates, St. Louis, MO, 2006 Search PubMed.
- D. J. Harrington, K. Adachi and W. E. Jr Royer, J. Mol. Biol., 1997, 272, 398–407 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 1.7.4, Schrodinger, LLC Search PubMed.
- M. K. Gupta and Y. S. Prabhakar, J. Chem. Inf. Model., 2006, 46, 93–102 CrossRef CAS PubMed.
- C. Hansch and A. Leo, in Substituent constants for correlation analysis in chemistry and biology, Wiley, York, 1979, ch. 6, pp. 48–63 Search PubMed.
- B. A. Solaja, D. Opsenica, K. S. Smith, W. K. Milhous, N. Terzic, I. Opsenica, J. C. Burnett, J. Nuss, R. Gussio and S. Bavari, J. Med. Chem., 2008, 51, 4388–4391 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-1: β-hematin inhibition dose dependent response, physicochemical characteristics of varying substitutions, Pf-HDP bioinformatics, and synthetic information. ESI-2: representative spectra. See DOI: 10.1039/c6ra00846a |
| This journal is © The Royal Society of Chemistry 2016 |