
Lewis-acid catalyzed synthesis and characterization of novel castor fatty acid-based cyclic carbonates†
Naganna Narraac,
Badari Narayana Prasad Rachapudiac,
Sahithya Phani Babu Vemulapallibc and
Padmaja V. Korlipara*ac
aCentre for Lipid Research, CSIR-Indian Institute of Chemical Technology, Uppal Road, Hyderabad 500 007, India. E-mail: padmajak@iict.res.in; Fax: +91-40-27193370
bCentre for NMR and Structural Chemistry, CSIR-Indian Institute of Chemical Technology, Hyderabad 500 007, India
cAcademy of Scientific and Innovative Research, New Delhi, India
First published on 2nd March 2016
Abstract
Novel bio-based cyclic carbonates were synthesised from ricinoleic acid by intra molecular rearrangement of an epoxy carbonate ester with Lewis acids. The first step was the carbonate interchange reaction between methyl ricinoleate and dialkyl carbonates followed by epoxidation using the performic acid method to get methyl 8-(3-(2-(methoxy carbonyloxy)octyl)oxiran-2-yl)octanoate (3a) and methyl 8-(3-(2-(ethoxycarbonyloxy)octyl)oxiran-2-yl)octanoate (3b). These products on treatment with Lewis acids (Sc(OTf)3, Yb(OTf)3 and ZnBr2) formed a mixture of inseparable five and six membered cyclic carbonates through a spiroorthocarbonate (SOC) intermediate by intramolecular rearrangement. Whereas, BF3·OEt2 and AlCl3 formed only SOC. All the newly synthesized compounds were characterized by 1H NMR, 13C NMR, ESI-MS, GC-MS (EI), HRMS, FT-IR and studied by thermogravimetric analysis (TGA). This is the first report on the preparation of castor fatty acid based cyclic carbonates without the use of carbon dioxide.
Introduction
Vegetable oils are an important class of naturally occurring renewable resources. The wide availability of vegetable oils at low prices is considered to be an alternative to the non-renewable petroleum based raw materials.1 Synthesis of cyclic carbonates from vegetable oils has been one of the recent interests due to their fascinating applications such as electrolytes for lithium ion batteries, green solvents, polymer precursors. They can also be used as intermediates for the preparation of pharmaceuticals and fine chemicals.2,3 They are non-toxic and biodegradable. Synthesis of fatty acid based cyclic carbonates was reported in 1958 using phosgene as a reagent.4,5 However, usage of phosgene is not environmentally friendly due its corrosive nature and toxicity. Later, cyclic carbonate ring was introduced on to fatty chains from fatty ester chlorohydrins utilizing tetramethylammonium hydrogencarbonate.6 Synthesis of carbonated vegetable oil was conducted and applied in the preparation of polyurethanes.7 In another study by Doll and Erhan, synthesis of cyclic carbonates was performed using epoxidized fatty acids and carbon dioxide catalyzed by tetrabutylammonium bromide.8 The authors also prepared cyclic carbonates using supercritical carbon dioxide in shorter reaction time compared to other reported methods.9Boyer et al. synthesized terminal and internal cyclic carbonates from oleic acid methyl ester by the reaction of the epoxide precursor with CO2 in the presence of tetrabutylammonium bromide. These cyclic carbonates were used for the preparation of non-isocyanate polyurethanes.10 Linseed and soybean oil-based cyclic carbonates were prepared using tetrabutylammonium bromide and silica supported alkylpyridinium iodide to recover catalyst by simple filtration without using any solvents.11 Organic halides and polyoxometalates were employed during the carbonation of epoxy fatty acid esters and substrate dependent synergetic and antagonistic effects of these catalysts were also studied.12 Vegetable oil-based bis six membered cyclic carbonates for the first time was reported by Maisonneuve et al. from methyl undecenoate via malonate, 1,3-diol and monofunctional six membered cyclic carbonate intermediates. Metathesis or thiol-ene reactions were employed to produce two bis 6-membered cyclic carbonates.13 Recently, carbonation of epoxy fatty acid esters has been conducted using CO2, halide salts and phase transfer catalysts where high yields were achieved.14 Epoxidized linseed oil has been carbonated in another study employing organo catalysts in combination with hydrogen bond donors for obtaining cyclic carbonates.15
Among vegetable oils, castor oil is unique due to the presence of about 90% of ricinoleic acid, a bifunctional fatty acid. Castor oil is known for its medicinal value since ancient days and it has several potential industrial and biomedical applications.16,17 In the present work, 12-hydroxyl functional group of methyl ricinoleate is functionalized with dialkyl carbonate to form linear carbonates. In the following reaction, the 9,10-double bond is converted into epoxide. This epoxide is reacted with different Lewis acids to obtain a mixture of cyclic carbonates namely castor fatty methyl ester carbonates (CFAMEC) via spiroorthocarbonate (SOC) intermediate by intra-molecular rearrangement (Scheme 1). The objective of the present work is to synthesize castor fatty based cyclic carbonates. These carbonates can be potential precursors for the synthesis of non-isocyanate polyurethanes, the most promising substitutes for conventional polyurethanes used in paints, coatings and/or as biomaterials.
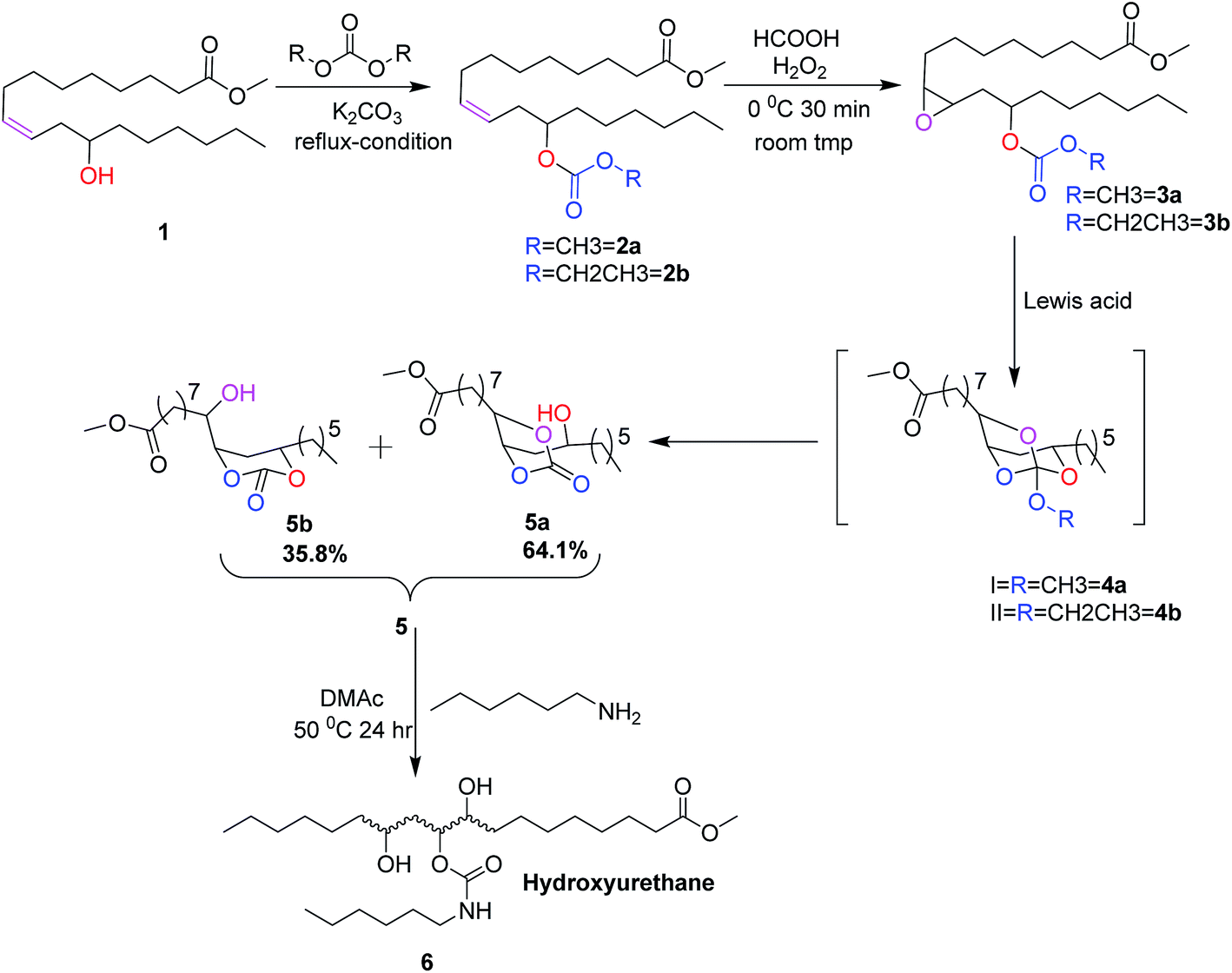 | ||
| Scheme 1 Synthetic pathway to cyclic carbonates from methyl ricinoleate. | ||
Materials and methods
Reagents
Castor oil was procured from M/s Ramcharan Industries Pvt. Ltd., Hyderabad. Dimethyl carbonate (DMC, 99%), diethyl carbonate (DEC, 99+%), scandium(III)trifluoro methanesulfonate (Sc(OTf)3) and ytterbium(III)trifluoro methanesulfonate (Yb(OTf)3) were purchased from Alfa Aesar (Hyderabad, India). Boron trifluoride diethyl etherate (BF3·OEt2), aluminum chloride, ferric chloride, titanium tetrachloride (TiCl4), zinc bromide (ZnBr2), zinc(II)trifluoromethanesulfonate (Zn(OTf)2), n-hexylamine, dimethylacetamide (DMAc) and copper(II)trifluoromethanesulfonate (Cu(OTf)2) were obtained from Sigma-Aldrich (St. Louis, USA). Potassium carbonate anhydrous, sodium sulphate, sulphuric acid, hydrogen peroxide, and formic acid were purchased from M/s SD Fine Chemicals (Mumbai, India). Silica gel (60–120 mesh) for column chromatography was purchased from M/s Acme synthetic chemicals (Mumbai, India) and pre-coated TLC plates (silica gel 60 F254) were purchased from M/s Merck (Darmstadt, Germany). Hexane, methanol, dichloro methane (DCM), acetonitrile (ACN) and ethyl acetate were purchased from Industrial Solvents and Chemicals Pvt. Ltd., (Mumbai, India). All other solvents and reagents were of analytical grade and used directly without purification.Instruments
1D-(1H, 13C) and 2D-(DQFCOSY, 13C–1H HSQC, and 13C–1H HMBC) NMR spectra were recorded on Bruker Avance 700/500 MHz and 175/125 MHz for 1H and 13C respectively. The NMR spectra were referenced to δ 7.26 ppm and δ 77.0 ppm in CDCl3 solvent for 1H and 13C, respectively. J-coupling pattern in 1H NMR spectra are described as: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet protons. Mass spectra were recorded using electron spray ionization on Waters e2695 Separators module (Waters, Milford, MA, USA) mass spectrometer. FT-IR spectra were recorded in chloroform on a Perkin-Elmer Fourier Transform (FT-IR) Spectrum BX instrument (Model: Spectrum BX; Connecticut, USA). HRMS spectra were obtained by Exactive Orbitrap mass spectrometer (Thermo Scientific, Waltham, MA, USA). The fatty acid composition and five and six carbonates were analyzed using Gas Chromatograph 6890 N series of Agilent make USA using an HP-1 column (30 m × 0.25 mm × 0.25 μm). The injector and flame ionization detector were at 280 and 300 °C respectively. The oven temperature was programmed at 150 °C for 2 min and then increased to 300 °C at 10 °C min−1 and final temperature hold for 20 min. The carrier gas used was nitrogen at a flow rate of 1.0 mL min−1. GC-MS (EI) analysis was used to study the mass spectral pattern of the synthesized castor methyl ester carbonates. GC-MS was performed using Agilent 6890 gas chromatograph connected to Agilent 5973 mass spectrometer at 70 eV (m/z 50–600; source at 230 °C and quadruple at 150 °C) with HP-1 MS capillary column (30 m × 0.25 mm × 0.5 μm) in the EI mode. The oven temperature was programmed at 150 °C for 2 min and then increased to 300 °C at 10 °C min−1 and 20 min at 300 °C. The carrier gas used was helium at a flow rate of 1.0 mL min−1. Computational calculations: all the geometries were optimized at B3LYP/6-31G* level of theory in Gaussian 09 program.Thermo gravimetric analysis (TGA)
TGA measurements were performed with a TA Q500 (TA Instruments, Inc., New Castle, DE, U.S.A.) instrument in non-isothermal mode under nitrogen atmosphere. About 4.0 mg of the sample was taken in α-Al2O3 crucible applying a heating rate of 10 °C min−1, in a temperature range of 30–500 °C.Experimental section
Preparation of methyl ricinoleate (1)
Methyl ricinoleate was prepared from castor oil as described previously.18 Briefly, transesterification of castor oil resulted in castor oil methyl esters which were purified by column chromatography to obtain pure methyl ricinoleate.(Z)-Methyl 12-(methoxy carbonyloxy)octadec-9-enoate (2a)
To a stirred mixture of methyl ricinoleate (6 g, 0.019 mol) and DMC (69.1 g, 0.768 mol), K2CO3 (3.9 g, 0.0288 mol) was added and refluxed for 35 h. The progress of the reaction was monitored by TLC using hexane/ethyl acetate (90/10 v/v). The reaction mixture was filtered to remove K2CO3 and the filtrate was concentrated under vacuum to remove excess DMC and methanol. The crude product was purified by silica gel column chromatography using hexane/ethyl acetate (98/2 v/v) to obtain 2a (6.4 g, yield 90%) as colourless liquid. The product was characterized by 1H NMR, 13C NMR, ESI-MS, GC-MS (EI) and FT-IR spectral analysis. Similar procedure was followed for the preparation of (Z)-methyl 12-(ethoxy carbonyloxy)octadec-9-enoate (2b) with diethyl carbonate in 89% yield.1H NMR (CDCl3, 500 MHz): δ ppm = 5.49 (m, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 5.37 (m, –CHCH–), 4.70 (m, –CH–C(O)OCH3), 3.76 (s, –OCH3), 3.67 (s, –OCH3), 2.36 (m, –COCH–CH2–CHCH–), 2.30 (t, –CH2–CH2–C(O)OCH3), 2.02 (m, –CHCH–CH2–CH2–), 1.62 (m, –CH2–CH2–CH2–), 1.31–1.24 (multiple signals), 0.88 (t, –CH2–CH3). 13C NMR (CDCl3, 125 MHz): δ ppm = 174.20 (–C(O)OCH3), 155.60 (–OC(O)OCH3), 132.98 (–CHCH–), 123.62 (–CHCH–), 78.51 (–CH–OC(O)OCH3), 54.47 (–OC(O)OCH3), 51.39 (–OC(O)OCH3), 34.13 (–CH2–C(O)OCH3), 22.52–34.24 (multiple signals), 13.91 (end carbon of fatty chain). FT-IR: (neat, cm−1) 3007, 2928, 2856, 1742, 1268, 1172, 792. ESI-MS: m/z 393 [M + Na]+. GC-MS (EI): m/z 349 ([M − OCH3]+), m/z 311 ([M − (CO)OCH3]+), m/z 294 ([M − HO(CO)OCH3]+), m/z 263 ([M − OCH3 − HO(CO)OCH3]+), m/z 220 ([M − McL − HO(CO)OCH3]+).
CH–), 5.37 (m, –CHCH–), 4.70 (m, –CH–C(O)OCH3), 3.76 (s, –OCH3), 3.67 (s, –OCH3), 2.36 (m, –COCH–CH2–CHCH–), 2.30 (t, –CH2–CH2–C(O)OCH3), 2.02 (m, –CHCH–CH2–CH2–), 1.62 (m, –CH2–CH2–CH2–), 1.31–1.24 (multiple signals), 0.88 (t, –CH2–CH3). 13C NMR (CDCl3, 125 MHz): δ ppm = 174.20 (–C(O)OCH3), 155.60 (–OC(O)OCH3), 132.98 (–CHCH–), 123.62 (–CHCH–), 78.51 (–CH–OC(O)OCH3), 54.47 (–OC(O)OCH3), 51.39 (–OC(O)OCH3), 34.13 (–CH2–C(O)OCH3), 22.52–34.24 (multiple signals), 13.91 (end carbon of fatty chain). FT-IR: (neat, cm−1) 3007, 2928, 2856, 1742, 1268, 1172, 792. ESI-MS: m/z 393 [M + Na]+. GC-MS (EI): m/z 349 ([M − OCH3]+), m/z 311 ([M − (CO)OCH3]+), m/z 294 ([M − HO(CO)OCH3]+), m/z 263 ([M − OCH3 − HO(CO)OCH3]+), m/z 220 ([M − McL − HO(CO)OCH3]+).
(Z)-Methyl 12-(ethoxycarbonyloxy)octadec-9-enoate (2b)
1H NMR (CDCl3, 500 MHz): δ ppm = 5.47 (m, –CHCH–), δ 5.36 (m, –CHCH–), 4.69 (m, CH–C(O)OCH2CH3), 4.17 (q, –C(O)OCH2CH3), 3.66 (s, OCH3), 2.39 (m, –COCH–CH2–CHCH–), 2.30 (t, –CH2–CH2–C(O)OCH3), 2.04 (m, –CHCH–CH2–CH2–), 1.63 (m, –CH2–CH2–CH2–), 1.32–1.29 (multiple signals), 0.88 (t, –CH2–CH3). 13C NMR (CDCl3, 125 MHz): δ ppm = 174.13 (–C(O)OCH3), 155.96 (–OC(O)OCH2CH3), 132.79 (–CHCH–), 123.78 (–CHCH–), 78.12 (–CH–OC(O)–OCH2–CH3), 63.61 (–OC(O)O–CH2–CH3), 51.40 (–OCH3), 22.49–34.04 (multiple signals), 14.23 (–OC(O)O–CH2–CH3), 13.87 (end carbon of fatty chain). FT-IR: (neat, cm−1) 3007, 2929, 2856, 1742, 1259, 1172, 792. ESI-MS: m/z 407 [M + Na]+. GC-MS (EI): m/z 294 ([M − HO(CO)OCH2CH3]+), m/z 263 ([M − OCH3 − HO(CO)OCH2CH3]+), m/z 220 ([M − McL − HO(CO)OCH2CH3]+).
Methyl 8-(3-(2-(methoxycarbonyloxy)octyl)oxiran-2-yl)octanoate (3a)
A mixture of 2a (5 g, 0.013 mol) and formic acid (0.9 g, 0.020 mol) in a three necked round bottomed flask equipped with an overhead stirrer, 50% aqueous hydrogen peroxide (1.83 g, 0.027 mol) was slowly added to the contents at 0 °C for a period of 30 min. After addition, the contents were stirred at room temperature for a period of 8 h. The progress of the reaction was monitored by TLC using hexane/ethyl acetate (80/20 v/v). The reaction mixture was extracted with ethyl acetate (50 mL) and washed with water at (3 × 50 mL) until it was acid free and dried over anhydrous Na2SO4. The epoxidized product was concentrated and dried under reduced pressure. The crude product was purified by silica gel column chromatography using hexane/ethyl acetate (96/4 v/v) to obtain 3a (4.6 g, 90% yield) as colourless liquid. The formation of the epoxy ring was confirmed by 1H NMR, 13C NMR, ESI-MS, GC-MS (EI) and FT-IR. Similar experimental procedure was followed for the epoxidation of 2b to get methyl 8-(3-(2-(ethoxy carbonyloxy)octyl)oxiran-2-yl)octanoate (3b) in 90% yield.1H NMR (CDCl3, 500 MHz): δ ppm = 4.89 (m, –CH–C(O)OCH3), 3.79 (s, –OCH3), 3.67 (s, –OCH3), 3.01 (m, –CHOCH–), 2.90 (m, –CHOCH–), 2.30 (t, –CH2–CH2–C(O)OCH3), 1.73 (m, –CH2–CHOCH–), 1.6 (m, –CHOCH–CH2–CH2–), 1.46 (m, –CH2–CH2–CH2–), 1.32–1.29 (multiple signals), 0.88 (t, –CH2–CH3). 13C NMR (CDCl3, 125 MHz): δ ppm = 173.89 (–C(O)OCH3), 155.31 (–OC(O)OCH3), 76.74 (–CH–OC(O)OCH3), 56.69 (–CHOCH–), 53.05 (–CHOCH–), 54.36 (–OC(O)OCH3), 51.18 (–C(O)OCH3), 34.18 (–CH2C(O)CH3), 22.47–34.28 (multiple signals), 13.87 (end carbon of fatty chain). FT-IR: (neat, cm−1) 2929, 2857, 1742, 1171, 841, 791. ESI-MS: m/z 409 [M + Na]+. GC-MS (EI): m/z 387 ([MH]+), m/z 355 ([M − OCH3]+), m/z 310 ([M − OH(CO)OCH3]+), m/z 279 ([M − OCH3 − HO(CO)OCH3]+), m/z 225 ([M − OHC(O)OCH3 − (CH2)5CH3]+).
Methyl 8-(3-(2-(ethoxycarbonyloxy)octyl)oxiran-2-yl)octanoate (3b)
1H NMR (CDCl3, 500 MHz): δ ppm = 4.89 (m, –CH–C(O)OCH2CH3), 4.20 (q, –C(O)OCH2CH3), 3.89 (s, –OCH3), 3.02 (m, –CHOCH–), 2.89 (m, –CHOCH–), 2.30 (t, –CH2–CH2–C(O)OCH3), 1.89 (m, –CH2–CHOCH–), 1.78 (m, –CHOCH–CH2–CH2–), 1.69 (m, –CH2–CH2–CH2–), 1.29–1.23 (multiple signals), 0.88 (t, –CH2–CH3). 13C NMR (CDCl3, 125 MHz): δ ppm = 174.18 (–C(O)OCH3), 154.92 (–OC(O)OCH2CH3), 76.67 (–CH–OC(O)OCH2–CH3), 63.38 (–OC(O)OCH2–CH3), 56.89 (–CHOCH–), 53.75 (–CHOCH–), 51.39 (–C(O)OCH3), 22.51–34.46 (multiple signals), 14.23 (–OC(O)OCH2–CH3), 13.97 (end carbon of fatty chain). FT-IR: (neat, cm−1) 2930, 2857, 1742, 1176, 841, 790. ESI-MS: m/z 423 [M + Na]+. GC-MS (EI): m/z 401 ([MH]+), m/z 369 ([M − OCH3]+), m/z 310 ([M − OH(CO)OCH2CH3]+), m/z 279 ([M − OCH3 − HO(CO)OCH2CH3]+), m/z 225 ([M − OHC(O)OCH2CH3 − (CH2)5CH3]+).Methyl 8-(3-hexyl-2,7,8-trioxa-bicyclo(3,2,1)octan-6-yl)octanoate (4a)
To a solution of 3a (2 g 0.00518 mol) in DCM (10 mL), BF3·OEt2 (0.3 g, 0.00259 mol) was added and the mixture was stirred at room temperature for 10 min. After 10 min, color change from colorless to golden yellow was observed and the completion of the reaction was confirmed by TLC using hexane/ethyl acetate (80/20 v/v). The reaction mixture was dissolved in to DCM (15 mL) and washed with water (3 × 15 mL) and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by silica gel column chromatography using hexane/ethyl acetate (98/2 v/v) to obtain 4a (1.8 g, 90% yield) as colourless liquid. The formation of the cyclic ring was confirmed by 1H NMR, 13C NMR, ESI-MS, GC-MS (EI), HRMS and FT-IR. Similar procedure was followed for the preparation of methyl 8-(1-ethoxy-3-hexyl-2,7,8-trioxa-bicyclo-(3,2,1)octan-6-yl)octanoate (4b) in 89% yield by reacting 3b with BF3·OEt2.1H NMR (CDCl3, 500 MHz): δ ppm = 4.87 (m, 1H cyclic hydrogen), 4.00 (m, 1H cyclic hydrogen), 3.78 (m, 1H cyclic hydrogen), 3.67 (s, –OCH3), 2.3–2.4 (m, Hd), 2.29 (t, –CH2–CH2–C(O)OCH3), 1.69 (m, H′d), 1.56–1.23 (multiple signals), 0.88 (t, –CH2–CH3). 13C NMR (CDCl3, 125 MHz): δ ppm = 174.25 (–C(O)OCH3), 154.79 (–OOCOO–), 78.22–83.67 (bicyclic ring carbons), 63.96 (–OCH3), 51.31 (–OCH3), 34.16 (CH2–C(O)OCH3), 22.61 (bicyclic ring middle carbon), 14.04 (end carbon of fatty chain). FT-IR: (neat, cm−1) 2930, 2856, 1742, 1200, 1172, 1107, 774. ESI-MS: m/z 409 [M + Na]+. GC-MS (EI): m/z 387 ([MH]+), m/z 355 ([M − OCH3]+), m/z 310 ([M − OH(CO)OCH3]+), m/z 279 ([M − OCH3 − HO(CO)OCH3]+), m/z 225 [M − OHC(O)OCH3 − (CH2)5CH3]+. HRMS: calcd for C21H39O6, [M + H]+, m/z 387.27412; found, m/z 387.27349.
Methyl 8-(1-ethoxy-3-hexyl-2,7,8-trioxa-bicyclo(3,2,1)octan-6-yl)octanoate (4b)
1H NMR (CDCl3, 500 MHz): δ ppm = 4.82 (m, 1H cyclic hydrogen), 4.20 (q, –OCH2–CH3), 3.99 (m, 1H cyclic hydrogen), 3.98 (m, 1H cyclic hydrogen), 3.66 (s, –OCH3), 2.4–2.4 (m, He), 2.30 (t, –CH2–CH2–COOCH3), 1.69 (m, H′e), 1.43–1.24 (multiple signals), 0.88 (t, –CH2–CH3). 13C NMR (CDCl3, 125 MHz): δ ppm = 174.25 (–C(O)OCH3), 155.41 (–OOCOO–), 78.25–83.67 (bicyclic ring carbons), 63.96 (–OCH2CH3), 51.37 (–OCH3), 34.01 (CH2–C(O)OCH3), 22.52 (bicyclic ring middle carbon), 14.20 (OCH2CH3), 14.01 (end carbon of fatty chain). FT-IR: (neat, cm−1) 2930, 2857, 1742, 1200, 1172, 1107, 774. ESI-MS: m/z 423 [M + Na]+. GC-MS (EI): m/z 401 ([MH]+), m/z 369 ([M − OCH3]+), m/z 310 ([M − OH(CO)OCH2CH3]+), m/z 279 ([M − OCH3 − HO(CO)OCH2CH3]+), m/z 225 [M − OHC(O)OCH2CH3 − (CH2)5CH3]+. HRMS: calcd for C22H41O6, [M + H]+, m/z 401.28977; found, m/z 401.28887.Castor fatty methyl ester carbonates (CFAMEC) (5)
3a (2 g, 0.00518 mol) was dissolved in DCM (10 mL) and Sc(OTf)3 (1.2 g, 0.0025 mol) was added and the mixture was stirred at room temperature for a period of 8 h. The progress of the reaction was monitored by TLC using hexane/ethyl acetate (70/30 v/v). The reaction mixture was dissolved in DCM (15 mL) and washed with water (3 × 15 mL) and dried over anhydrous Na2SO4. The cyclic carbonate product was concentrated and dried under reduced pressure. The crude product was purified by silica gel column chromatography using hexane/ethyl acetate (94/6 v/v) to obtain a mixture of 5a and 5b (1.7 g, 90% yield) as colourless liquids. The formation of cyclic carbonate group was confirmed by 1H NMR, 13C NMR, ESI-MS, GC-MS (EI), HRMS, GC and FT-IR.1H NMR (CDCl3, 500 MHz): δ ppm = 4.52–4.26 (cyclic carbonate ring protons), 3.84–3.77 (m, 1H, –CH–OH), 3.67 (s, –OCH3), 2.29 (t, –CH2–CH2–C(O)OCH3), 1.30–1.93 (multiple signals), 0.88 (t, –CH2–CH3). 13C NMR (CDCl3, 125 MHz): δ ppm = 174.25 (–C(O)OCH3), 154.61 and 154.56 (cyclic carbonyl carbon), 82.43–79.79 (cyclic ring carbons), 68.22–67.96 (–CH–OH), 51.43 (–OCH3), 34.71 (–CH2–C(O)OCH3), 41.34–24.58 (multiple signals), 22.54 (cyclic ring middle carbon), 14.00 (end carbon of fatty chain). FT-IR: (neat, cm−1) 3497 (hydroxy), 2929, 2857, 1801 (cyclic carbonate), 1742, 1461, 1438, 1174, 1053, 775. ESI-MS: m/z 395 [M + Na]+. GC-MS (EI): m/z 373 ([MH]+), m/z 341 ([M − OCH3]+), m/z 287 ([M − (CH2)5CH3]+), m/z 255 ([M + H − CO2 − McL]). HRMS: calcd for C20H37O6, [M + H]+, m/z 373.25730; found, m/z 373.25847; elemental analysis: calcd (%) for C20H36O6: C 64.49, H 9.74, O 25.77; found: C 64.51, H 9.67, O 25.80.
Confirmation of cyclic carbonate by hydroxyurethane formation (6)
A solution of 5 (0.5 g, 0.00134 mol) and n-hexylamine (1.3 g, 0.00134 mol) dissolved in DMAc (4 mL) was stirred at 50 °C for a period of 24 h. The progress of the reaction was monitored by TLC using hexane/ethyl acetate (70/30 v/v). The reaction mixture was dissolved in ethyl acetate (10 mL) and washed with water (3 × 10 mL) and dried over anhydrous Na2SO4. After removal of the solvent, the crude product was purified by silica gel column chromatography using hexane/ethyl acetate (92/8 v/v) to obtain 6 (0.5 g, 88% yield). The formation of 6 was confirmed by 1H NMR, 13C NMR, ESI-MS, GC-MS (EI), HRMS and FT-IR.1H NMR (CDCl3, 500 MHz): δ ppm = 4.82 (m, –CH–OC(O)NH–), 4.67 (m, –CH–OH), 3.88 (m, –CH–OH), 3.17 (m, –C(O)NH–CH2–CH2–), 2.29 (t, –CH2–CH2–C(O)OCH3), 1.30–1.93 (multiple signals), 0.88 (m, –CH2–(CH3)2). 13C NMR (CDCl3, 125 MHz): δ ppm = 174.25 (–C(O)OCH3), 156.92 and 156.72 (–OC(O)NH–), 72.96 (–CH–OH), 73.97 (–CHOH), 70.17 (–COC(O)NH), 51.43 (–OCH3), 41.08 (–CH–NHC(O)O–), 34.08 (–CH2C(O)OCH3), 24.58–32.12 (multiple signals), 14.07 (end carbon of fatty chain), 13.99 (–CH2–CH3). FT-IR: (cm−1, KBr) 3497 (OH), 3347 (NH), 2929, 2857, 1738, 1688, 1540, 1461, 1438, 1174, 1053, 775. ESI-MS: m/z 496 [M + Na]+. GC-MS (EI): m/z 255 ([M − C7H14NO2 − McL]+). HRMS: calcd for C21H39O6, [M + Na], m/z 496.3581; found, m/z 496.3609.
Results and discussion
Methyl ricinoleate was prepared from castor oil by transesterification using sulphuric acid in methanol. Linear carbonates of methyl ricinoleate (2a and 2b) were prepared by dialkyl carbonate interchange reaction between methyl ricinoleate and dialkyl carbonates (dimethyl/diethyl carbonate) catalyzed by K2CO3 without any solvent in 90% yields. Different catalysts were reported for dialkyl carbonate interchange reaction.19–22 However, we have chosen K2CO3 due to its effectiveness as a catalyst in synthesizing linear alkyl carbonates and the same was also reported by Rokicki.22The structure of the compounds 2a and 2b were confirmed by FT-IR, 1H, 13C NMR spectroscopy and GC-MS (EI). The FT-IR spectra of linear carbonates showed a band at 1742 cm−1 for –OC(O)O–, which was overlapped with the carbonyl ester function band. The absence of hydroxyl group band at 3448 cm−1 indicated complete conversion of hydroxyl to carbonate groups (Fig. 1).
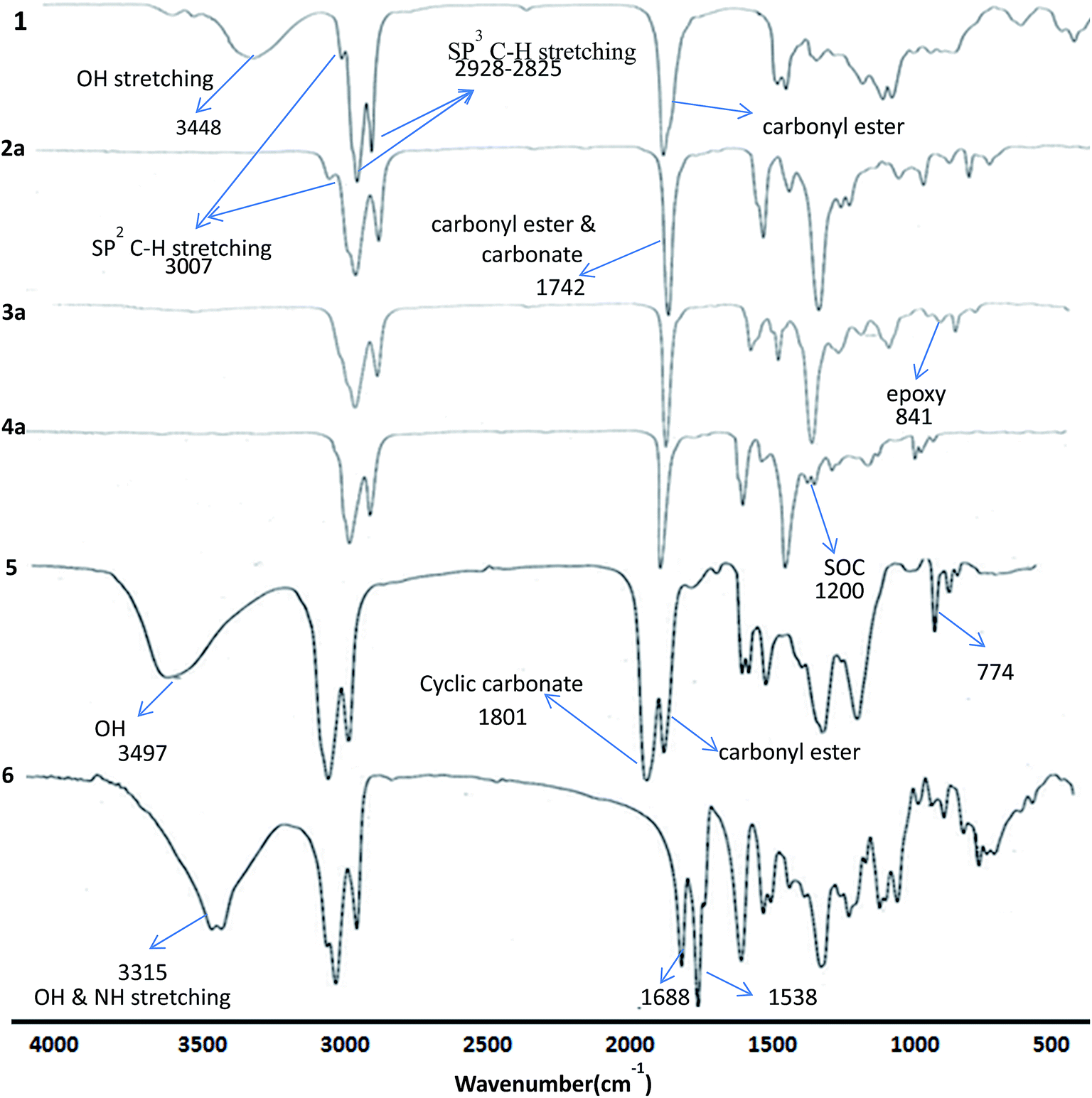 | ||
| Fig. 1 Stacked FT-IR spectra of methyl ricinoleate 1, linear carbonate (2a), epoxycarbonate (3a), spiroorthocarbonate (4a), cyclic carbonates (5) and hydroxy urethane (6). | ||
Formation of linear carbonates was also confirmed from their 1H NMR spectra. The methine proton shift from 3.6 to 4.7 ppm confirmed the formation of carbonate moiety in 2a (Fig. 2) and 2b (Fig. S1, ESI†). A singlet at 3.6 ppm for new methoxy protons (–OCH3) for 2a and a quartet which appeared at 4.2 ppm corresponding to methylene protons adjacent to carbonate linkage for 2b in 1H NMR spectra confirmed the linear carbonate structure (Fig. S1, ESI†). Multiplets at 5.3–5.5 ppm indicated 9,10-double bond protons in both the compounds. The remaining protons for alkyl chain well matched in 1H NMR spectra. In 13C NMR spectra, the carbonate carbon appeared at 155 ppm whereas ester carbon appeared at 173 ppm (Fig. 3). This is due to the electron releasing effect of the additional oxygen atom compensating for electron withdrawal of the carbonyl oxygen.23 The appearance of chemical shift at 76 ppm confirmed the complete conversion of hydroxyl to carbonate group. The olefinic carbons appeared at 133 ppm and 126 ppm indicating the presence of double bond. The compounds 2a and 2b showed characteristic fragment ion of m/z 294 due to cleavage around the carbonate moiety, confirming its presence. These carbonates did not give any ions corresponding to the M.W. (Fig. S2, ESI†).
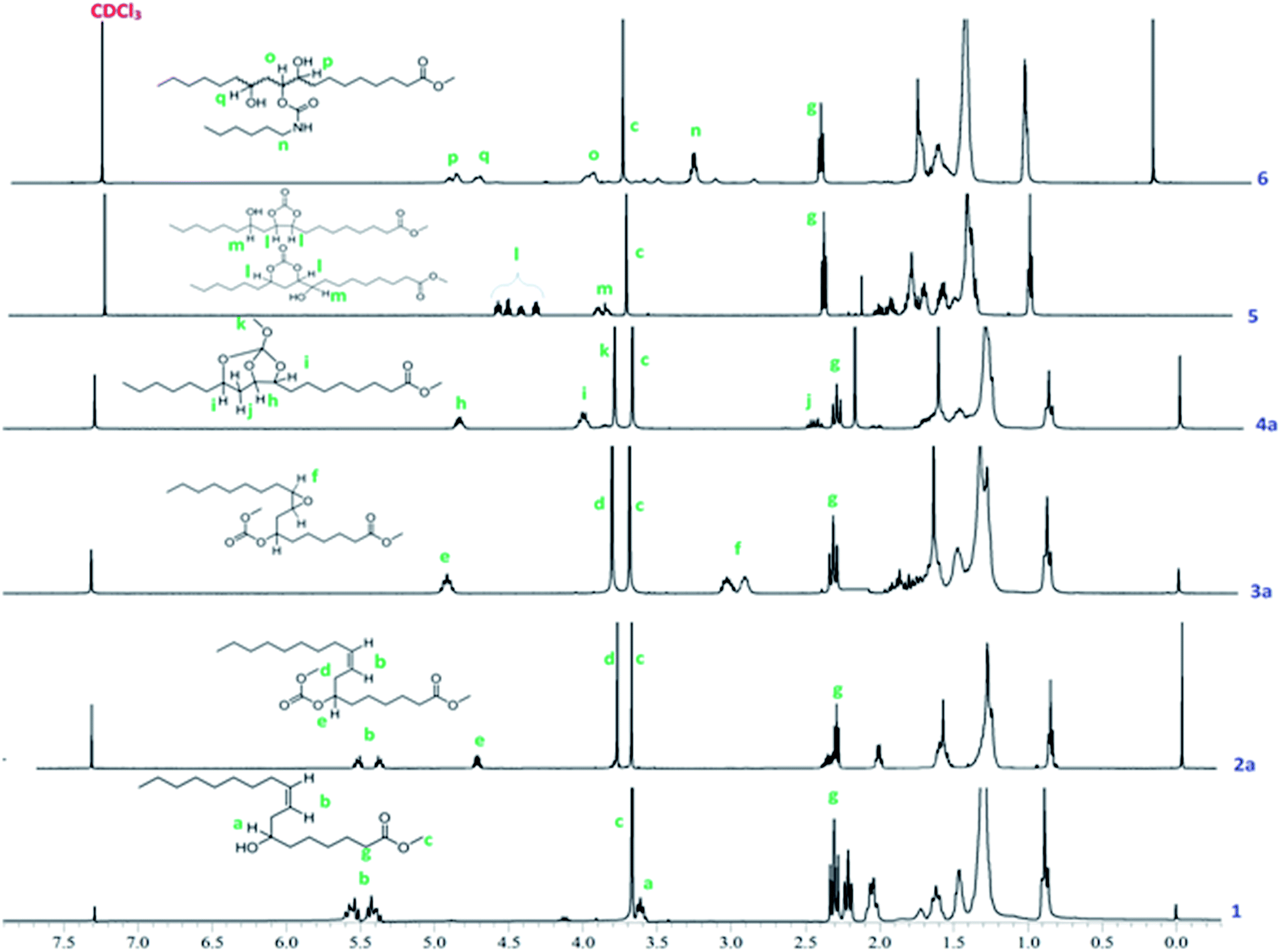 | ||
| Fig. 2 Stacked 1H NMR spectra of methyl ricinoleate 1, 2a, 3a, 4a, 5 and 6. | ||
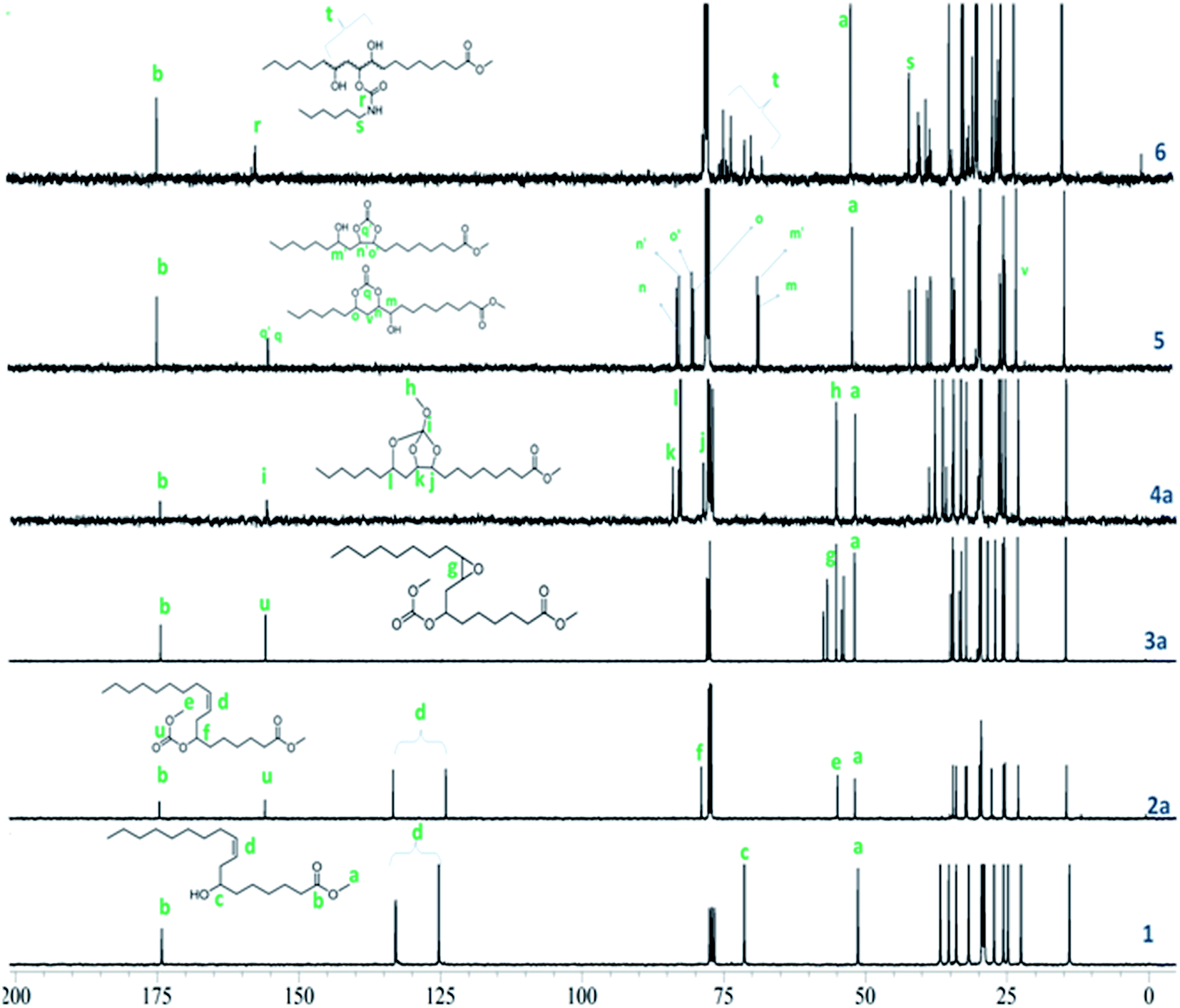 | ||
| Fig. 3 Stacked 13C NMR spectra of methyl ricinoleate 1, 2a, 3a, 4a, 5 and 6. | ||
The epoxidation of double bond in 2a and 2b was carried out using the modified performic acid method to obtain 3a and 3b.24–27 The structure of epoxy products 3a and 3b was confirmed by FT-IR, 1H NMR, 13C NMR studies and GC-MS (EI). The formation of the epoxy group was observed in the FT-IR spectra at 841 cm−1 (Fig. 1) and also observed disappearance of double bond band at 3007 cm−1 confirmed complete conversion of unsaturation into epoxy group. The protons on the epoxy group carbons appeared at 2.8–2.9 ppm for 3a (Fig. 2) and at 2.9–3.0 ppm for 3b in 1H NMR spectra confirmed the presence of epoxy group (Fig. S3, ESI†). New peaks were observed in 13C NMR spectra of epoxy carbons at 53–56 ppm in both the products (Fig. 3). The mass spectrum of 3a and 3b at m/z 310 correspond to cleavage at the same bond locations as observed for linear carbonates. Also observed [MH]+ ions such as m/z 387 and 401 for both the compounds (Fig. S4, ESI†).
The epoxy carbonate esters (3a and 3b) were treated with Lewis acids such as Sc(OTf)3, Cu(OTf)2, Zn(OTf)2, Yb(OTf)3, ZnBr2, BF3·OEt2, AlCl3, FeCl3 and TiCl4. Among these Lewis acids, triflates and ZnBr2 gave cyclic carbonates through SOC intermediate by intramolecular rearrangement. Whereas, BF3·OEt2 and AlCl3 ended the reaction only with SOC. Neither SOC nor cyclic carbonates were formed with FeCl3, and TiCl4. We obtained good yields of cyclic carbonates with Sc(OTf)3, Yb(OTf)3 and ZnBr2. Whereas, other catalysts such as copper triflate and zinc triflate resulted in incomplete conversions even after 10 h. The yields of the reaction products after column chromatography purifications are given in Table 1. The methodology was optimized with DCM. However, usage of ACN is greener compared to DCM. Due to this reason, we have also conducted experiments in ACN. However, no conversion to either cyclic carbonates or spiroorthocarbonates was observed with BF3·OEt2 and AlCl3 using ACN as solvent (Table 2). This could be due to the involvement of lone pair of electrons on the nitrogen of ACN with these Lewis acids rendering them deactivated. The cyclic carbonates and SOC were characterized by 1H NMR, 13C NMR, ESI-MS, GC-MS (EI), HRMS and FT-IR.
| Entry | Lewis acid | Equiv. | Time (h) | SOC (%) | CFAMEC (%) | ||
|---|---|---|---|---|---|---|---|
| Ib | IIb | IIIc | IVc | ||||
| a All reactions performed in DCM at room temperature.b Spiroorthocarbonates.c Cyclic carbonates. | |||||||
| 1 | Sc(OTf)3 | 0.5 | 1 | — | — | 90 | 80 |
| 2 | Sc(OTf)3 | 0.1 | 10 | — | — | 85 | 73 |
| 3 | Yb(OTf)3 | 0.5 | 1 | — | — | 83 | 70 |
| 4 | Yb(OTf)3 | 0.1 | 10 | — | — | 80 | 60 |
| 5 | ZnBr2 | 0.5 | 8 | — | — | 75 | 63 |
| 6 | ZnBr2 | 0.1 | 10 | — | — | 60 | 50 |
| 7 | Cu(OTf)2 | 0.5 | 8 | — | — | 20 | 10 |
| 8 | Zn(OTf)2 | 0.5 | 10 | — | — | 30 | 20 |
| 9 | BF3·OEt2 | 0.5 | 0.1 | 90 | 89 | — | — |
| 10 | BF3·OEt2 | 0.1 | 6 | 70 | 68 | — | — |
| 11 | AlCl3 | 0.5 | 10 | 85 | 80 | — | — |
| 12 | AlCl3 | 0.1 | 6 | 60 | 52 | — | — |
| 13 | FeCl3 | 0.5 | 6 | — | — | — | — |
| 14 | TiCl4 | 0.5 | 6 | — | — | — | — |
The Lewis acid catalyzed reaction is expected to proceed through the activation of epoxide by a Lewis acid. Thus activated epoxide undergoes nucleophilic ring opening with carbonyl group of the carbonate moiety resulting in the formation of oxocarbenium ion. A subsequent attack of anion (–OLA) on oxocarbenium ion would give the in situ intermediate spiroorthocarbonate (Scheme 2, 4a and 4b), which is subsequently converted into a mixture of five and six membered cyclic carbonates. We propose two pathways of ring-opening of the intermediate SOC resulting in five and six membered carbonates. The five membered cyclic carbonate (5a) was formed due to the intramolecular rearrangement of ring alkoxy to carbonyl there by the secondary alcohol formation is observed on C12 carbon. Whereas, six membered cyclic carbonate (5b) was formed by the intramolecular rearrangement of ring alkoxy to carbonyl resulting in the secondary alcohol formation at C9 carbon (Scheme 3). The existence of these two products was confirmed by 1H and 13C NMR spectroscopy (Fig. 2 and 3). When this mixture was analyzed by GC, two peaks were observed adjacent to each other with retention times 16.689 and 16.808 min and the peak area percentages were found to be 35.8% and 64.1% respectively. Computational studies were also carried out, where the –OC(O)O– bond angle calculations of five membered cyclic carbonate (110.12°) and six membered cyclic carbonate (118.57°) revealed that carbonyl group induces more strain in six membered ring compared to five membered ring carbonate (Fig. S5, ESI†). Five membered cyclic carbonate is stable than six membered cyclic carbonate because of less ring strain. From the computational studies, we assumed that the peak percentages 35.8% and 64.1% could be for six and five membered cyclic carbonates respectively. The reactions of epoxy esters and SOCs with Lewis acids generally give the corresponding spiroortho esters and cyclic carbonates,28,29 whereas the present reaction of 3a and 3b with Sc(OTf)3, Yb(OTf)3 and ZnBr2 resulted in cyclic carbonates. However, similar reactions performed with BF3·OEt2 and AlCl3 resulted in spiroorthocarbonates 4a and 4b quantitatively without the formation of cyclic carbonates. Sc(OTf)3 is a new type of a Lewis acid that is different from other Lewis acids such as AlCl3, BF3, TiCl4 and FeCl3 while [Ln(OTf)3] have similar properties. The catalytic activity of Sc(OTf)3 is higher compared to other lanthanide triflates.30
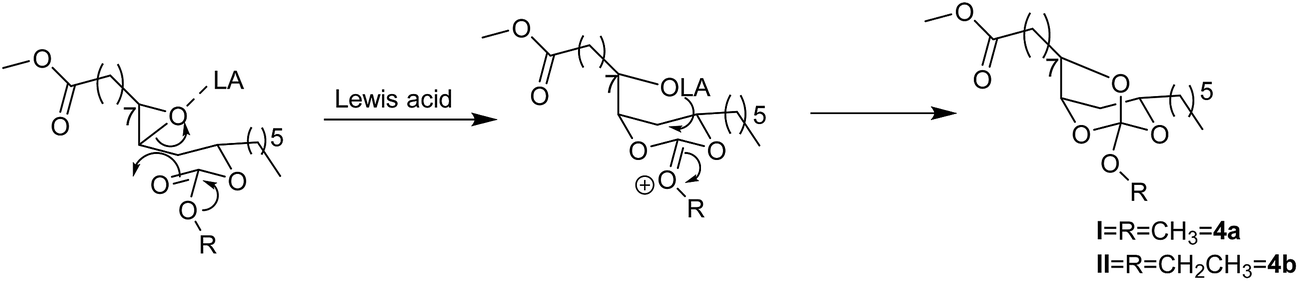 | ||
| Scheme 2 Lewis acid catalysed rearrangement reaction. | ||
 | ||
| Scheme 3 Cyclic carbonate for Lewis acid-induced epoxide rearrangement. | ||
The chemical structures of the cyclic carbonates were also confirmed by NMR analyses, as illustrated in Fig. 2 for five and six membered castor cyclic carbonate esters. The disappearance of proton NMR signals at 2.8, 3.02, and 3.63 ppm corresponding to epoxy protons and alkoxy carbonated protons and appearance of new proton NMR signals at 4.52, 4.26 (cyclic ring protons) and 3.77 ppm (CH–OH) confirmed the cyclic carbonate structure (Fig. 2). The 13C NMR spectroscopic data also revealed that the cyclic carbonate ester mixture contained both five and six membered rings. 13C NMR spectrum (Fig. 3) showed the presence of two carbonyl carbon atoms in adjacent position at 154.23, 154.41 ppm and two carbonate rings carbon signals in at 82.43, 82.02 ppm and 79.81, 79.47 ppm. This proved the presence of the two cyclic carbonates rings in the product mixture. The NMR values are well in agreement with the reported values for five membered fatty acid-based cyclic carbonate compounds.6–12,14 The other carbon chain values were in the range of 41.34–14.00 ppm. The cyclic carbonates formation was also confirmed from their mass spectral analysis, showing m/z 373 [M + H]+ and molecular formula of C20H37O6 obtained from HRMS and elemental analysis. FT-IR spectroscopy showed the presence of two bands at 1801 and 774 cm−1 corresponding to the cyclic carbonyl functions of five and six membered cyclic carbonates respectively. This also matched well with the reported data.6–12,14 Besides this, a large band appeared at 3497 cm−1 which is attributed to the OH vibrations (Fig. 1). The mass spectrum of cyclic carbonates showed a prominent ion at m/z 255 due to [M − CO2 − McL] + H indicating the presence of cyclic carbonates and [MH]+ ion (Fig. S6, ESI†).
Further, following the reported procedure,31 the carbamate (urethane) formation from cyclic carbonates in a reaction with n-hexylamine in DMAc at 50 °C confirmed their structure. The formation of carbamate was confirmed from their 1H NMR, 13C NMR, ESI-MS, GC-MS (EI), HRMS and FT-IR spectral analysis. The formation of the urethane linkage was confirmed by the presence of a signal at 3.17 ppm in proton NMR and at 41.08 ppm in carbon NMR corresponding to CH2–NHCOO of the urethane function (Fig. 2 and 3). Cyclic methine protons of cyclic carbonates at 4.26–4.52 ppm disappeared and new peaks appeared at 4.67, 4.82 ppm due to methine proton attached to hydroxyl group (–CH–OH). Also carbon NMR values at 70.1, 72.9, and 73.9 ppm confirmed the opening of cyclic methine carbons. The band at 1688 cm−1 and 1538 cm−1 in FT-IR spectrum proved the urethane formation from cyclic carbonates. Broad bands at 3497–3315 cm−1 were attributed to the NH and OH vibrations32 (Fig. 1). In the mass spectrum, the fragment ion was observed at m/z 255 which is attributed to the loss of [C7H14NO2]+ moiety with McLafferty rearrangement, indicating the presence of urethane bond (Fig. S7, ESI†).
The SOC was characterized with 1D and 2D-NMR spectroscopy, ESI-MS, HRMS, and FT-IR. 1H NMR spectra of SOC clearly showed proton signals for methine at 3.98, 3.99, and 4.70 ppm, and cyclic ring carbons appeared at 78.2–83.6 ppm in 13C NMR spectra (Fig. 2 and 3). Absence of epoxy ring protons at 2.9–3.0 ppm in 1H NMR and carbons at 53–56 ppm in 13C NMR confirmed the formation of SOCs. The 13C–1H HSQC spectra of 4a and 4b (Fig. S8 and S9, ESI†) indicated the presence of three methine protons 4.82, 3.99 and 3.98 ppm, one methyl group 0.86 ppm, one methoxy group at 3.77 ppm, and one ester methoxy group at 3.65 ppm. Through bond correlations were made by using the observed COSY cross peaks between He/Hf, He/Hd, He/Hd′, Hc/Hd, and Hc/Hd′ protons (Fig. S10 and S11, ESI†). Formation of new quaternary centre in compounds 4a and 4b was established from the observed characteristic long range 13C–1H HMBC correlations between Cb–He and Cb–Ha (Fig. S12 and S13, ESI†).
Absence of an epoxy band peak at 841 cm−1 and formation of a new band peak at 1200 cm−1 in FT-IR spectrum confirmed the formation of SOC for both 4a and 4b. These values matched well with the reported data.33 Mass spectral analysis of these compounds 4a, and 4b showed the molecular ion peaks (M + H) at m/z 387 and m/z 401 respectively. This was also further confirmed from their HRMS analysis and obtained the molecular formulas as C21H39O6 (387.27349) and C22H41O6 (401.28887) respectively for 4a, and 4b matched well with calculated values 387.27412 and 401.28977. Further, the mass spectrum showed the fragment ions for spiroorthocarbonates 4a and 4b at m/z 310 corresponding to the cleavage at the same bond locations as observed for epoxy carbonates. (Fig. S14, ESI†).
Thermo gravimetric analysis (TGA)
Thermo gravimetric analysis (TGA) of the synthesized cyclic carbonates (5) along with linear carbonates (2a) and epoxycarbonates (3a) was conducted to evaluate their thermal stability. We found that cyclic carbonates were more stable compared to their precursor epoxidised linear carbonates and linear carbonates. 50% weight loss decomposition temperature (Td50) of cyclic carbonate was found to be at 260 °C, whereas for linear carbonates and epoxy carbonates, Td50 was observed at 189, and 200 °C respectively (Fig. 4).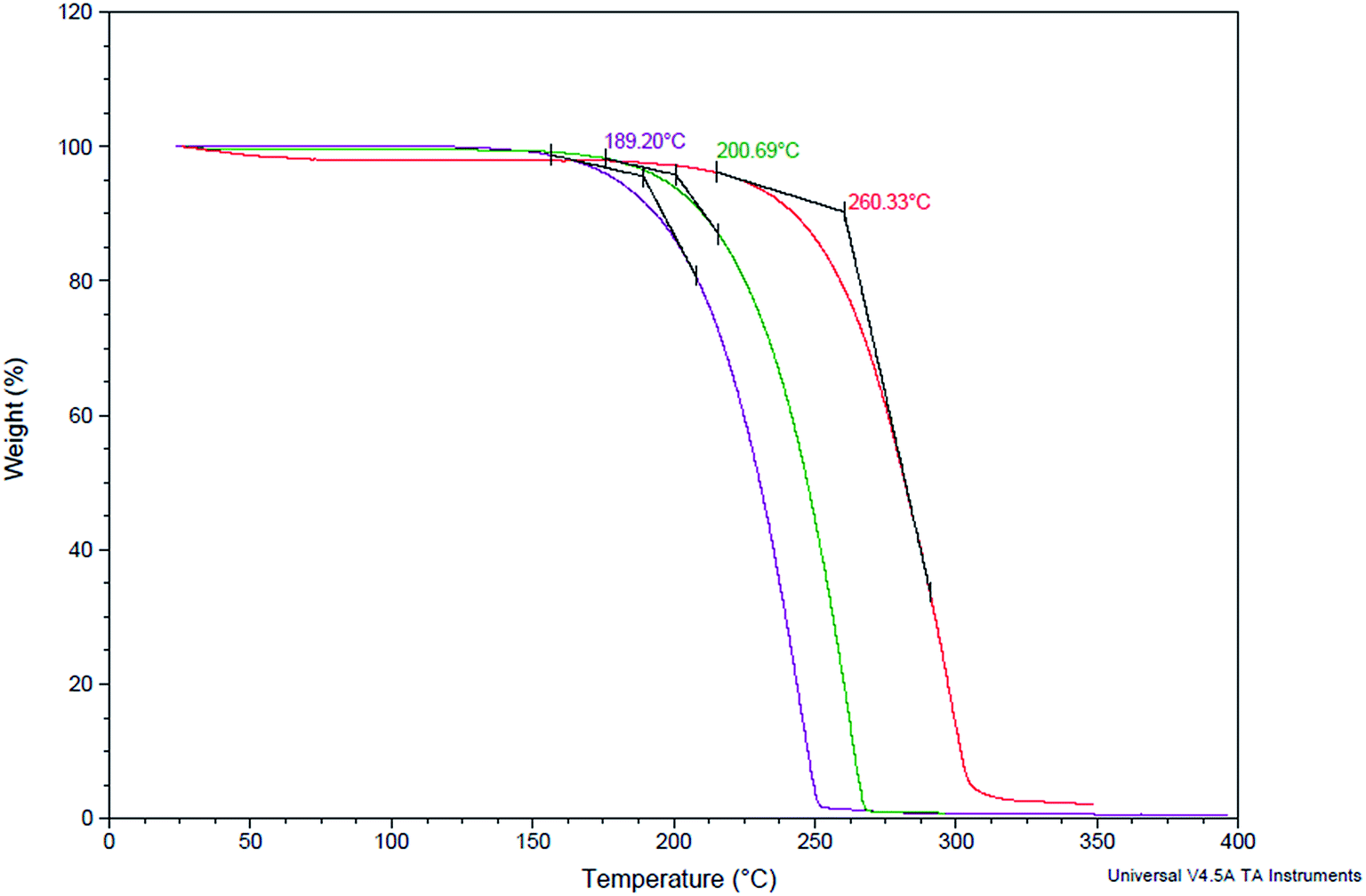 | ||
| Fig. 4 TGA curves of cyclic carbonated ester (red line), epoxy carbonate ester (green line), and linear carbonate ester (pink line). The samples show at 260, 200, and 189 °C, weight loss to 50% (heating rate 10 °C min−1, N2). | ||
Conclusions
In this article, we report synthesis of castor fatty acid methyl ester-based five and six membered cyclic carbonates following a three-step procedure for the first time. The first stage was the interchange reaction followed by epoxidation and finally Lewis acid catalyzed rearrangement. A number of Lewis acids were screened for the rearrangement reaction. Five Lewis acids were found to be promising with three of them resulting in good yields. The prepared novel carbonates can be potential precursors for biodegradable polymers.Acknowledgements
Naganna Narra thanks the University Grants Commission (UGC), New Delhi, India, for financial support through a Senior Research Fellowship (SRF). We would like to thank Dr Y. Soujanya and M. Srikanth for helpful suggestions in our computational calculations.References
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- B. H. Xu, J. Q. Wang, J. Sun, Y. Huang, J. P. Zhang, X. P. Zhang and S. J. Zhang, Green Chem., 2015, 17(1), 108–122 RSC.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12(9), 1514–1539 RSC.
- W. I. Riedeman, U.S. Pat., 2,826,591, March 11, 1958.
- W. I. Riedeman, U.S. Pat., 2,858,286, Oct 28, 1958.
- J. A. Kenar and I. D. Tevis, Eur. J. Lipid Sci. Technol., 2005, 107, 135–137 CrossRef CAS.
- B. Tamami, S. Sohn and G. L. Wilkes, J. Appl. Polym. Sci., 2004, 92, 883–891 CrossRef CAS.
- K. M. Doll and S. Z. Erhan, J. Agric. Food Chem., 2005, 53, 9608–9614 CrossRef CAS PubMed.
- K. M. Doll and S. Z. Erhan, Green Chem., 2005, 7, 849–854 RSC.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205–2213 RSC.
- M. Bähr and R. Mülhaupt, Green Chem., 2012, 14, 483–489 RSC.
- J. Langanke, L. Greiner and W. Leitner, Green Chem., 2013, 15, 1173–1182 RSC.
- L. Maisonneuve, A. L. Wirotius, C. Alfos, E. Grau and H. Cramail, Polym. Chem., 2014, 5, 6142–6147 RSC.
- B. Schäffner, M. Blug, D. Kruse, M. Polyakov, A. Köckritz, A. Martin, P. Rajagopalan, U. Bentrup, A. Brückner, S. Jung, D. Agar, B. Rüngeler, A. Pfennig, K. Müller, W. Arlt, B. Woldt, M. Grass and S. Buchholz, ChemSusChem, 2014, 7, 1133–1139 CrossRef PubMed.
- M. Alves, B. Grignard, S. Gennen, C. Detrembleur, C. Jerome and T. Tassaing, RSC Adv., 2015, 5(66), 53629–53636 RSC.
- H. Mutlu and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2010, 112, 10–30 CrossRef CAS.
- K. R. Kunduru, A. Basu, M. Haim Zada and A. J. Domb, Biomacromolecules, 2015, 16(9), 2572–2587 CrossRef CAS PubMed.
- A. Sammaiah, K. V. Padmaja and R. B. N. Prasad, Eur. J. Lipid Sci. Technol., 2015, 117, DOI:10.1002/ejlt.201500109.
- H. Mutlu, J. Ruiz, S. C. Solleder and M. A. R. Meier, Green Chem., 2012, 14, 1728–1735 RSC.
- A. Takagaki, K. Iwatani, S. Nishimura and K. Ebitani, Green Chem., 2010, 12, 578–581 RSC.
- A. S. More, D. V. Palaskar, E. Cloutet, B. Gadenne, C. Alfos and H. Cramail, Polym. Chem., 2011, 2, 2796–2803 RSC.
- K. M. Tomczyk, P. A. Guńka, P. G. Parzuchowski, J. Zachara and G. Rokicki, Green Chem., 2012, 14, 1749–1758 RSC.
- J. A. Kenar, G. Knothe and A. L. Copes, J. Am. Oil Chem. Soc., 2004, 81, 285–291 CrossRef CAS.
- K. M. Doll, G. B. Bantchev and R. E. Murray, ACS Sustainable Chem. Eng., 2013, 1, 39–45 CAS.
- B. K. Sharma, K. M. Doll and S. Z. Erhan, Green Chem., 2007, 9, 469–474 RSC.
- T. W. Findley, D. Swern and J. T. Scanlan, J. Am. Chem. Soc., 1945, 67, 412–414 CrossRef CAS.
- W. R. Schmitz and J. G. Wallace, J. Am. Oil Chem. Soc., 1954, 31, 363–365 CrossRef CAS.
- A. Ahmed, S. D. R. Christie, M. R. J. Elsegood and G. J. Pritchard, Chem. Commun., 2013, 49, 7489–7491 RSC.
- G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS.
- S. Kobayashi, Eur. J. Org. Chem., 1999, 15–27 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 162–168 CrossRef CAS.
- L. Maisonneuve, A. S. More, S. Foltran, C. Alfos, F. Robert, Y. Landais, T. Tassaing, E. Grau and H. Cramail, RSC Adv., 2014, 4, 25795–25803 RSC.
- J. Ge, M. Trujillo-Lemon and J. W. Stansbury, Macromolecules, 2006, 39, 8968–8976 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra00880a |
| This journal is © The Royal Society of Chemistry 2016 |